
Abstract
Hybrid lethality, a type of postzygotic reproductive barrier, is important for species. Discovering novel hybrid lethality cases and analyzing corresponding causal genes may provide new insights into the establishment and maintenance of reproductive isolation. In this study, we observed the hybrid lethality phenomena in a cross between two cabbage inbred lines, 09-211 and 09-222. Genetic analysis revealed that the hybrid lethality was controlled by two complementary dominant genes, BolC.HL1.a and BolC.HL2.a, from 09-211 and 09-222, respectively. Further analysis indicated that the two genes conform to the Bateson-Dobzhansky-Muller model. Fine mapping of hybrid lethal genes revealed that BolC.HL1.a was located on the C01 chromosome by Indels HL132 and HL134, with a genetic distance of 0.2 and 0.1 cM, respectively. The interval distance between the two markers was 101 kb. BolC.HL2.a was fine-mapped on the C04 chromosome by HL235 and HL234 at a distance of 0.3 and 0.3 cM, respectively. The physical distance was 70 kb. These findings lay the foundation for cloning the hybrid lethality genes in the future and contribute to our understanding of the molecular and evolutionary mechanisms of hybrid lethality in Brassica oleracea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reproductive isolation, which plays a vital role in speciation, restricts gene flow and accelerates genetic divergence between species (Yamamoto et al. 2010). These restrictive mechanisms can be divided into two types, namely pre-zygotic and post-zygotic barriers. Hybrid lethality is a postzygotic barrier that occurs after mating or fertilization and causes F1 hybrid death. In plants, hybrid lethal cases mainly occur in interspecific crosses such as Crepis (Hollingshead 1930), Nicotiana (East 1935; Kostoff 1930; Deverna et al. 1987; Yamada et al. 1999, 2000; Tezuka and Marubashi 2006; Tezuka 2012), Gossypium (Silow 1941; Brown and Menzel 1952; Phillips and Merritt 1972), Triticum(Sears 1944), Lycopersicon (Sawant 1956), Oryza (Chu and Oka 1972) and Lactuca (Jeuken et al. 2009); however, several lethal cases have also been observed in intraspecific crosses such as Triticum (Sax 1921; Caldwell and Compton 1943; Tsunewaki 1970; Nishikawa et al. 1974), Hordeum (Wiebe 1934), Vigna (Saunders 1952), Oryza (Sato and Hayashi 1983) and Arabidopsis (Bomblies et al. 2007; Chae et al. 2014). In this study, we report a causal case in which plants from a cross between two inbred cabbage lines (Brassica oleracea L.) died at the seedling stage. Symptoms of the observed hybrid lethality included yellowing, wilting, chlorosis, growth retardation, and death.
The genetic mechanism of hybrid lethality is theoretically explained by the Bateson-Dobzhansky-Muller (BDM) model (Dobzhansky 1937; Muller 1942). The BDM model includes a one locus and two loci model. Most hybrid lethal cases are caused by two complementary genes, which fit the two loci BDM model. The simple explanation of the two loci BDM model begins with a species that carries an “aabb” genotype. During evolution, the species splits into two isolated populations: in one population, an A allele becomes fixed and the genotype becomes “AAbb”, while in the other population, a B allele becomes fixed and the genotype becomes “aaBB”. The interactions between the two alleles A and B may cause lethality in their F1 progeny. The lethal mechanism is caused by genes that come from a common ancestor but that have diverged in the progeny (Song et al. 2009). In the common ancestor, these genes may work together without harm, but evolution has resulted in antagonism between the two (Dobzhansky 1937; Muller 1942; Brideau et al. 2006; Bomblies and Weigel 2007).
In addition to the research on genetic mechanisms, investigation of molecular mechanisms will provide a deeper understanding of hybrid lethality. The identification of lethal hybrid genes in Drosophila has revealed that hybrid male rescue (Hmr) in Drosophila simulans interacts with lethal hybrid rescue (Lhr) in D. melanogaster causing lethality in F1 hybrid males (Barbash et al. 2003; Brideau et al. 2006). Hybrid lethality in Arabidopsis reported by Bomblies et al. (2007) is caused by a NB-LRR disease resistance gene “homolog” and an allele of the DM2 locus. The deeper research of DM2 by Chae et al. (2014) revealed the DM2 locus is a highly variable cluster of NLR genes.
The completion of the B. oleracea genome sequence (Liu et al. 2014) provides insights into the dynamics of Brassica genome evolution and divergence and serves as an important resource for Brassica vegetable and oilseed crop breeding. The BRAD database (http://brassicadb.org/brad/) serves as an infrastructure to study the molecular function of genes and comparative genomics and to promote advances in molecular breeding within Brassica.
