
Abstract
Main conclusion
This study shows that NgRBP suppresses both local and systemic RNA silencing induced by sense- or double-stranded RNA, and the RNA binding activity is essential for its function.
To counteract host defence, many plant viruses encode viral suppressors of RNA silencing targeting various stages of RNA silencing. There is increasing evidence that the plants also encode endogenous suppressors of RNA silencing (ESR) to regulate this pathway. In this study, using Agrobacterium infiltration assays, we characterized NgRBP, a glycine-rich RNA-binding protein from Nicotiana glutinosa, as an ESR. Our results indicated that NgRBP suppressed both local and systemic RNA silencing induced by sense- or double-stranded RNA. We also demonstrated that NgRBP could promote Potato Virus X (PVX) infection in N. benthamiana. NgRBP knockdown by virus-induced gene silencing enhanced PVX and Cucumber mosaic virus resistance in N. glutinosa. RNA immunoprecipitation and electrophoretic mobility shift assays showed that NgRBP bound to GFP mRNA, dsRNA rather than siRNA. These findings provide the evidence that NgRBP acts as an ESR and the RNA affinity of NgRBP plays the key role in its ESR activity. NgRBP responds to multiple signals such as ABA, MeJA, SA, and Tobacco mosaic virus infection. Therefore, it could participate in the regulation of gene expression under specific conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
RNA silencing is a conserved surveillance mechanism that plays a key role in defending plants against invasive nucleic acids (Hannon 2002; Himber et al. 2003). Replication intermediates or folded viral RNAs activate RNA silencing. They are then cleaved by Dicer-like proteins (DCL) into small interfering RNAs (siRNAs) (Lichner et al. 2003; Baulcombe 2004; Susi et al. 2004). These siRNAs are incorporated into the AGO-containing RNA-induced silencing complex (RISC). Sequence-specific cleavage of target RNAs then follows (Schwarz et al. 2003; Tomari and Zamore 2005). In higher plants, RNA silencing plays a critical role in antiviral defence. To counteract RNA silencing, viruses encode RNA silencing suppressors (Baulcombe 2002). To date, RNA-silencing suppressors (RSSs) have been reported which target various effector molecules in RNA silencing (Moissiard and Voinnet 2004; Csorba et al. 2015; Daròs 2017; Yang and Li 2018). For instance, the p19 protein encoded by Tomato bushy stunt virus is a potent viral suppressor of RNA silencing (VSR) preventing the spread of the silencing signal by binding 21–25 nt siRNA duplexes (Vargason et al. 2003; Ye et al. 2003). In this way, it inhibits a downstream step involving the cleavage of cognate RNAs (Silhavy et al. 2002). P6, a VSR encoded by Cauliflower mosaic virus, interferes with viral siRNA production by interacting with double-stranded RNA-binding protein 4 (DRB4), an essential partner of DCL4 (Haas et al. 2008). Plants also encode endogenous RNA-silencing suppressors (ESRs) which function in the defence–counterdefence arm race between host plants and viruses. The first-characterized ESR, Ntrgs-CaM, was proven to interact with HC-Pro, a VSR encoded by Tobacco etch virus, using the yeast two-hybrid system (Anandalakshmi et al. 2000). Over the following decade, several plant ESRs were identified, including RLI2, FIERY1, XRN2, XRN3 and XRN4 from Arabidopsis thaliana (Gazzani et al. 2004; Sarmiento et al. 2006; Gy et al. 2007), and rgs-CaM from Nicotiana benthamiana (Li et al. 2014). Nevertheless, little is known about their modes of action.
Certain RSSs bind siRNA duplexes or double-stranded RNA (dsRNA) (Lakatos et al. 2006; Mérai et al. 2006). This process probably can be regulated by RNA-binding proteins (RBPs). RBPs are commonly referred to as RNA chaperones (Lorsch 2002). They have multiple members and are ubiquitous among various plant species. They directly or indirectly guide various aspects of post-transcriptional modification by interacting with specific targeted mRNAs. These interactions always require several conserved RNA-binding domains such as the RNA recognition motif (RRM), the K homology domain (KH), and the double-stranded RNA-binding domain (dsRBD) (Burd and Dreyfuss 1994). Glycine-rich RNA-binding proteins (GRPs) have two distinct conserved domains: RRM and glycine-rich domain (GD) (Gómez et al. 1988). The RRM has an octamer, ribonucleoprotein domain I (RNPI), and a hexamer, RNPII. Both of these are highly homologous. GD typically contains 2–5 glycine repeats bordered by tyrosine and/or arginine. It has been reported to be involved in protein–protein interactions (Steinert et al. 1991). The first GRP was found in maize (Gómez et al. 1988). Since then, the genes encoding homologous proteins have been consecutively isolated from a broad range of plant species such as Medicago sativa (Ferullo et al. 1997), A. thaliana (Carpenter et al. 1994) and Sorghum bicolor (Aneeta et al. 2002). Studies show that the circadian clock regulating RNA-binding protein AtGRP7, feedback regulates the expression of itself and its homologous gene AtGRP8. The binding of AtGRP7 with its own precursor mRNA (pre-mRNA) promotes its alternative splicing, and the alternative transcripts are degraded by nonsense-mediated mRNA decay (NMD). It is believed that AtGRP7 also acts on other target genes through a similar mechanism (Staiger et al. 2003; Schöning et al. 2008). In addition, AtGRP7 also participates in cold response as an RNA chaperone, and the mutant plants are more sensitive to cold. OsGRP1 and OsGRP4 can restore the growth defects of Atgrp7 under cold stress, while OsGRP6 can enhance the frost resistance of Atgrp7. This indicates that the function of GRPs is somewhat conserved in dicotyledon and monocotyledon (Kim et al. 2010).
Compelling evidence indicates that GRPs mediate post-transcriptional regulation of RNA metabolism, including pre-mRNA splicing, mRNA transport, and mRNA translation. These findings present new regulatory strategies for both pathogen infection and plant defence (Fu et al. 2007; Qi et al. 2010; Jeong et al. 2011). Naqvi et al. (1998) discovered that Tobacco mosaic virus (TMV) induced the glycine-rich RNA-binding protein gene from N. glutinosa NgRBP at 24 h post-inoculation. Therefore, NgRBP may participate in plant-virus interactions. In this study, we showed that NgRBP could suppress the RNA silencing induced by sense RNA or dsRNA and prevent silencing from spreading systemically. We revealed that the RSS activity of NgRBP is associated with its RNA binding ability, and the 47th arginine residue is essential for its function.
