
Abstract
Main conclusion
Transcriptome analysis was performed on the roots of susceptible and resistant sweetpotato cultivars infected with the major root-knot nematode species Meloidogyne incognita. In addition, we identified a transcription factor-mediated defense signaling pathway that might function in sweetpotato–nematode interactions.
Root-knot nematodes (RKNs, Meloidogyne spp.) are important sedentary endoparasites of many agricultural crop plants that significantly reduce production in field-grown sweetpotato. To date, no studies involving gene expression profiling in sweetpotato during RKN infection have been reported. Therefore, in the present study, transcriptome analysis was performed on the roots of susceptible (cv. Yulmi) and resistant (cv. Juhwangmi) sweetpotato cultivars infected with the widespread, major RKN species Meloidogyne incognita. Using the Illumina HiSeq 2000 platform, we generated 455,295,628 pair-end reads from the fibrous roots of both cultivars, which were assembled into 74,733 transcripts. A number of common and unique genes were differentially expressed in susceptible vs. resistant cultivars as a result of RKN infection. We assigned the differentially expressed genes into gene ontology categories and used MapMan annotation to predict their functional roles and associated biological processes. The candidate genes including hormonal signaling-related transcription factors and pathogenesis-related genes that could contribute to protection against RKN infection in sweetpotato roots were identified and sweetpotato–nematode interactions involved in resistance are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sweetpotato (Ipomoea batatas L.), one of the most important root crop plants worldwide, serves as a major food source, especially in Asia and Africa. This crop also plays an important role in sustainable agriculture, as it serves as a useful source of nutrients and biofuel, enhances global food security, is amenable to sustainable fertilization, and enhances agricultural productivity (Woolfe 1992; Diaz et al. 2014; Grace et al. 2014). However, the narrow genetic base of the cultivated sweetpotato gene pool, together with its hexaploid nature and its genomic complexity, pose a challenge to breeding efforts aimed at developing sweetpotato cultivars with useful traits and resistance to multiple environmental stresses along with high quality and yields.
Sweetpotato yields are affected by various biotic environmental factors worldwide, such as viruses, fungal diseases, and root-knot nematodes (RKNs) (Clark and Moyer 1998; Kreuze 2002; Kistner et al. 1993; Palomares-Rius and Kikuchi 2013). Among RKNs, members of the genus Meloidogyne, such as Meloidogyne incognita, M. arenaria, M. javanica, and M. hapla, represent a major threat to many agricultural crops including sweetpotato (Castagnone-Sereno et al. 2013). M. incognita is a destructive RKN and the most common nematode species found in agricultural regions worldwide. Sweetpotato is highly susceptible to RKNs, especially M. incognita, which causes severe damage to the plant roots and occurs in tropical regions throughout the world (Bridge and Starr 2010).
Although many sources of genetic resistance to RKNs have been identified and characterized in many crop plants (Williamson and Kumar 2006), the molecular, physiological, and genetic mechanisms of RKN resistance in sweetpotato are poorly understood. Early attempts to address this question have provided insights into the basis of susceptibility and resistance to RKN in sweetpotato. However, recently developed genomic technologies and resources used to quantify changes in gene expression during RKN development have not yet been applied towards improving our understanding of these complex interactions. A large number of putative RKN-resistance-related genes are present in sweetpotato, and this has been used as the basis to argue that RKN resistance in this crop might be inherited as multiple molecular genetic factors (Cervantes-Flores et al. 2008). Thus, it appears that the biological processes underlying resistance to RKN in sweetpotato are quite unusual, which might also be reflected in the structure of the sweetpotato transcriptome.
A previous study of Korean sweetpotato accessions maintained in South Korea showed that the sweetpotato cultivar Yulmi is highly sensitive and specific to M. incognita, whereas Juhwangmi is highly resistant (Choi et al. 2006; Ha et al. 2017). Although each sweetpotato cultivar can be classified as being either resistant or sensitive to M. incognita, the molecular mechanisms of RKN resistance have not yet been reported. Therefore, we performed transcriptome profiling of two sweetpotato cultivars with contrasting responses to RKN M. incognita. The aim of this study was to investigate the molecular defense mechanism in governing sweetpotato responses RKN during M. incognita infestation using transcriptomic analysis. The results of this study increases our understanding of plant defense mechanisms against RKN infestation in RKN-sensitive and resistant sweetpotato cultivars.