In this study, we report the identification of hybrid lethality from a cross between the inbred cabbage lines 09-211 and 09-222. Our genetic analyses revealed the genetic mechanisms and fine mapping of the hybrid lethal genes in cabbages.
Materials and methods
Plant materials
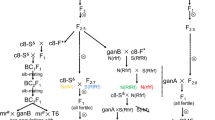
Six cabbage lines were used in this study: (1) The two inbred lines, 09-211 and 09-222, carrying the hybrid lethality gene, developed from commercial F1 varieties from Chinese Taipei and Japan, respectively; (2) the other four inbred lines without hybrid lethality genes were 87-534, 11-196, 96-109, and 96-100. The phenotypes, deductive genotypes, and origins of the inbred lines are presented in table S1. For the genetic analysis of the lethal hybrid genes, 13 different cross combinations were constructed. Likewise, for the mapping of lethal genes, five large segregation populations were obtained by a three-way cross. The segregation populations for BolC.HL1.a mapping were constructed by crossing 09-222 and the F1, which was first obtained by crossing 09-211 and 87-534. Likewise, 09-222 was first crossed with 87-534 to obtain the F1, which was then crossed with 09-211 to construct the segregation populations for BolC.HL2.a mapping. Additionally, the reciprocal cross between 09-211 and 09-222 was performed for lethal symptom observations.
Genetic analysis
The hybrid lethality phenotype was evaluated from the first true leaf stage and continued until the plants had reached maturity. Appearance of death characterized by chlorosis and wilting or seedling death was assigned to the lethal phenotype. Normal plants were marked as the normal phenotype. For all of the field tests, the corresponding F1 and the two parents were used as controls. The numbers of lethal and normal plants were counted in the F1 and segregation populations. The lethal ratios of the F1 and segregation populations were analyzed by a chi-squared test using SAS software (SAS Institute 2001).
DNA extraction, PCR amplification, and electrophoresis
Genomic DNA was extracted from the leaves of each individual from the mapping populations and their parents according to the modified cetyltrimethylammonium bromide protocol (Murray and Thompson 1980). The DNA concentration was estimated using a spectrophotometer (BioDrop, UK) and diluted to 40–50 ng/μl. PCR was performed in a reaction volume of 20 μl containing 4 μl template DNA (40–50 ng/μl), 1.6 μl dNTP (2.5 mM each), 2 μl 10 × PCR buffer (Mg2+ included), 0.8 μl forward and reverse primers (10 μM), 0.4 μl Taq DNA polymerase (2.5 U/μl), and 10.4 μl ddH2O. Amplification was carried out in an ABI Thermocycler (ABI) under the following conditions: 5 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 55 °C, and 45 s at 72 °C, and a final extension of 7 min at 72 °C. PCR products were separated in 8.0 % agarose gels at 160 V for 1.5 h, followed by silver staining.
BSA and InDel primer design
Twelve normal and 12 lethal plants were selected from the two lethal segregation populations to construct the pools. DNA was extracted from each plant to perform pooled segregant analysis (BSA) (Michelmore et al. 1991). Equal amounts of DNA from each of 12 samples were combined into a single pooled sample for genotyping; 1049 InDel primers and 1062 SSR primers were chosen for preliminary screening. 707 pairs of InDel primers were from Lv et al. (2014); 978 pairs of EST-SSR primers were from Chen et al. (2010); 342 pairs of InDel primers were designed in our laboratory according to re-sequencing data of two cabbage inbred lines 87-534 and 96-100, and 84 pairs of SSR primers were designed by the ‘02-12’ reference genome sequence of B. oleracea (http://brassicadb.org/brad/) (table S2). Markers showing polymorphisms between the two pools were used to analyze the normal plants of the lethal segregation populations. After BSA, the polymorphic markers were used for gene mapping.
The sequencing-by-synthesis method (Ronaghi et al. 1998) was used to re-sequence the parents at 10× coverage over the cabbage genome. This work was completed at Beijing Genomics Institute (BGI) (Shenzhen, China). The Indel primers were designed by comparing the re-sequencing data from 09-211 and 09-222 in comparison with the reference genome sequence ‘02-12’ from BRAD (http://brassicadb.org/brad/) (Liu et al. 2014). Using the sequencing-by-synthesis method, a total of 7.68 Gb Illumina pair-end reads were generated from parental line 09-211 as well as 7.74 Gb pair-end reads from parental line 09-222. The process used to detect InDel polymorphisms involved three steps according to Liu et al. (2013). Primers were designed with amplicon lengths of 80–160 bp, GC contents of 40–50 %, and Tm values of 52–56 °C.