Materials and methods
Plant materials and plasmid constructs
Wild-type Nicotiana glutinosa, N. benthamiana, and GFP-transgenic N. benthamiana 16c line (from Dr. David Baulcombe, University of Cambridge, UK) (Voinnet et al. 1998) were raised in a greenhouse at 24 °C under a 16-h light/8-h dark photoperiod. The full-length open reading frame (ORF) 471-bp segment of NgRBP (Accession no. AF005359) was amplified from N. glutinosa total RNA by RT-PCR using DNA Polymerase High Fidelity (HiFi) (TransGen, Beijing, China). The PCR product was ligated to a pMD-18T vector to generate pMD-NgRBP. All NgRBP mutants including ΔRNPI, ΔRNPII and R47A were produced by reverse PCR from the entire plasmid pMD-NgRBP, using the primer pairs that contained the corresponding nucleotide substitution or deletion (Table S1). The constructs were cloned into the binary vector pBI121 between the 35S promoter and the Nos terminator (Chen et al. 2003). For transient expression in 16c plants, 35S-p19, 35S-GFP, and 35S-dsGFP were constructed as previously described (Jing et al. 2011). To generate NgRBP carrying Potato Virus X (PVX) vector, NgRBP was inserted into the Cla I/Not I site in the PVX vector (pGR106) (Voinnet et al. 2000). All fragments generated by PCR were confirmed by DNA sequencing. The recombined plasmids were transformed by the freeze–thaw method into Agrobacterium strain GV3101 which contains the helper plasmid pJIC SA Rep (Höfgen and Willmitzer 1988).
Co-infiltration and GFP imaging
The 16c plants expressing GFP were infiltrated at five- or six-leaf stage with Agrobacterium GV3101 carrying the aforementioned constructs using the previously described method (Brigneti et al. 1998). Each Agrobacterium culture (OD600 = 1.0) was incubated for 3 h and then mixed with other culture (s) in a 1:1 (v/v) ratio prior to infiltration. Local and systemic RNA silencing were determined by observing GFP fluorescence both in the infiltrated and newly emerging leaves under long-wavelength (365 nm) UV light (Spectroline Model SB-100P/A; Spectronics Corporation, Lexington, KY, USA) and photographed with a Fujifilm FinePix S8000fd digital camera (Fujifilm Holdings Corporation). More than five plants were tested per experimental condition.
Virus-induced gene silencing (VIGS)
For the NgRBP VIGS, TRV-based vectors were used as previously described (Chung et al. 2004). To ensure specific silencing of NgRBP, the full-length CDS was inserted into the TRV2 vector to generate the pTRV2:NgRBP, and there is no homologous gene in N. glutinosa by NCBI Blast. The pTRV1, pTRV2:00 (empty vector), and pTRV2:NgRBP constructs were transformed into Agrobacterium GV3101. The Agrobacterium cultures were resuspended in infiltration buffer containing 10 mM MgCl2, 10 mM Mes, and 200 mM acetosyringone (pH 5.6; OD600 = 0.8). Agrobacterium culture harbouring pTRV1 was mixed with pTRV2:00 or pTRV2:NgRBP in a 1:1 ratio and then infiltrated into the lower leaves of 3-week-old N. glutinosa plants. The infiltrated plants were then placed in an illuminated incubator at 24 °C and 70% RH under a long-day (16-h light/8-h dark) photoperiod. After 15 days post-inoculation (dpi), the empty vector control plants and the NgRBP-silenced plants were used in Real-time quantitative polymerase chain reaction (RT-qPCR) and PVX or CMV inoculation assays.
For virus inoculation, 1 g of PVX (PVX-HM isolate, GenBank GQ863228) or CMV (CMV-SD1 isolate, GenBank AY792596)-infected N. tabacum leaves were ground in 1 mL of 5 mM phosphate buffer, pH 7.2. Control plants and the NgRBP-silenced plants at six-leaf stage were inoculated by rubbing leaves with freshly prepared sap. Inoculated plants were grown in an insect-free greenhouse at 24 °C and the viral symptom was monitored. Each experiment was replicated three times and each experiment included five independent plants.
RT-qPCR analysis
Total RNAs were isolated from leaves using Trizol reagent (TaKaRa, Shiga, Japan) according to the manufacturer′s instructions and treated with DNase I at 37 °C for 30 min prior to reverse transcription. cDNA was synthesized from 1 μg of total RNA using TIANScript RT Kit (Tiangen, Beijing, China). RT-qPCR was performed using Talent SYBR Green Kit (Tiangen, Beijing, China). The ssRUBP gene was used as the internal reference. Each reaction was conducted in triplicate and repeated three times. The results were analysed by Bio-Rad CFX Manager software (Bio-Rad, California, USA).
Total RNA and siRNA Northern blot analysis
Total and low molecular weight RNAs were extracted from leaves as described previously (Jing et al. 2011). 20 µg total RNA aliquot of each sample was separated on 1% formaldehyde agarose gels and transferred to Hybond-N+ membranes (GE Healthcare, Marborough, USA) by upward capillary transfer in 20 × SSC buffer. The membranes were hybridized with digoxigenin (DIG)-labelled probes corresponding to the full-length ORFs of GFP, NgRBP, PVX-CP and CMV-2b, respectively. For siRNA detection, 15 µg low molecular weight RNAs were separated on 15% polyacrylamide–7 M urea gel and transferred to a Hybond-N+ membrane in 0.5 × TBE at 0.8 mA cm−2 for 1 h. After being UV-crosslinked and incubated at 80 °C for 2 h, the membrane was hybridized with DIG-labelled probe GFP mRNA. Chemiluminescent detection was conducted using a DIG Northern Starter Kit (Roche, Basel, Switzerland) according to the manufacturer′s instructions.
Electrophoretic mobility shift assay (EMSA)
His-tagged NgRBP, R47A, NS1 and p19 proteins were expressed in Escherichia coli strain BL21 and purified by His-Tagged Protein Purification Kit (CwBio, Beijing, China). The GFP mRNA and dsRNA were created by T7 RiboMAXTM Express RNAi System (Promega, Wosconsin, USA) using specific primers GFP sense RNA F/R and GFP anti-sense RNA F/R in Table S1. Two complementary 21 nt GFP siRNAs with 2-nt 3′ overhangs (GFP siRNA-F, CUGUCCACACAAUCUGCCCUU; GFP siRNA-R, GGGCAGAUUGUGUGGACAGUU) were synthesized by Takara. GFP siRNAs were annealed to obtain double-stranded siRNAs. EMSA assay was performed using a Light Shift RNA EMSA Optimization and Control Kit (Thermo, Massachusetts, USA) according to the manufacturer’s instructions. The binding reactions were incubated for 30 min at room temperature. The products were separated on an 8% polyacrylamide gel and transferred to a nylon membrane. The membrane was then hybridized with DIG-labelled GFP mRNA probes. Chemiluminescent detection was conducted using a DIG Northern Starter Kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions.