Materials and methods
Plant materials, M. incognita treatment, and analysis
The two sweetpotato cultivars used in this study, including the highly RKN-susceptible cultivar, Yulmi (cv. Yulmi, Y), and the highly RKN-resistant cultivar, Juhwangmi (cv. Juhwangmi, J), were obtained from the Bioenergy Crop Research Center, National Crop Research Institute (RDA, Muan, Jeonnam, Korea). Sweetpotato plants were cultivated and treated with M. incognita as described by Lee et al. (2012) and Ha et al. (2017). Cuttings (approximately 10 cm long) with three leaves attached were transplanted into plastic pots (24 cm width, 18 cm length, and 12 cm height) filled with sandy loam soil infested with M. incognita at a density of 1,154 ± 176 s-stage juveniles per 300 g of soil. As a control, the soil was steam-sterilized at 115 °C and 1.5 atmospheric pressure for 20 min. Both Juhwangmi and Yulmi plants were grown in the same pots under homogeneous infection conditions. The sweetpotato cuttings were grown under controlled-green house conditions (16 h photoperiod, 30/22 °C day/night temperatures, and 70% relative humidity) with a light intensity of 200 µmol s−1 m−2 provided by fluorescent and metal halide lamps. The plants were harvested 90 days after planting the cuttings, washed with tap water, and then, the RKN egg masses were determined. Each experiment was repeated three times with ten replicates per experiment. Root samples were ground to a fine powder in liquid nitrogen using a pestle and mortar and stored at − 70 °C until further analysis. The egg masses formed by M. incognita on the fibrous roots of sweetpotato plants were dyed with Phloxin B solution and counted (Viaene et al. 2012). The experiment was performed using a completely randomized design, with ten replicates in three independent experiments. The data were subjected to analysis of variance (ANOVA), and means were separated using an LSD multiple range test.
RNA extraction, cDNA library preparation, and sequencing
Total RNA was isolated from fibrous sweetpotato roots using a Trizol RNA Isolation kit (Invitrogen). Samples with an RNA integrity number (RIN) value > 8 were used for library preparation. Each paired-end cDNA library was prepared according to the TruSeq RNA Sample Preparation Guide (Illumina, San Diego, CA, USA). The cDNA libraries were sequenced on the HiSeq 2000 platform. Each analysis was performed independently three times.
RNA-seq data processing and de novo assembly
Paired-end reads were cleaned using prinseq-lite version 0.20.4 with the following parameters: min_len 50; min_qual_score 10; min_qual_mean 20; derep 14; trim_qual_left 20; and trim_qual_right 20. The clean paired-end reads were subjected to the Trinity assembly pipeline (Trinity Release v. 2.3.2) using default parameters for de novo transcriptome assembly. Candidate coding transcripts were predicted using TransDecoder Release v. 3.0.1 (http://transdecoder.github.io). Transcripts sequences were clustered using CD-HIT v. 4.6.1 (Fu et al. 2012), and the longest sequence from each cluster was retained for downstream analysis.
Transcript quantification and differential expression analysis
The pre-processed reads from each sample were back aligned to the final reference transcriptome using Bowtie. The RSEM 1.3.0 software was used to obtain read counts and TMM-normalized FPKM (i.e., trimmed mean of M values–normalized fragments per kb of exon per million reads mapped) values for each transcript. For differential expression analysis, EdgeR version 3.16.5 was used to calculate the negative binomial dispersion across conditions. Genes were determined to be significantly differentially expressed if the expression levels showed a > 4 fold change with an FDR-adjusted P value < 0.001.
Functional annotation
Functional annotation of differentially expressed genes (DEGs) was performed via sequence similarity searches using the BLAST program against the Arabidopsis thaliana protein database with an e value threshold of 0.05. Gene Ontology (GO) classification was performed using DAVID. Putative sweetpotato transcription factors (TFs) were classified into families using TAIR annotation. For MapMan analysis, homologous Arabidopsis IDs and fold changes of DEGs in the two sweetpotato cultivars were mapped to biotic stress pathways. Pictorial representations of the biotic stress pathways were uploaded from the MapMan website.