Data analysis
Five segregation populations that derived from the three cross were employed to map the lethal genes. The first two segregation populations (09-211 × 87-534) × 09-222 and (09-222 × 87-534) × 09-211, consisting of 1840 and 1744 individuals, respectively, were used to map the lethal genes. The other three segregation populations (09-211 × 11-196) × 09-222, (09-222 × 11-196) × 09-211, and (09-222 × 96-100) × 09-211 consisting of 3049, 2915, and 2000 individuals, respectively, served as verification populations. To accurately count the recombinant plants, normal plants were chosen to avoid death as a result of environmental and human factors.
To analyze the phenotype and genotype, plants with normal phenotypes matching normal genotypes were recorded as “a” while plants with normal phenotypes but lethal genotypes were recorded as “h”. Genetic distances between markers were calculated by the Kosambi map function (Kosambi 1943). The genetic map was constructed in MapDraw (Liu and Meng 2003).
Results
Identification of the hybrid lethality phenomenon in cabbages
In the autumn of 2009, a number of cabbage crosses were made among various inbred lines, and their seeds were sown in the field in Beijing and characterized for a breeding program. Although at first development appeared normal, one cross between two elite inbred lines 09-211 and 09-222 exhibited 100 % seedling mortality, whereas the parents and F1 hybrids from other crosses all produced normal plants. To investigate this seedling mortality phenomenon, the same cross 09-211 × 09-222 and the reciprocal one 09-222 × 09-211 were obtained. Mortality occurred in both crosses at the seedling stage in two subsequent autumns. Therefore, this is an intraspecific hybrid lethality case in B. oleracea. To further investigate hybrid lethality symptoms, we sowed the F1 seeds from the 09-211 and 09-222 cross in the autumn in the next 2 years. The crossed seed itself appeared to be normal in size, plumpness, and color, as far as could be determined by eye. Additionally, the germination rate of 09-222 × 09-211, 09-222 and 10-260 × 09-211 was 100 ± 0.0, 92.7 ± 5.7, and 94.9 ± 4.2 %, respectively. There were no significant differences among the cross lethal combination with the other inbred lines or cross combinations. The seedlings from the cross initially grew and developed normally to the cotyledon stage. However, the lethal symptoms, e.g., retarded growth, wilting, and chlorosis, gradually appeared in all F1 seedlings; most died at the four-leaf stage and the remaining ones before the rosette stage in autumn. When the seeds of the lethal cross were sown in spring, seedling mortality mainly occurred at the two-leaf stage; 09-211 and 09-222 were developed from autumn cabbage F1 hybrids from Chinese Taipei and Japan, respectively. Fifteen inbred lines derived from Asian autumn cabbage F1 varieties were crossed with both 09-211 and 09-222, and the hybrids were screened for lethal genes, but no novel hybrid lethal line was found.
Genetic analysis of hybrid lethality
To investigate the genetic mechanism of hybrid lethality in B. oleracea, five cabbage inbred lines, 09-211,09-222, 87-534, 96-109, and 11-196, along with their resulting F1, F2, and other three-way crossed progenies, were grown in the field in the autumn of 2013. All of the parent plants and F1 hybrid crosses, except for the 09-211 and 09-222 reciprocal crosses, exhibited normal growth, as did the F2 plants derived from the normal F1 hybrids. The reciprocal F1 hybrids from 09-211 and 09-222 exhibited the same lethal phenotype with 100 % seedling mortality occurring before the rosette stage, indicating that the lethal trait is controlled by the interaction of nuclear genes from 09-211 and 09-222, while cytoplasmic genes were excluded. The six three-way crosses, e.g., F1 of 09-211 × 87-534 to 09-222, showed the same segregation results (Table 1). The segregation of 324 plants from (09-211 × 87-534) × 09-222 revealed 160 lethal and 164 normal individuals, which fits the expected ratio of 1:1, indicating that a pair of complementary dominant genes control the hybrid lethality. Likewise, the other three-way crossed populations, (96-109 × 09-211) × 09-222, (96-109 × 09-222) × 09-211, (09-222 × 87-534) × 09-211, (11-196 × 09-222) × 09-211, and (11-196 × 09-211) × 09-222, also segregated in a 1:1 (lethal:normal) ratio, as expected under the two dominant complementary genes model. The four-way crossed hybrid (96-109 × 09-211) × (11-196 × 09-222) segregated in a 1:3 (lethal:normal) ratio and also fit the expected genetic model (Table 1).