RNA immunoprecipitation assay (RIP)
Three grams of leaves co-infiltrated with Agrobacterium carrying 35S-GFP with 35S-His-NgRBP, 35S-His-R47A or empty vector were put in 50-mL conical centrifuge tubes containing 37 mL of 1% formaldehyde and 2.5 mL of 2 M glycine separately, and vacuumed for 15 min in a closed container attached to a vacuum pump. Then the samples were ground and 30 mL extraction buffer 1 was added (0.4 M sucrose, 10 mM Tris–HCl, 5 mM BME, 0.1 mM PMSF). Solutions were filtered through two layers of 200 mesh stainless steel filter screen, and centrifuged at 1252g for 20 min at 4 °C. Pellets were resuspended in 1 mL extraction buffer 2 (0.25 M sucrose, 10 mM Tris–HCl, 10 mM MgCl2, 1% Triton X-100, 5 mM BME, 0.1 mM PMSF), and centrifuged at 9469g for 10 min at 4 °C. Then pellets were resuspended in 600 μL extraction buffer 3 (1.7 M sucrose, 10 mM Tris–HCl, 0.15% Triton X-100, 2 mM MgCl2, 5 mM BME, 0.1 mM PMSF), spined at the top speed in for 1 h at 4 °C, and resuspended in 500 μL nuclear lysis buffer (50 mM Tris–HCl, 10 mM EDTA, 1% SDS, 0.1 mM PMSF). Sample solutions were ultrasonicated on ice for 30 min. 60 μL supernatants are added to 540 μL RIP dilution buffer (1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl, 167 mM NaCl) and 25 μL equilibrated protein A beads and mixed on a shaker at 4 °C for 1 h. Then 5 μL His antibody (TransGen, Beijing, China) was added to each tube, and incubated on a shaker rotating mixer for 4 h at 4 °C. Immune complexes were eluted with 500 μL elution buffer (100 mM NaHCO3, 1% SDS) for 30 min at 65 °C. Crosslinking was reversed at 65 °C for 1 h with 20 μg proteinase K (Invitrogen, California, USA). RNA was purified by acidic phenol, chloroform and ethanol precipitation. RT-qPCR was used to detect the enriched levels of each segment of GFP mRNAs using the primers in Table S1.
Results
Expression profiles of NgRBP gene in N. glutinosa
NgRBP gene encodes a GRP harbouring a single copy of RRM which comprising highly conserved RNPI (R-G-F-G-F-V-T-F) and RNPII (C-F-V-G-G-L) at the N terminus and a GD at the C terminus. Sequence alignment indicated that a high level of amino acid similarity spanning the whole protein sequence exists among NgRBP homologs, suggesting that these proteins are evolutionarily conserved (Fig. 1a). Several GRPs were also isolated in other plant species, including A. thaliana, N. tabacum and Solanum lycopersicum. Phylogenetic analysis indicated that NgRBP shared a high amino acid similarity (94.3%) to NtGRP1 (Accession no. ACD03270) (Fig. 1b), a negative regulator of gene expression through binding to DNA or RNA under flooding stress (Lee et al. 2009). A well-studied glycine-rich RNA-binding protein AtGRP7 (Accession no. Q03250) shows 76% similarity to NgRBP (Fig. 1b), having an RRM in N terminal and a GD in C-terminal.

Structure and conservation of NgRBP. a Pairwise alignment of GRBPs from N. glutinosa (NgRBP, AF005359), N. tabacum (NtGRP1, EU569289), S. lycopersicum (SlGRP1b, JQ613216), A. thaliana (AtGRP7, Q03250 and AtGRP8, Q03251) using DNAStar. Identical and similar regions were indicated by a black background. The N-terminal RRM harbouring two highly conserved regions, RNPI and RNPII, was denoted by a solid line above its sequences. The C-terminal GD was represented by dotted lines. The 47th amino acid was marked with an asterisk. b Phylogenetic analysis showed the relationship of NgRBP and its homologues from various plant species. The tree was generated in MEGA with the neighbour-joining algorithm (1000 replicates) using nucleotide sequences (see supporting information). Bootstrap values were shown near the internal nodes. AtRBP1, a glycine-rich RNA-binding protein from Arabidopsis unrelated to NgRBP in sequence, was used as an out group
The tissue-specific expression of NgRBP gene was analysed by Northern blot. As shown in Fig. 2a, NgRBP gene is constitutively expressed in all examined organs, and clearly expressed at higher levels in root and stem compared with leaf and flower. To further study the expression regulation of NgRBP gene, a 982-bp fragment of its 5′-flanking region was isolated from N. glutinosa genomic DNA using inverse PCR technique. Three types of cis-acting element were predicted to be related to environment response: ABA-responsive element, methyl jasmonate (MeJA)-responsive element and light-responsive element. In addition, the cis-acting element required for endosperm expression was also predicted within the promoter region of NgRBP (Fig. S1). Expression profiles by Northern blot analysis proved that the NgRBP expression was indeed induced by ABA and MeJA in N. glutinosa leaves. The NgRBP transcripts were detectable at 4 h, reached peak level at 8 h and gradually decreased by 12 h after ABA treatment (Fig. 2b), while the NgRBP expression gradually increased from 4 to 24 h after MeJA treatment (Fig. 2b). In addition, Naqvi et al. (1998) showed that NgRBP was also induced by salicylic acid (SA; Naqvi et al. 1998). It suggests NgRBP might be involved in the responses to biotic and abiotic stresses.