Expression analysis by qRT-PCR
Quantitative real-time PCR analysis was performed in a Bio-Rad CFX96 thermal cycler (Bio-Rad) using EvaGreen fluorescent dye according to the manufacturer’s instructions. The interexperimental quality control comparisons of repeated samples were assessed using CT values between three replications. Samples that yielded differential values greater than 1.5 were removed from the data set. In addition, quality control of the reaction by gel electrophoresis verified the presence of a single product of the correct size, and samples with multiple peaks in the dissociation graph were also eliminated to exclude unspecific PCR reactions. Linear data were normalized to the mean CT of the ubiquitin extension protein (UBI) and ADP-ribosylation factor (ARF) genes as a reference genes (Park et al. 2012). The gene-specific PCR primer sets are listed in Table S1.
Results
RKN resistance differs between the two sweetpotato cultivars
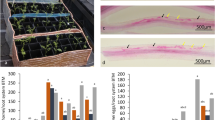
According to reports from the Bioenergy Crop Research Center and our previous study on Korean sweetpotato accessions maintained in South Korea, sweetpotato cultivar Yulmi is highly sensitive and specific to M. incognita, whereas Juhwangmi is highly resistant. Therefore, we compared the resistance of Juhwangmi and Yulmi to M. incognita by measuring nematode egg mass formation after infestation under greenhouse conditions. M. incognita formed 932 ± 174 egg masses in Yulmi plants, but only 5 ± 2.7 egg masses in Juhwangmi plants (Fig. 1). Therefore, similar to the previous reports (Ha et al. 2017), our results demonstrate that compared with Yulmi, Juhwangmi plants are highly resistant to M. incognita under greenhouse conditions.

Effect of the root-knot nematode M. incognita on Yulmi and Juhwangmi sweetpotato plants grown under greenhouse conditions for 3 months. a Yulmi and Juhwangmi cultivars infected by M. incognita. The plants were grown in steam-sterilized (control) and non-sterilized sand. Egg masses in non-sterilized sand containing M. incognita are also shown. b Quantification of egg masses formed by M. incognita. Egg masses formed in non-sterilized sand containing M. incognita, but not in sweetpotato plants cultured in sterilized soil. Data represent mean ± SD of ten replicates. Statistical significance of differences between the control and treated plant groups was determined by one-way ANOVA with LSD post hoc test (*P < 0.05; **P < 0.01)
Sequencing and de novo assembly of the transcriptomes of fibrous sweetpotato roots in response to RKN
To perform de novo assembly and transcriptome analysis of fibrous roots from both sweetpotato cultivars under M. incognita-infected and control conditions, we isolated total RNA and used it to synthesize cDNA libraries. The cDNA libraries were subjected to sequencing on the Illumina HiSeq 2000 platform, generating 455,295,628 raw reads (45,984,858,428 bp). After filtering out low-quality and unpaired reads using the PRINSEQ software, we obtained 408,279,602 high-quality reads, which we used for de novo assembly of the transcriptomes of both sweetpotato cultivars (Table S2). All raw reads were deposited at NCBI and can be accessed under SRA accession number SRP128609 (PRJNA429283). A total of 1332 and 77,334 transcripts were obtained from Yulmi and Juhwangmi with 754 bps and 735 bps of N50, respectively. A total of 74,733 unigenes (61,340,286 nucleotides) were obtained by assembling high-quality reads using the Trinity program, with an average length of 820 bp and an N50 length of 1041 bp (Table 1), which generated more high-quality transcriptome data than single cultivar de novo assembly. In this respect, we selected the reference unigene set as the de novo transcriptome, which was assembled using both cultivars in this study. Open reading frames were identified from all transcripts, including 23,566 full length, 18,463 5′-partial, 21,217 internal partial, and 11,487 3′-partial transcripts (Table 2).