All of these data indicate that the lethal phenotype in the B. oleracea hybrids is controlled by two dominant complementary genes: one derived from 09-211 and the other from 09-222. According to the gene nomenclature proposed by Ostergaard and King (2008), the two complementary lethal genes were putatively named BolC.HL1.a from 09-211 and BolC.HL2.a from 09-222.
Fine mapping of BolC.HL1.a
To map the lethal BolC.HL1.a gene from 09-211, BSA was conducted on 324 individuals derived from the segregation population (09-211 × 87-534) × 09-222 to identify the polymorphic markers linked to the BolC.HL1.a gene. Among 1049 Indel primers and 1062 SSR primers that were distributed over the nine chromosomes, six pairs of primers including D25, D35, LTSSR44, D103, D105, and D107 (table S2) on chromosome C01 demonstrated a significant polymorphism between the pooled DNA samples from normal and lethal plants. The D107 exhibited a significant polymorphism between the two pooled DNA samples and performed consistently in each individual (Fig. 1). Besides, we used 324 individuals from segregation population (09-211 × 87-534) × 09-222 to confirm whether or not the polymorphic primers were in accordance with the phenotypes. It turned out that the genotypes of the six primers were in accordance with the phenotypes. The D107 surveyed all the 324 individuals between the two bulks, with 3 recombinants in 23 individuals (Fig. 2). Using the six markers, linkage analysis revealed that the lethal BolC.HL1.a gene was located on one side of D107. According to the reference genome (http://brassicadb.org/brad/), the chromosome C01 was 38.76 Mbp and the position of D107 was from 37,498,448 to 37,498,562 bp on the chromosome C01. Furthermore, comparing the whole-genome re-sequencing data for 09-211 and 09-222, 704 pairs of Indel primers were designed on chromosome C01. All of the primers were designed with either the insertion or deletion between 4 and 5 bp. According to the flanking marker D107, 58 pairs of primers (table S4) were chosen from the region between D107 and the chromosome C01 terminus. All of the primers were used in the BSA screening and 32 pairs of primers were polymorphic. The segregation population was subsequently used to examine the polymorphic markers to choose the co-dominant markers with good and stable amplifications for further analysis.

Polymorphisms of D107 in gene pools and 12 normal and lethal pooled individuals from the segregation population. M represents the 200-bp DNA ladder, NP normal pool, LP lethal pool. Lanes 1–12 normal pooled individuals; lanes 13–24 lethal pooled individuals

Polymorphisms of D107 in gene pools and partial individuals from the normal segregation population. M represents the 100-bp DNA ladder, NP normal pool, LP lethal pool. Lanes 1–23 normal individuals, lanes 1, 11, and 18 are three recombinants
To further refine the position of BolC.HL1.a, an additional large mapping population (09-211 × 87-534) × 09-222 containing a total of 1840 plants was chosen. The population containing 896 normal and 944 lethal plants fit the lethal ratio of 1:1 (χ 2 = 1.25 < χ 20.05 ). The flanking markers D107 and the farthest markers HL134 were used to survey all 896 normal individuals, which resulted in the detection of 19 recombinants. The polymorphic markers HL088, HL099, HL104, HL062, HL116, HL 130, HL132, and HL133 between D107 and HL134 were chosen to survey the 19 recombinants, which resulted in the detection of three recombinants in HL133 and one in HL134. Moreover, 1571 normal plants from population (09-211 × 11-196) × 09-222 were used to examine the accuracy of the result, which resulted in the detection of three recombinants in HL132 and two in HL134. Finally, the lethal gene BolC.HL1.a was fine mapped between HL132 and HL134.
According to the marker locations in the reference genome, BolC.HL1.a was located on chromosome C01. A genetic map comprising five InDel markers and the corresponding physical map was constructed (Fig. 3) using MapDraw (Liu and Meng 2003). The InDel markers HL132 and HL134 were closest to BolC.HL1.a, flanking the gene with genetic distances of 0.2 cM and 0.1 cM, respectively. The marker interval was 101 kb.

Genetic map of BolC.HL1.a on the left, units: cM; the corresponding physical map on the right, units: Mb
Fine mapping of BolC.HL2.a
The same strategy was applied to fine map BolC.HL2.a, which was found in 09-222. BSA was conducted on 336 plants from the segregation population (09-222 × 87-534) × 09-211 to identify the polymorphic markers linked to the BolC.HL2.a gene. Among 1049 Indel and 1062 SSR primers, 6 pairs of primers including InDel0110, InDel0112, InDel0114, D349, LTSSR342, and LTSSR346 (table S2, S3) on chromosome C04 exhibited significant polymorphism between the pooled DNA samples. Using the six pairs of primers to locate the lethal BolC.HL2.a gene, linkage analysis revealed that BolC.HL2.a was located between LTSSR346 and InDel0114.