NgRBP expression profiles in N. glutinosa. a Tissue-specific NgRBP expression was detected by Northern blot analysis using total RNAs extracted from the roots, stems, leaves, and flowers of N. glutinosa plants. Ethidium bromide-stained rRNA was used as a loading control. b Northern blot analysis of the NgRBP expression induced by ABA and MeJA. Total RNAs were extracted from leaves at the indicated time after treatment with 100 µM ABA and 1 mM MeJA, respectively. Control plants were sprayed with water. Ethidium bromide-stained rRNA was used as a loading control
NgRBP blocks both local and systemic RNA silencing triggered by GFP sense RNA or dsRNA
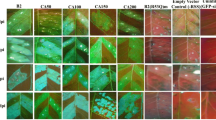
The effect of NgRBP gene on RNA silencing was investigated using Agrobacterium-mediated transient expression system in GFP-transgenic N. benthamiana line 16c plants. Agrobacterium strain harbouring NgRBP was mixed with the Agrobacterium carrying GFP, and infiltrated into the leaves of 16c plants. The co-infiltrations of GFP with an empty vector or with p19 were performed as negative and positive controls. GFP fluorescence was hardly observed in the infiltrated leaves with empty vector at 6 dpi under UV light, but the patches infiltrated with NgRBP displayed high-intensity GFP fluorescence at that time, similar to the infiltration of p19 (Fig. 3a). Northern blot analysis revealed that, both at 3 dpi and 6 dpi, the GFP mRNA levels of the leaves infiltrated with NgRBP plus GFP or p19 plus GFP were obviously higher than the leaves infiltrated with GFP plus empty vector. The siRNA blots also confirmed that NgRBP drastically reduced the GFP-specific siRNAs both at 3 dpi and 6 dpi, similar to p19 (Fig. 3b).

NgRBP blocks both local and systemic RNA silencing triggered by sense GFP RNA. a Suppression of local GFP silencing in GFP-transgenic N. benthamiana line 16c. Leaf patches were co-infiltrated with Agrobacterium cultures expressing GFP (35S-GFP) and a vector control, NgRBP or TBSV p19. Photographs of 16c leaves were taken at 3 dpi and 6 dpi under handheld long-wave ultraviolet lamp. b Northern blot analysis of GFP mRNA and siRNA extracted at 3 dpi and 6 dpi from patches co-infiltrated with the various strains indicated above each lane. Ethidium bromide-stained rRNA and tRNA were used as loading controls for mRNA and siRNA, respectively. c Photographs of 16c plants infiltrated with Agrobacterium harbouring 35S-GFP and a vector control, NgRBP or TBSV p19 under handheld long-wave ultraviolet lamp at 12 dpi. d Northern-blot analysis of GFP mRNA isolated from systemic leaves of plants with the different strains indicated above each lane at 12 dpi. Ethidium bromide-stained rRNA was used as a loading control
Furthermore, the effect of NgRBP on systemic spread of RNA silencing was studied. 16c plants infiltrated with above Agrobacterium combinations were further observed to monitor GFP expression in the newly emerged leaves at 12 dpi. As previously reported, systemic GFP silencing was visualized as the disappearance of the GFP fluorescence in the GFP-infiltrated plants (Voinnet et al. 1998; Silhavy et al. 2002). Most of the newly emerged leaves of plants infiltrated with NgRBP plus GFP still maintained GFP fluorescence, similar to that of p19 (Fig. 3c). The levels of steady-state GFP mRNA in the newly emerged leaves were consistent with the GFP fluorescence observed under UV light (Fig. 3d). Taken together, these results demonstrate that NgRBP has a bona fide ESR activity and inhibits RNA silencing induced by sense GFP RNA both at local and systemic levels.
RNA silencing is a multi-step process (Lichner et al. 2003; Baulcombe 2004; Susi et al. 2004). To determine in which step NgRBP targets RNA silencing, we tested the effect of NgRBP on GFP dsRNA-triggered silencing. Leaves of 16c plants were co-infiltrated with the Agrobacterium carrying NgRBP, GFP plus dsGFP. We found that the patches co-infiltrated with GFP plus dsGFP showed red fluorescence under UV light at 6 dpi. But GFP fluorescence still sustained when NgRBP was added (Fig. 4a). High GFP mRNA levels and negligible GFP-specific siRNA were detected by Northern blot analysis in the infiltrated leaves with NgRBP (or p19) addition (Fig. 4b). To further demonstrate whether NgRBP interferes with systemic RNA silencing triggered by dsGFP, we co-infiltrated 16c leaves with GFP and dsGFP plus NgRBP. Our results showed that, at 12 dpi, systemic RNA silencing occurred in the dsGFP infiltrated plants with trace amounts of GFP mRNA accumulated. But GFP fluorescence was maintained in the newly emerged leaves of the infiltrated plants with NgRBP or p19 addition (Fig. 4c), which was consistent with the levels of GFP mRNA detected by Northern blot analysis (Fig. 4d). This indicated that NgRBP also suppressed local and systemic RNA silencing triggered by dsRNA.

NgRBP suppresses local and systemic RNA silencing triggered by dsGFP RNA. a 16c leaves were co-infiltrated with three strains of Agrobacterium carrying 35S-GFP, 35S-dsGFP, and either empty vector, NgRBP or TBSV p19. The GFP expression in the infiltrated tissue was monitored at 3 dpi and 6 dpi under handheld long-wave ultraviolet lamp. b Northern-blot analysis of GFP mRNA and siRNA extracted from the patches with the different strains indicated above each lane at 3 dpi and 6 dpi. Ethidium bromide-stained rRNA and tRNA were used as the loading controls for mRNA and siRNA, respectively. c Photographs of 16c leaves infiltrated with Agrobacterium harbouring 35S-GFP, 35S-dsGFP and either vector control NgRBP or TBSV p19 under handheld long-wave ultraviolet lamp at 12 dpi. d Northern blot analysis of GFP mRNA isolated from systemic leaves of the plants with the different strains indicated above each lane at 12 dpi. Ethidium bromide-stained rRNA was used as a loading control
NgRBP increases the pathogenicity of PVX and NgRBP silencing confers enhanced resistance to PVX and CMV
To investigate whether NgRBP increases viral pathogenicity, the PVX vector pGR106 was utilized to express NgRBP in N. benthamiana plants (Fig. 5a). At 10 dpi, plants infected with PVX-NgRBP showed more severe disease symptoms along the veins than those receiving PVX vector alone (Fig. 5b). Viral RNAs accumulated to a higher level in PVX-NgRBP-inoculated plants than those infected with PVX (Fig. 5c).