Global statistical evaluation of samples used for comparative transcriptome analysis
To perform a statistical evaluation of the samples used for comparative transcriptome analyses, we mapped high-quality reads to the assembled transcriptome and calculated transcript abundance. We identified DEGs based on pairwise sample comparisons and visualized the distribution of the DEGs in MA plots (Fig. 2a). In pairwise comparisons, the biological replicates were more highly correlated within samples than between samples, and identical cultivars were more closely clustered together than the other cultivars, regardless of M. incognita infection (Fig. 2b).

Statistical analysis of differentially expressed genes across samples. a MA plots showing pairwise comparisons of transcript levels across samples. YC Yulmi control, YT Yulmi treatment, JC Juhwangmi control, JT Juhwangmi treatment. Log2 fold change (logFC) between the two samples is plotted on the Y-axis, and the Log2 average of the counts normalized by size factor is shown on the X-axis. The red dots indicate transcripts for which the logFC was significantly higher than 2 or lower than − 2. The black dots indicate transcripts for which the logFC was between − 2 and 2. b Clustered heatmap showing the Pearson correlation matrix for pairwise sample comparisons. The color key was adjusted based on the log2-centered values for optimal visual detection of the differences, and the dendrogram illustrates the relationship distance between samples
Identification of DEGs in pairwise sample comparisons
In total, we identified 4479 putative unique transcripts as reliable DEGs (using a cutoff > 4 fold change and < 0.001 of Kal’s z test FDR P value) in pairwise sample comparisons (Fig. S1a). Initially, we examined the transcriptional responses against RKNs in both cultivars (Fig. S1b). Among the DEGs, 294 and 587 were significantly up- and downregulated after M. incognita infection in Yulmi, respectively. In the RKN-resistant cultivar, Juhwangmi, 929 putative transcripts were differentially expressed in response to M. incognita, including 179 upregulated and 750 downregulated DEGs. We also compared the transcriptomes of uninfected cultivars to examine the pre-existing resistance mechanism in Juhwangmi. We identified 663 DEGs, including 331 that were upregulated and 332 that were downregulated in Juhwangmi compared with Yulmi under control (uninfected) conditions. Finally, we compared the transcriptomes of both cultivars after M. incognita infection to uncover the underlying molecular mechanism that causes their differential responses to RKN. We identified 1403 transcripts as reliable DEGs, including 743 that were upregulated and 660 that were downregulated in Juhwangmi compared with Yulmi.
Characterization of the DEGs based on biological processes
To functionally characterize the DEGs, we identified their encoded gene products via comparisons with an A. thaliana protein database and performed GO (biological process) enrichment analysis of the annotated genes (with Benjamini–Hochberg-adjusted P values < 0.05). Several biological processes (response to cadmium ion, response to salt stress, response to cytokinin, and response to chitin) were enriched in the DEGs between Yulmi plants that were infected vs. uninfected with M. incognita (Fig. S2a). Biological processes such as response to cadmium ion, tricarboxylic acid cycle, and oxidation–reduction process were enriched in the DEGs between Juhwangmi plants with and without RKN infection (Fig. S2b). Biological processes related to response to cadmium ion, response to salt stress, tricarboxylic acid cycle, and ethylene (ET)-activated signaling pathway were enriched in the DEGs between Yulmi and Juhwangmi uninfected with RKN (control) (Fig. S2c), whereas biological processes related to response to wounding, response to cadmium ion, ET-activated signaling pathway, response to chitin, response to abscisic acid (ABA), oxidation–reduction process, and response to jasmonic acid (JA) were enriched in the DEGs between Yulmi and Juhwangmi infected with RKN (Fig. S2d).