According to the whole-genome re-sequencing results for 09-211 and 09-222, 879 pairs of Indel primers were designed on chromosome C04. According to the flanking marker LTSSR346 and InDel0114, 73 pairs of primers (table S5) were used in the BSA and obtained 33 polymorphic markers with good and stable amplifications for further analysis.
To further refine the position of BolC.HL2.a, an additional large mapping population (09-222 × 87-534) × 09-211 containing total 1744 plants was chosen. The population containing 926 normal plants and 818 lethal plants differed slightly from the lethal ratio of 1:1 (χ 2 = 6.69). The flanking marker LTSSR346 and the farthest marker HL249 were used to survey all 926 normal plants, which resulted in detection of 37 recombinants. The survey of the 37 recombinants with the polymorphic markers HL204, HL206, HL207, HL213, HL223, HL227, HL235, and HL249 between LTSSR346 and InDel01114 resulted in the detection of two recombinants in HL235 and one in HL213. Furthermore, 1388 normal plants from population (09-222 × 11-196) × 09-211 and 882 normal plants from (09-222 × 96-100) × 09-211 were used to narrow the region, which resulted in the detection of three recombinants in HL235 and two in HL234.
Finally, the lethal BolC.HL2.a gene was located between HL235 and HL234 by fine mapping. According to the marker locations in the reference genome, BolC.HL2.a was located at chromosome C04. A genetic map comprising seven InDel markers and the corresponding physical map was constructed (Fig. 4) using MapDraw (Liu and Meng 2003). The InDel markers HL235 and HL234 were closest to BolC.HL2.a, flanking the gene with genetic distances of 0.3 cM and 0.3 cM, respectively. The marker interval was 70 kb.

Genetic map of BolC.HL2.a on the left, units: cM; the corresponding physical map on the right, units: Mb
Discussion
Hybrid lethality in B. oleracea
Hybrid lethality plays an important role in reproductive isolation and speciation (Orr and Presgraves 2000). To date, hybrid lethality cases in plants have mainly been observed in interspecifics such as Gossypium and Nicotiana, and several intraspecific lethal cases occur in self-pollinated species such as Triticum, Hordeum, Vigna, and Arabidopsis. Cabbages (B. oleracea L. var. capitata L.) are cross-pollinated species and are one of the most cultivated vegetables worldwide. In this study, we identified intraspecific hybrid lethality between two inbred lines in B. oleracea. The lethal cross 09-211 × 09-222 exhibited symptoms of yellowing, wilting, chlorosis, and growth retardation mainly at the four-leaf stage, with 100 % seedling mortality occurring before the rosette stage in autumn in Beijing. The variation in the occurrence of seedling mortality within a cross indicated that the hybrid lethality might also be influenced by environmental factors. Temperature sensitivity has been reported in hybrid seedlings from lethal crosses; generally, hybrid lethality is suppressed at elevated temperatures, as observed in many plant species, including Nicotiana (Yamada et al. 1999; Yamada and Marubashi 2003; Tezuka and Marubashi 2006), Gossypium (Phillips 1977), Oryza (Saito et al. 2007), Lactuca (Jeuken et al. 2009), Arabidopsis (Bomblies et al. 2007), and intergeneric hybrids between Pyrus pyrifolia Nakai and Malus domestica Borkh (Inoue et al. 2003). We sowed the seeds from the lethal cross in spring and found that the lethal symptoms appeared at the two-leaf stage, which was a little earlier than observed in autumn. We speculate that the lower temperature accelerated hybrid seedling mortality in B. oleracea, as has been reported in other plant species. However, temperature sensitivity in hybrid lethality in B. oleracea is still unclear. For finding novel hybrid lethality materials to disclose the distribution of causal genes in B. oleracea, we are screening more inbred lines by crossing with 09-211 and 09-222, respectively.