NgRBP enhances the pathogenicity of chimeric PVX. a Schematic representation of recombinant PVX variants carrying NgRBP. b Symptoms in N. benthamiana plants infected with the recombinant PVX variants. More than 5 plants that were tested. Mock plants were infiltrated with buffer only. Systemic leaves (bottom panels) were photographed at 10 dpi. c Northern blot analysis of the accumulation of PVX genomic RNA in the systemic leaves at 10 dpi. The bottom panels show rRNA with ethidium bromide staining as a loading control
We investigated the role of NgRBP in plant defence responses using the Tobacco rattle virus (TRV)-based VIGS technique. NgRBP silencing resulted in milder symptoms than that of the controls (Fig. 6a). The RT-qPCR showed that the NgRBP mRNA level in TRV:NgRBP plants was ~ 70% lower than that of the empty vector controls (TRV:00) at 15 dpi (Fig. 6b), indicating that NgRBP was effectively silenced in N. glutinosa plants. Systematically NgRBP-silenced plants were then inoculated with PVX and CMV, respectively. Symptoms were visibly different between the NgRBP-silenced plants and the controls. The latter were relatively more susceptible to PVX and CMV infection than the former. Only slight mosaicism and downward leaf curling were observed in the NgRBP-silenced plants. UV illumination revealed that the control plants infected with PVX and CMV accumulated more phenolic compounds than the NgRBP-silenced plants did (Fig. 6c). Northern blot analysis showed that the levels of PVX genomic RNA and CMV RNA2 were higher in the control plants than in the NgRBP-silenced plants at 10 dpi (Fig. 6d). These results indicate that NgRBP interferes with antiviral RNA silencing in host plants.

NgRBP silencing in N. glutinosa plants with enhanced resistance to PVX and CMV. a Phenotype of NgRBP-silenced and empty vector control plants. Photographs were taken at 10 dpi. b RT-qPCR analysis of NgRBP expression in the empty vector control (TRV: 00) and the NgRBP-silenced (TRV:NgRBP) plants. c Visible- and ultraviolet lamp-illuminated disease symptoms developed on the empty vector control and the NgRBP-silenced plants infected with PVX (left panel) and CMV (right panel). Photographs were taken at 10 dpi. d Northern blot analysis of the accumulation of PVX genome RNA and CMV RNA2 at 10 dpi. Ethidium bromide-stained rRNAs were used as loading controls
The RNA binding activity of NgRBP is essential for its RNA silencing suppressor function
Much evidences support the idea that the silencing suppression activity of RSSs commonly requires the ability of RNA binding (Lakatos et al. 2006; Mérai et al. 2006). Two conserved RNA-binding motifs in RRM domain of GRPs, RNPI and RNPII, particularly the R47 residue of RNPI, play the key role in RNA recognition and binding (Khan et al. 2014). Therefore, two deletion mutants ΔRNPI and ΔRNPII, and a point mutant R47A in which the 47th arginine being substituted by alanine were constructed to explore the functional domain and loci of NgRBP using Agrobacterium transient expression system. Strong GFP fluorescence was observed in the tissues co-infiltrated with 35S-GFP and ΔRNPII constructs at 3 dpi, similar to that of NgRBP co-infiltration. The ΔRNPI and R47A constructs yielded weak fluorescence at 3 dpi (Fig. 7a). Then, to explore whether the RSS function of NgRBP is related to its RNA binding activity, NgRBP and R47A proteins were purified (Fig. S2) and incubated with GFP mRNA. EMSA assays showed that NgRBP rather than R47A bound to GFP mRNA (Fig. 7b). To study dsRNA binding ability of NgRBP, NS1 was used as a positive control in the EMSA assay, which is an Avian influenza virus-encoded RSS with siRNA and longer dsRNA affinity (Lin et al. 2007; Yu et al. 2018). The result showed that NgRBP could bind to GFP dsRNA, but R47A could not. Furthermore, RIP assays showed that NgRBP protein could enrich about fivefold 3′ end fragments of GFP mRNA compared with the control and the R47A protein (Fig. 7c). siRNA affinity of NgRBP was also detected by EMSA with p19 as a positive control (Silhavy et al. 2002). The result showed that NgRBP does not bind to siRNA which usually was taken as target by many VSRs (Fig. 7d). These results suggested that RNA binding activity of NgRBP is essential for its RSS function, and the 47th arginine located in RNPI motif is the key functional site.

NgRBP binds to the 3′ end of GFP mRNA and dsRNA but not siRNA. a Co-infiltration in N. benthamiana leaves with GFP and empty vector, NgRBP, △RNPI, △RNPII, R47A or p19, respectively. Images were obtained under handheld long-wave ultraviolet lamp at 3 dpi. b EMSA analysis of samples contained 1 μM GFP mRNA or GFP dsRNA and in addition 0, 130, 390, 650, 1300, and 2600 μM His-NgRBP and His- R47A protein, respectively. 390 μM NS1 was used as a positive control. c RIP analysis of GFP mRNA extracted from patches co-infiltrated GFP with His-NgRBP, His-R47A or empty vector at 3 dpi. d EMSA assay of samples contained 1 μM GFP siRNA and in addition 0, 390, 1300, and 2600 μM His-NgRBP, respectively. The free probe bands correspond to the siRNA duplexes. 390 μM p19 was used as a positive control
Discussion
A study on N. tabacum rgs-CaM provided the earliest clue that host genes served as negative regulators of RNA silencing (Anandalakshmi et al. 2000). Later, several ESRs or endogenous negative factors involved in RNA silencing have been discovered by genetic screening. It is believed that ESRs always induce the expression of certain silenced genes depending on developmental requirements. They may also be subverted by viruses in counterattacks against host antiviral responses (Trinks et al. 2005; Sarmiento et al. 2006; Gy et al. 2007; Endres et al. 2010). Recent studies showed that rgs-CaM had opposite effect on RNA silencing-mediated antiviral defence compared with its ESR function. So rgs-CaM was proposed as a counter-VSR factor that quenched certain VSRs containing dsRBD (Nakahara et al. 2012). HC-Pro was proved to upregulate the antiviral silencing potentially antagonists, such as Arabidopsis FRY1 and CML38 (a homologue of rgs-CaM) which both being defined as ESRs. rgs-CaM, FRY1, and CML38 are thought to be the downstream mediators of HC-Pro (Anandalakshmi et al. 2000; Gy et al. 2007; Endres et al. 2010). These findings suggested that the endogenous silencing pathway was subject to negative feedback regulation in certain cases. VSRs may require an ESR either to activate their components or to be able to work together to block silencing.