Transcriptional regulation of TF genes in response to RKN infection
Transcription factors (TFs) are highly important, as the induction of genes in response to stress occurs primarily at the level of transcription. Spatiotemporal regulation of the expression of TF genes plays an important role in downstream signaling pathways to initiate protective defense responses in various plant species, including sweetpotato. The major TFs identified in sweetpotato were classified into ten families, with the most abundant TFs related to biotic stress responses, including MYB (R2R3-type MYB domain protein, 21%), bHLH (basic helix–loop–helix domain protein, 18%), WRKY (WRKY domain protein, 14%), ERF (ethylene responses factor protein, 13%), bZIP (basic leucine zipper domain protein, 12%), NAC (NAM/ATAF/CUC protein, 10%), HD-ZIP (homeodomain–leucine zipper protein, 7%), ARF (auxin response factor protein, 5%), and ZF-HD (zinc finger-homeodomain protein, 0.5%) (Fig. S3a). The four most highly expressed TF families in this study, MYB, bHLH, WRKY, and ERF, which are implicated in many physiological and biochemical processes, are also involved in various plant–pathogen interactions through their effects on regulating pathogen resistance and cell death. We also identified differentially expressed TFs in pairwise sample comparisons, with the proportions of differentially expressed TFs as follows: ERF (24.8%), bHLH (16.4%), MYB (15.2%), WRKY (12.9%), NAC (10.9%), bZIP (9.1%), HD-ZIP (5.1%), ARF (5.0%), and ZF-HD (0.6%; Fig. S3b). Among these, ERF TFs produced the highest proportion of differentially expressed transcripts and might, therefore, play important roles in the response to M. incognita infection in sweetpotato. Therefore, perhaps, the differentially expressed TF genes identified in this study have closely overlapping functions and might activate or repress genes through cis-acting sequences that respond to M. incognita infection.
Differential regulation of hormonal signaling-related TF and pathogenesis-related genes in response to RKN infection
We identified DEGs encoding TFs involved in various hormonal signaling-related pathways during RKN infection (Fig. 3a). Putative SA-dependent WRKY53/WRKY70 genes were not expressed or were slightly induced under RKN infection in both cultivars. By contrast, putative ET-dependent ERF1/ERF2 and JA-dependent MYC2/MYC4 genes were induced or highly upregulated in Juhwangmi compared with Yulmi during RKN infection. Next, we identified various pathogenesis-related (PR) genes among DEGs during RKN infection (Fig. 3b). PR1, PR2 (beta-1,3-glucanase), PR4 (chitinase), and PR5 (thaumatin) were not expressed or were slightly induced under RKN infection in both cultivars. By contrast, the expression of PR3 (chitinase), PR5 [protease (trypsin) inhibitor], and PR10 (RNase) increased moderately or strongly in Juhwangmi compared with Yulmi during RKN infection.

Comparison of the relative expression levels of selected candidate genes in two sweetpotato cultivars under control and RKN treatment. a DEGs including SA-dependent WRKY53 and WRKY70, ET-dependent ERF1 and ERF2, and JA-dependent MYC2 and MYC4. b DEGs including SA-dependent PR1, PR2, and PR5 and ET/JA-dependent PR3, PR4, PR6, and PR10. The heatmap, which was constructed using MultiExperiment Viewer v. 4.9 (MEV4.9), displays the normalized transcript levels of each gene
Expression profiling of RKN-resistant PR genes by qRT-PCR analysis
We analyzed the expression patterns of four randomly selected candidate RKN-responsive PR genes, including two PR6 (Ibatatas_LOC24172, Ibatatas_LOC34060) and two PR10 (Ibatatas_LOC62265, Ibatatas_LOC62267) genes, in RKN-infected root tissues of the two sweetpotato cultivars by qRT-PCR analysis (Fig. 4). Both qRT-PCR and RNA-Seq analysis revealed significant DEGs between the two cultivars: PR6 and PR10 were highly upregulated in Juhwangmi during RKN infection compared with Yulmi.

Comparison between qRT-PCR results and RNA-Seq expression profiles. Relative mRNA levels of DEGs including PR6 (Ibatatas_LOC24172, Ibatatas_LOC34060) and two PR10 (Ibatatas_LOC62265, Ibatatas_LOC62267) genes in two sweetpotato cultivars under RKN infection, as analyzed by qRT-PCR (left Y-axis) and RNA-Seq (right Y-axis)
Expression profiling of hormonal signaling-related marker genes during the early period of RKN infection
To identify changes in expression of hormone signaling-related genes (e.g., JA, ET, SA, and ABA) in response to RKN infection during the early stage of treatment, we performed qRT-PCR analysis of two different sweetpotato cultivars (Fig. 5). In general, transcription of genes involved in JA signaling (LOX1 and MYC2) increased in the Juhwangmi cultivar upon infection with RKN, whereas that of the JA-repressor JAZ1 fell. In the case of ET signaling, we noted a slight increase in transcription of ET signaling-related genes such as EIN4 and ERF1 during RKN infection. However, changes in SA and ABA signaling-related genes were relatively less pronounced in both cultivars. Detailed examination of the qRT-PCR data revealed that infection by RKN triggered changes in expression of hormonal signaling-related genes, but that the regulatory mechanism in the two sweetpotato cultivars was different.