In our study, genetic analysis indicated that the lethal trait was controlled by two dominant complementary genes: one BolC.HL1.a on C01 in 09-211 and the other BolC.HL2.a on C04 in 09-222. These two genes, BolC.HL1.a and BolC.HL2.a, conform to the Bateson-Dobzhansky-Muller model in which hybrid incompatibilities, including hybrid death and sterility, are caused by genes that have evolved from a common ancestor but diverged in each of the species (Dobzhansky 1937; Muller 1942; Brideau et al. 2006; Bomblies and Weigel 2007). The genetic model of hybrid lethality in B. oleracea is similar to that in Arabidopsis (Bomblies et al. 2007) and Gossypium (Song et al. 2009). According to the BDM model, we proposed a model in which the causal genes in B. oleracea evolved from a common ancestor, similar to the normal inbred lines such as 87-534 and 96-109, with the genotype “aabb.” During evolution, “aa” diverged in 09-211 to “AA” and “bb” diverged in 09-222 to “BB”. 09-211 and 09-222 are elite cabbage inbred lines and grow normally, while resulting in seedling mortality in the F1 hybrids. To our knowledge, this is the first intraspecific hybrid lethality case in B. oleracea, and the interaction of causal genes may provide new insights into the hybrid lethality mechanism. For cabbage breeders, hybrid lethality presents a barrier to the introduction of desirable genes into other inbred lines. If hybrid lethality can be overcome, a larger germplasm pool will be available in breeding programs (Tezuka 2012). In addition, the development of lethal molecular markers will avoid random crosses causing lethality.
Mechanisms of hybrid lethality
Cloning of the causal genes can help to clarify the molecular mechanisms of hybrid lethality. As an initial step to clone the causal genes following the BDM model, some of the lethal genes have been located on linkage maps, including Oryza (Ichitani et al. 2001, 2007), Triticum (Chu et al. 2006), Arabidopsis (Bomblies et al. 2007; Chae et al. 2014), Gossypium (Song et al. 2009), and Lactuca (Jeuken et al. 2009). In animals, several BDM genes have been cloned in Drosophila that exhibit the common features of rapid evolution and positive Darwinian selection (Barbash et al. 2003; Presgraves et al. 2003; Coyne and Orr 2004; Orr 2005; Brideau et al. 2006). The causative genes for hybrid lethality in plants have been identified as follows: an allele of the DANGEROUS MIX 1 (DM1) locus that encodes a disease resistance (R) gene interacts with an allele of DM2 locus, which is a highly variable cluster of NLR genes in Arabidopsis, indicates an autoimmunity-like response as a potential gene-flow barrier in plants (Bomblies et al. 2007; Bomblies and Weigel 2007; Chae et al. 2014); the gene Rin4 in Lactuca interacts with multiple R gene products, and this interaction results in hypersensitive resistance to Pseudomonas syringae (Jeuken et al. 2009). These two findings have revealed that the hybrid lethality mechanism is related to disease resistance in plants. However, whether or not other hybrid lethality cases are explained by disease resistance is uncertain.
Different hybrid lethality examples can provide more evidence to help us understand the evolution of post-zygotic reproductive isolation and speciation. In cabbages, no hybrid lethality-causing genes have been tagged with molecular markers. The current study is the first to locate two BDM genes on different chromosomes in B. oleracea. According to the cabbage reference genome in the BRAD database (http://brassicadb.org/brad/), this mapping region of BolC.HL1.a contains 17 genes including 11 annotated and 6 unknown genes; the annotation and orthologous gene analyses are presented in table S6. Likewise, the BolC.HL2.a mapping region contains nine genes including four annotated and five unknown genes; the analyses of these genes are presented in table S7. Based upon the literature, the causal genes may have diverse functions, and the hybrid lethality mechanisms are potentially diverse. Therefore, annotation analysis of these genes from BRAD cannot yet determine the candidate BolC.HL1.a and BolC.HL2.a genes. Cloning of the BolC.HL1.a and BolC.HL2.a genes might lead to a better understanding of the molecular mechanism controlling hybrid lethality and the evolutionary dynamics of reproductive isolation genes in B. oleracea. In the future, we plan to clone the causal BolC.HL1.a and BolC.HL2.a genes by narrowing the mapped region and comparing the gene sequences.