In this study, we characterized NgRBP, an RNA-binding protein from N. glutinosa, as a novel ESR. NgRBP could suppress local and systemic RNA silencing induced by sense RNA or dsRNA. NgRBP belongs to the class IVa of GRPs with the typical structure including an RRM in N terminal and a GD in C-terminal. Our data showed that when RNPI, one of the conserved motifs of RRM in NgRBP, was deleted, NgRBP lost its RSS activity. R47 mutation in RNPI motif brought the same effect on its function, and the mutant protein no longer bound to GFP mRNA. It is suggested that RNA binding ability of NgRBP effectively contributes to its RSS activity. A recent report revealed that in Arabidopsis both 5′-3′ and 3′-5′ cytoplasmic RNA decay pathways act as repressors of transgene and endogenous PTGS to safeguard plant transcriptome and development (Zhang et al. 2015; Zhang and Guo 2017). AtXRN4 which being in charge of 5′-3′ mRNA degradation antagonizes RNA silencing possibly by degrading the template for RdRp or interacting with HC-Pro (Gazzani et al. 2004; Li and Wang 2018). Our results showed that NgRBP could bind to the 3′ end of GFP mRNA and dsRNA, it is speculated that NgRBP might prevent RdRp from dsRNA synthesis at the early stage of RNA silencing and, therefore, impede the formation of RISC complex, or inhibit the step of Dicer processing by competitive dsRNA binding, respectively. Similar strategies are adopted by some viral RNA silencing repressors, such as Infectious bursal disease virus (IBDV) VP3 (Valli et al. 2012), Turnip crinkle virus (TCV) p38 (Iki et al. 2017), and Flock house virus (FHV) B2 (Lingel et al. 2005).
Why would a host plant have evolved proteins to thwart its own defence mechanisms? The ESR regulatory mechanism is required to prevent exaggerated RNA silencing, ensuring that appropriate PTGS in the plants. ESRs may potentially impair the host silencing machinery controlling cellular RNA levels so that they are normally suppressed until required. This postulate is supported by the observation that viral infection or wounding induces certain ESRs (Naqvi et al. 1998; Tadamura et al. 2012). Plants GRPs have been identified in a variety of plant species, and have been demonstrated to be regulated by a number of external stimuli including hormones and pathogens (Kang et al. 2013). As a TMV-induced GRP, NgRBP was induced by ABA, MeJA, SA as well (Naqvi et al. 1998; Czolpinska and Rurek 2018). Therefore, the suppressor function of NgRBP is most likely closely associated with the defence or multiple stress responses. Previous researches showed that virus attack induced GRPs expression in tobacco, petunia and rice, and some of them contributed to virus resistance (van Kan et al. 1988; Linthorst et al. 1990; Fang et al. 1991; Ueki and Citovsky 2002). AtGRP7, a typical GRP with 76% similarity to NgRBP, plays a positive role in the defence against TMV (Lee et al. 2012) and is described to preferentially bind to 3′UTR of target genes (Staiger et al. 2003; Meyer et al. 2017). It is similar to our findings that NgRBP could bind to the 3′ end of GFP mRNA. Both AtGRP7 and AtGRP8, the NgRBP homologs, were proved to be involved in an interlocked feedback loop through which they could autoregulate and reciprocally cross-regulate by coupling unproductive splicing to nonsense-mediated mRNA decay (NMD; Schöning et al. 2008). NMD is a host RNA control pathway that removes aberrant transcripts due to premature termination of codons that always arises through defective splicing. RNA viruses often contain internally located stop codons that should also be prime targets for NMD (May et al. 2018). We observed that NgRBP transient overexpression in N. glutinosa enhanced TMV resistance. Further studies need to explore the exact mechanisms of NgRBP involved in viral defence and stress response.
Author contribution statement
HL designed the research and edited the paper; XH and RY performed the main experiments and drafted the paper; WL, LG, XJ and CZ revised the paper. All authors have read and approved the final version of the manuscript.
Abbreviations
- CMV:
-
Cucumber mosaic virus
- EMSA:
-
Electrophoretic mobility shift assay
- ESR:
-
Endogenous suppressor of RNA silencing
- GD:
-
Glycine-rich domain
- GRP:
-
Glycine-rich RNA-binding protein
- PVX:
-
Potato virus X
- RBP:
-
RNA-binding protein
- RIP:
-
RNA immunoprecipitation
- RRM:
-
RNA recognition motif
- RSS:
-
RNA-silencing suppressor
- TMV:
-
Tobacco mosaic virus
- VSR:
-
Viral suppressor of RNA silencing
References
Anandalakshmi R, Marathe R, Ge X, Herr J, Mau C, Mallory A, Pruss G, Bowman L, Vance VB (2000) A calmodulin-related protein that suppresses posttranscriptional gene silencing in plants. Science 290(5489):142–144
Aneeta Sanan-Mishra N, Tuteja N, Kumar Sopory S (2002) Salinity- and ABA-induced up-regulation and light-mediated modulation of mRNA encoding glycine-rich RNA-binding protein from Sorghum bicolor. Biochem Biophys Res Commun 296(5):1063–1068
Baulcombe D (2002) Viral suppression of systemic silencing. Trends Microbiol 10(7):306–308
Baulcombe D (2004) RNA silencing in plants. Nature 431(7006):356–363
Brigneti G, Voinnet O, Li WX, Ji LH, Ding SW, Baulcombe DC (1998) Viral pathogenicity determinants are suppressors of transgene silencing in Nicotiana benthamiana. EMBO J 17(22):6739–6746
Burd CG, Dreyfuss G (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science 265(5172):615–621
Carpenter CD, Kreps JA, Simon AE (1994) Genes encoding glycine-rich Arabidopsis thaliana proteins with RNA-binding motifs are influenced by cold treatment and an endogenous circadian rhythm. Plant Physiol 104(3):1015–1025
Chen PY, Wang CK, Soong SC, To KY (2003) Complete sequence of the binary vector pBI121 and its application in cloning T-DNA insertion from transgenic plants. Mol Breed 11(4):287–293
Chung E, Seong E, Kim YC, Chung EJ, Oh SK, Lee S, Park JM, Joung YH, Choi D (2004) A method of high frequency virus induced gene silencing in chili pepper (Capsicum annuum L. cv. Bukang). Mol Cells 17(2):377–380
Csorba T, Kontra L, Burgyán J (2015) Viral silencing suppressors: tools forged to fine-tune host-pathogen coexistence. Virology 479–480:85–103
Czolpinska M, Rurek M (2018) Plant glycine-rich proteins in stress response: an emerging, still prospective story. Front Plant Sci 9:302
Daròs JA (2017) Viral suppressors: combatting RNA silencing. Nat Plants 3:17098
Endres MW, Gregory BD, Gao Z, Foreman AW, Mlotshwa S, Ge X, Pruss GJ, Ecker JR, Bowman LH, Vance V (2010) Two plant viral suppressors of silencing require the ethylene-inducible host transcription factor RAV2 to block RNA silencing. PLoS Pathog 6(1):e1000729
Fang RX, Pang Z, Gao DM, Mang KQ, Chua NH (1991) cDNA sequence of a virus-inducible, glycine-rich protein gene from rice. Plant Mol Biol 17(6):1255–1257
Ferullo JM, Vézina LP, Rail J, Laberge S, Nadeau P, Castonguay Y (1997) Differential accumulation of two glycine-rich proteins during cold-acclimation alfalfa. Plant Mol Biol 33(4):625–633
Fu ZQ, Guo M, Jeong BR, Tian F, Elthon TE, Cerny RL, Staiger D, Alfano JR (2007) A type III effector ADP-ribosylates RNA-binding proteins and quells plant immunity. Nature 447(7142):284–288
Gazzani S, Lawrenson T, Woodward C, Headon D, Sablowski R (2004) A link between mRNA turnover and RNA interference in Arabidopsis. Science 306(5698):1046–1048
Gómez J, Sánchez-Martínez D, Stiefel V, Rigau J, Puigdomènech P, Pagès M (1988) A gene induced by the plant hormone abscisic acid in response to water stress encodes a glycine-rich protein. Nature 334(6179):262–264
Gy I, Gasciolli V, Lauressergues D, Morel JB, Gombert J, Proux F, Proux C, Vaucheret H, Mallory AC (2007) Arabidopsis FIERY1, XRN2, and XRN3 Are Endogenous RNA Silencing Suppressors. Plant Cell 19(11):3451–3461
Haas G, Azevedo J, Moissiard G, Geldreich A, Himber C, Bureau M, Fukuhara T, Keller M, Voinnet O (2008) Nuclear import of CaMV P6 is required for infection and suppression of the RNA silencing factor DRB4. EMBO J 27(15):2102–2112
Hannon GJ (2002) RNA interference. Nature 418(6894):244–251
Himber C, Dunoyer P, Moissiard G, Ritzenthaler C, Voinnet O (2003) Transitivity-dependent and-independent cell-to-cell movement of RNA silencing. EMBO J 22(17):4523–4533
Höfgen R, Willmitzer L (1988) Storage of competent cells for Agrobacterium transformation. Nucleic Acids Res 16(20):9877
Iki T, Tschopp MA, Voinnet O (2017) Biochemical and genetic functional dissection of the P38 viral suppressor of RNA silencing. RNA 23(5):639–654
Jeong BR, Lin Y, Joe A, Guo M, Korneli C, Yang H, Wang P, Yu M, Cerny RL, Staiger D (2011) Structure function analysis of an ADP-ribosyltransferase type III effector and its RNA-binding target in plant immunity. J Biol Chem 286(50):43272–43281
Jing XL, Fan MN, Jia G, Liu LW, Ma L, Zheng CC, Zhu XP, Liu HM, Wang XY (2011) A multifunctional protein encoded by Turkey herpesvirus suppresses RNA silencing in Nicotiana benthamiana. J Virol 85(23):12792–12803
Kang H, Park SJ, Kwak KJ (2013) Plant RNA chaperones in stress response. Trends Plant Sci 18(2):100–106
Khan F, Daniëls MA, Folkers GE, Boelens R, Saqlan Naqvi SM, van Ingen H (2014) Structural basis of nucleic acid binding by Nicotiana tabacum glycine-rich RNA-binding protein: implications for its RNA chaperone function. Nucleic Acids Res 42(13):8705–8718
Kim JY, Kim WY, Kwak KJ, Oh SH, Han YS, Kang H (2010) Glycine-rich RNA-binding proteins are functionally conserved in Arabidopsis thaliana and Oryza sativa during cold adaptation process. J Exp Bot 61(9):2317–2325
Lakatos L, Csorba T, Pantaleo V, Chapman EJ, Carrington JC, Liu YP, Dolja VV, Calvino LF, López-Moya JJ, Burgyán J (2006) Small RNA binding is a common strategy to suppress RNA silencing by several viral suppressors. EMBO J 25(12):2768–2780
Lee MO, Kim KP, Kim BG, Hahn JS, Hong CB (2009) Flooding stress-induced glycine-rich RNA-binding protein from Nicotiana tabacum. Mol Cells 27(1):47–54
Lee HJ, Kim JS, Yoo SJ, Kang EY, Han SH, Yang KY, Kim YC, McSpadden Gardener B, Kang H (2012) Different roles of glycine-rich RNA-binding protein 7 in plant defense against Pectobacterium carotovorum, Botrytis cinerea, and tobacco mosaic viruses. Plant Physiol Biochem 60:46–52
Li F, Wang A (2018) RNA decay is an antiviral defense in plants that is counteracted by viral RNA silencing suppressors. PLoS Pathog 14(8):e1007228
Li F, Huang C, Li Z, Zhou X (2014) Suppression of RNA silencing by a plant DNA virus satellite requires a host calmodulin-like protein to repress RDR6 expression. PLoS Pathog 10(2):e1003921
Lichner Z, Silhavy D, Burgyán J (2003) Double-stranded RNA-binding proteins could suppress RNA interference-mediated antiviral defences. J Gen Virol 84(4):975–980
Lin D, Lan J, Zhang Z (2007) Structure and function of the NS1 protein of influenza A virus. Acta Biochim Biophys Sin 39(3):155–162
Lingel A, Simon B, Izaurralde E, Sattler M (2005) The structure of the flock house virus B2 protein, a viral suppressor of RNA interference, shows a novel mode of double-stranded RNA recognition. EMBO Rep 6(12):1149–1155
Linthorst HJ, van Loon LC, Memelink J, Bol JF (1990) Characterization of cDNA clones for a virus-inducible, glycine-rich protein from petunia. Plant Mol Biol 15(4):671
Lorsch JR (2002) RNA chaperones exist and DEAD box proteins get a life. Cell 109(7):797–800
May JP, Yuan X, Sawicki E, Simon AE (2018) RNA virus evasion of nonsense-mediated decay. PLoS Pathog 14(11):e1007459
Mérai Z, Kerényi Z, Kertész S, Magna M, Lakatos L, Silhavy D (2006) Double-stranded RNA binding may be a general plant RNA viral strategy to suppress RNA silencing. J Virol 80(12):5747–5756