References
Barbash DA, Siino DF, Tarone AM, Roote J (2003) A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc Natl Acad Sci USA 100(9):5302–5307
Bomblies K, Weigel D (2007) Hybrid necrosis: autoimmunity as a common barrier to gene-flow in plants. Nat Rev Genet 8:382–393
Bomblies K, Lempe J, Epple P, Warthmann N, Lanz C, Dangl JL, Weigel D (2007) Autoimmune response as a mechanism for a Dobzhansky–Muller-type incompatibility syndrome in plants. PLoS Biol 5(9):1962–1972
Brideau NJ, Flores HA, Wang J, Maheshwari S, Wang X, Barbash DA (2006) Two Dobzhansky–Muller genes interact to cause hybrid lethality in Drosophila. Science 314:1292–1295
Brown MS, Menzel MY (1952) Additional evidence on the crossing behavior of Gossypium gossypioides. Bull Torrey Bot Club 79(4):285–292
Caldwell RM, Compton LE (1943) Complementary lethal genes in wheat: causing a progressive lethal necrosis of seedlings. J Hered 34:67–70
Chae E, Bomblies K, Kim ST, Karelina D, Zaidem M, Ossowski S, Martín-Pizarro C, Laitinen RAE, Rowan BA, Tenenboim H, Lechner S, Demar M, Habring-Müller A, Lanz C, Rätsch G, Weigel D (2014) Species-wide genetic incompatibility analysis identifies immune genes as hot spots of deleterious epistasis. Cell 159(6):1341–1351
Chen C, Zhuang M, Li K, Liu Y, Yang L, Zhang Y, Chen F, Sun P, Fang Z (2010) Development and utility of EST-SSR marker in cabbage. Acta Hortic Sin 37(2):221–228 (in Chinese)
Chu YE, Oka HI (1972) The distribution and effects of genes causing F1 weakness in Oryza breviligulata and O glaberrima. Genetics 70:163–173
Chu CG, Faris JD, Friesen TL, Xu SS (2006) Molecular mapping of hybrid necrosis genes Ne1 and Ne2 in hexaploid wheat using microsatellite markers. Theor Appl Genet 112:1374–1381
Coyne JA, Orr AH (2004) Speciation. Sinauer Associates Inc, Sunderland
Deverna JW, Myers JR, Collins GB (1987) Bypassing prefertilization barriers to hybridization in Nicotiana using in vitro pollination and fertilization. Theor Appl Genet 73:665–671
Dobzhansky TH (1937) Genetics and the origin of species. Columbia University Press, New York
East EM (1935) Genetic reactions in Nicotiana. I. compatibility. Genetics 20:0403–0413
Hollingshead LA (1930) A lethal factor in Crepis effective only in an interspecific hybrid. Genetics 15:114–140
Ichitani K, Fukuta Y, Taura S, Sato M (2001) Chromosomal location of Hwc2, one of the complementary hybrid weakness genes, in rice. Plant Breed 120:523–525
Ichitani K, Namigoshi K, Sato M, Taura S, Aoki M, Matsumoto Y, Saitou T, Marubashi W, Kuboyama T (2007) Fine mapping and allelic dosage effect of Hwc1, a complementary hybrid weakness gene in rice. Theor Appl Genet 114:1407–1415
Inoue E, Sakuma F, Kasumi M, Hara H, Tsukihashi T (2003) Effect of high-temperature on suppression of the lethality exhibited in the intergeneric hybrid between Japanese pear (Pyrus pyrifolia Nakai) and apple (Malus × domestica Borkh.). Sci Hortic 98:385–396
Jeuken MJW, Zhang NW, Mchale LK, Pelgrom K, den Boer E, Lindhout P, Michelmore RW, Visser RG, Niks RE (2009) Rin4 causes hybrid necrosis and race-specific resistance in an interspecific lettuce hybrid. Plant Cell 21:3368–3378
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Eugen
Kostoff D (1930) Ontogeny, genetics and cytology of Nicotiana hybrids. Genetica 12:33–139
Liu R, Meng J (2003) Map draw: a Microsoft excel macro for drawing genetic linkage maps based on given genetic linkage data. Hereditas 25:317–321 (in Chinese)
Liu B, Wang Y, Zhai W, Deng J, Wang H, Cui Y, Cheng F, Wang X, Wu J (2013) Development of InDel markers for Brassica rapa based on whole-genome re-sequencing. Theor Appl Genet 126:231–239
Liu S, Liu Y, Yang X et al (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. doi:10.1038/ncomms4930
Lv H, Wang Q, Zhang Y, Yang L, Fang Z, Wang X, Liu Y, Zhuang M, Lin Y, Yu H, Liu B (2014) Linkage map construction using InDel and SSR markers and QTL analysis of heading traits in cabbage. Mol Breed 34:87–98
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA 88:9828–9832
Muller HJ (1942) Isolating mechanisms, evolution, and temperature. Biol Symp 6:71–124
Murray HG, Thompson WF (1980) Rapid isolation of high molecular weight DNA. Nucleic Acids Res 8:4321–4325
Nishikawa K, Mori T, Takami N, Furuta Y (1974) Mapping of progressive necrosis genes, Ne1 and Ne2 of common wheat by the telocentric method. Japan J Breed 24:277–281
Orr HA (2005) The genetic theory of adaptation: a brief history. Nat Rev Genet 6:119–127
Orr HA, Presgraves DC (2000) Speciation by postzygotic isolation: forces, genes and molecules. BioEssays 22:1085–1094
Ostergaard L, King G (2008) Standardized gene nomenclature for the Brassica genus. Plant Methods 4:10
Phillips LL (1977) Interspecific incompatibility in gossypium. IV. Temperature-conditional lethality in hybrids of G. klotzschianum. Bot Soc Am 64:914–915
Phillips LL, Merritt JF (1972) Interspecific Incompatibility in Gossypium. I. Stem histogenesis of G. hirsutum × G. gossypioides. Am J Bot 59:203–208
Presgraves DC, Lakshmi B, Abmayr SM, Allen OH (2003) Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature 423:715–719
Ronaghi M, Uhlén M, Nyren P (1998) A sequencing method based on real-time pyrophosphate. Science 281:363–365
Saito T, Ichitani K, Suzuki T, Marubashi W, Kuboyama T (2007) Developmental observation and high temperature rescue from hybrid weakness in a cross between Japanese rice cultivars and Peruvian rice cultivar ‘Jamaica’. Breed Sci 57:281–288
SAS Institute (2001) SAS/SYSTAT user’s guide. Version 8. SAS Inst Inc, Cary
Sato YI, Hayashi K (1983) Distribution of the complementary genes causing F1 weakness in the common rice and its wild relatives I. L-2-a gene in Asian native cultivars. Jpn J Genet 58:411–418
Saunders AR (1952) Complementary lethal genes in the cowpea. S Afr J Sci 48:195–197
Sawant AC (1956) Semilethal complementary factors in a tomato species hybrid. Evolution 10:93–96
Sax K (1921) Sterility in wheat hybrids. I. Sterility relations and endosperm development. Genetics 6:399–416
Sears ER (1944) Inviability of intergeneric hybrids involving Triticum monococcum and T. aegilopoides. Genetics 29:113–127
Silow RA (1941) The comparative genetics of Gossypium anomalum and the cultivated Asiatic cottons. J Genet 42:259–358
Song L, Guo WZ, Zhang TZ (2009) Interaction of novel Dobzhansky–Muller type genes for the induction of hybrid lethality between Gossypium hirsutum and G. barbadense cv. Coastland R4–4. Theor Appl Genet 119:33–41
Tezuka T (2012) Hybrid lethality in the genus Nicotiana. Botany Intech, pp 191–210
Tezuka T, Marubashi W (2006) Hybrid lethality in interspecific hybrids between Nicotiana tabacum and N. suaveolens: evidence that the Q chromosome causes hybrid lethality based on Q chromosome-specific DNA markers. Theor Appl Genet 112:1172–1178
Tsunewaki K (1970) Necrosis and chlorosis genes in common wheat and its ancestral species. Seiken Ziho 22:67–75
Wiebe GA (1934) Complementary factors in barley giving a lethal progeny. J Hered 25:272–274
Yamada T, Marubashi W (2003) Overproduced ethylene causes programmed cell death leading to temperature-sensitive lethality in hybrid seedlings from the cross Nicotiana suaveolens × N. tabacum. Planta 217:690–698
Yamada T, Marubashi W, Niwa M (1999) Detection of four lethality types in interspecific crosses among Nicotiana species through the use of three rescue methods for lethality. Breed Sci 49:203–210
Yamada T, Marubashi W, Niwa M (2000) Apoptotic cell death induces temperature-sensitive lethality in hybrid seedlings and calli derived from the cross of Nicotiana suaveolens × N. tabacum. Planta 211:614–622
Yamamoto E, Takashi T, Morinaka Y, Lin SY, Wu JZ, Matsumoto T, Kitano H, Matsuoka M, Ashikari M (2010) Gain of deleterious function causes an autoimmune response and Bateson–Dobzhansky–Muller incompatibility in rice. Mol Genet Genomics 283:305–315
Acknowledgments
We thank Dr. Xueyuan Cao at St. Jude Children’s Research Hospital, USA, for his comments and suggestions on writing the manuscript. This work was financially supported by grants from the National Natural Science Foundation of China (31372071, 31572139), the earmarked fund for the Modern Agro-Industry Technology Research System, China (nycytx-35-gw01). This work was performed in the Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, Beijing 100081, People’s Republic of China.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hu, Y., Xue, Yq., Liu, Js. et al. Hybrid lethality caused by two complementary dominant genes in cabbage (Brassica oleracea L.). Mol Breeding 36, 73 (2016). https://doi.org/10.1007/s11032-016-0498-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0498-3