Meyer K, Köster T, Nolte C, Weinholdt C, Lewinski M, Grosse I, Staiger D (2017) Adaptation of iCLIP to plants determines the binding landscape of the clock-regulated RNA-binding protein AtGRP7. Genome Biol 18(1):204
Moissiard G, Voinnet O (2004) Viral suppression of RNA silencing in plants. Mol Plant Pathol 5(1):71–82
Nakahara KS, Masuta C, Yamada S, Shimura H, Kashihara Y, Wada TS, Meguro A, Goto K, Tadamura K, Sueda K, Sekiguchi T, Shao J, Itchoda N, Matsumura T, Igarashi M, Ito K, Carthew RW, Uyeda I (2012) Tobacco calmodulin-like protein provides secondary defense by binding to and directing degradation of virus RNA silencing suppressors. Proc Natl Acad Sci USA 109(25):10113–10118
Naqvi SS, Park KS, Yi SY, Lee HW, Bok SH, Choi D (1998) A glycine-rich RNA-binding protein gene is differentially expressed during acute hypersensitive response following tobacco mosaic virus infection in tobacco. Plant Mol Biol 37(3):571–576
Qi Tsuda K, Joe A, Sato M, Nguyen LV, Glazebrook J, Alfano JR, Cohen JD, Katagiri F (2010) A putative RNA-binding protein positively regulates salicylic acid–mediated immunity in Arabidopsis. Mol Plant Microbe Interact 23(12):1573–1583
Sarmiento C, Nigul L, Kazantseva J, Buschmann M, Truve E (2006) AtRLI2 is an endogenous suppressor of RNA silencing. Plant Mol Biol 61(1–2):153–163
Schöning JC, Streitner C, Meyer IM, Gao Y, Staiger D (2008) Reciprocal regulation of glycine-rich RNA-binding proteins via an interlocked feedback loop coupling alternative splicing to nonsense-mediated decay in Arabidopsis. Nucleic Acids Res 36(22):6977–6987
Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115(2):199–208
Silhavy D, Molnár A, Lucioli A, Szittya G, Hornyik C, Tavazza M, Burgyán J (2002) A viral protein suppresses RNA silencing and binds silencing-generated, 21- to 25-nucleotide double-stranded RNAs. EMBO J 21(12):3070–3080
Staiger D, Zecca L, Wieczorek Kirk DA, Apel K, Eckstein L (2003) The circadian clock regulated RNA-binding protein AtGRP7 autoregulates its expression by influencing alternative splicing of its own pre-mRNA. Plant J 33(2):361–371
Steinert PM, Mack JW, Korge BP, Gan SQ, Haynes SR, Steven AC (1991) Glycine loops in proteins: their occurence in certain intermediate filament chains, loricrins and single-stranded RNA binding proteins. Int J Bio Macromol 13(3):130–139
Susi P, Hohkuri M, Wahlroos T, Kilby NJ (2004) Characteristics of RNA silencing in plants: similarities and differences across kingdoms. Plant Mol Biol 54(2):157–174
Tadamura K, Nakahara KS, Masuta C, Uyeda I (2012) Wound-induced rgs-CaM gets ready for counterresponse to an early stage of viral infection. Plant Signal Behav 7(12):1548–1551
Tomari Y, Zamore PD (2005) Perspective: machines for RNAi. Genes Deve 19(5):517–529
Trinks D, Rajeswaran R, Shivaprasad PV, Akbergenov R, Oakeley EJ, Veluthambi K, Hohn T, Pooggin MM (2005) Suppression of RNA silencing by a geminivirus nuclear protein, AC2, correlates with transactivation of host genes. J Virol 79(4):2517–2527
Ueki S, Citovsky V (2002) The systemic movement of a tobamovirus is inhibited by a cadmium-ion-induced glycine-rich protein. Nat Cell Biol 4(7):478–486
Valli A, Busnadiego I, Maliogka V, Ferrero D, Castón JR, Rodríguez JF, García JA (2012) The VP3 factor from viruses of Birnaviridae family suppresses RNA silencing by binding both long and small RNA duplexes. PLoS One 7(9):e45957
van Kan JA, Cornelissen BJ, Bol JF (1988) A virus-inducible tobacco gene encoding a glycine-rich protein shares putative regulatory elements with the ribulose bisphosphate carboxylase small subunit gene. Mol Plant Microbe Interact 1(3):107–112
Vargason JM, Szittya G, Burgyán J, Hall TM (2003) Size selective recognition of siRNA by an RNA silencing suppressor. Cell 115(7):799–811
Voinnet O, Vain P, Angell S, Baulcombe DC (1998) Systemic spread of sequence-specific transgene RNA degradation in plants is initiated by localized introduction of ectopic promoterless DNA. Cell 95(2):177–187
Voinnet O, Lederer C, Baulcombe DC (2000) A viral movement protein prevents spread of the gene silencing signal in Nicotiana benthamiana. Cell 103(1):157–167
Yang Z, Li Y (2018) Dissection of RNAi-based antiviral immunity in plants. Curr Opin Virol 32:88–99
Ye K, Malinina L, Patel DJ (2003) Recognition of small interfering RNA by a viral suppressor of RNA silencing. Nature 426(6968):874–878
Yu R, Jing X, Li W, Xu J, Xu Y, Geng L, Zhu C, Liu H (2018) Non-structural protein 1 from avian influenza virus H9N2 is an efficient RNA silencing suppressor with characteristics that differ from those of Tomato bushy stunt virus p19. Virus Genes 54(3):368–375
Zhang X, Guo H (2017) mRNA decay in plants: both quantity and quality matter. Curr Opin Plant Biol 35:138–144
Zhang X, Zhu Y, Liu X, Hong X, Xu Y, Zhu P, Shen Y, Wu H, Ji Y, Wen X (2015) Suppression of endogenous gene silencing by bidirectional cytoplasmic RNA decay in Arabidopsis. Science 348(6230):120–123
Acknowledgements
We thank Dr. David Baulcombe for providing P19, PVX vector, and N. benthamiana line 16c seeds. This work was supported by Shandong Provincial Natural Science Foundation, China [ZR2015CM018, ZR2018LC009], Genetically Modified Organisms Breeding Major Projects of China [2016ZX08001-002], Shandong Medical and Health Science and Technology Development Plan Project [2017WS426].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, X., Yu, R., Li, W. et al. Identification and characterisation of a glycine-rich RNA-binding protein as an endogenous suppressor of RNA silencing from Nicotiana glutinosa. Planta 249, 1811–1822 (2019). https://doi.org/10.1007/s00425-019-03122-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-019-03122-5