Expression profiling of hormonal signaling-related marker genes. Relative mRNA levels of DEGs including JA marker (LOX1, JAZ1, and MYC2), ET marker (EIL1, EIN4, and ERF1), SA marker (EDS1, NPR1, and TGA1), and ABA marker (SNRK2, ABF3, and LEA14) genes in two sweetpotato cultivars under early RKN infection time
Discussion
To the best of our knowledge, this is the first comprehensive analysis of global gene expression profiles in RKN-infected sweetpotato roots under soil-grown conditions in both susceptible and resistant cultivars. We identified DEGs by comparing the expression profiles of infected vs. uninfected roots. Our approach identified a large set of DEGs, as well as their potential roles in resistance-related signaling during RKN development.
In addition, we constructed a model using MapMan analysis to visualize the dynamic changes in expression of genes involved in various biological processes in both sweetpotato cultivars during RKN infection based on a summary of our transcriptome data (Fig. 6). Using the MapMan annotation software to organize and display our data sets in the context of biological pathways, we identified genes related to biotic stress responses among the distinct DEGs, with many genes involved in defense signaling, including genes encoding TFs such as ERF, WRKY, and MYB; these genes were more responsive to RKN in Juhwangmi than in Yulmi under both control and RKN infection conditions. In the MapMan data set, ERF and ET signaling genes were more responsive under control conditions vs. RKN infection in Juhwangmi compared with Yulmi; these results are consistent with the results of GO and RNA-Seq analysis (Figs. 3, S2). JA, ABA, and SA signaling were also more responsive to RKN infection in Juhwangmi than in Yulmi. GO analysis also indicated that the categories JA signaling, JA response, and ABA response were more highly enriched in RKN-treated Juhwangmi than in RKN-treated Yulmi (Fig. S2d). RNA-Seq analysis showed that MYC TFs were more highly responsive to RKN infection in Juhwangmi than in Yulmi (Fig. 3a).

MapMan diagram of sweetpotato genes involved in the response to RKN (M. incognita) infection. Overview of DEG expression patterns (log2FC of fpkm + 1) in Juhwangmi roots relative to Yulmi roots. a YC and YT, b JC and JT, c YC and JC, and d YT and JT. Dots show the different paralogous genes encoding proteins related to a certain step in the defense response. Red dots indicate upregulation, and blue dots indicate downregulation. YC Yulmi control, YT Yulmi treatment, JC Juhwangmi control, JT Juhwangmi treatment
The basic helix–loop–helix (bHLH) TF MYC2 plays a role in multiple hormone signaling pathways in Arabidopsis under stress conditions (Fujita et al. 2006). AtMYC2, which was first identified as a transcriptional activator involved in the ABA-mediated drought stress signaling pathway (Abe et al. 2003), also upregulates the expression of genes involved in the JA-mediated wounding response and negatively regulates the expression of JA/ET-mediated defense signaling pathway genes during pathogen infection (Anderson et al. 2004; Boter et al. 2004; Lorenzo et al. 2004). Under biotic stress conditions, AtMYC2 either negatively or positively modulates JA-dependent signaling pathways (Kazan and Manners 2008). For example, AtMYC2 positively regulates the JA-mediated insect-resistance response, metabolic changes such as flavonoid metabolism, and the oxidative stress response (De Vos et al. 2005). By contrast, MYC2 negatively regulates JA-dependent indole glucosinolate biosynthesis and pathogen defense responses (Dombrecht et al. 2007). Therefore, our data suggest that MYC2 might be a key regulator of the crosstalk among hormonal signaling pathways between biotic and abiotic stress responses in RKN-resistant sweetpotato.
Other genes related to pathogen recognition and defense response, cell wall metabolism, proteolysis, redox state, and secondary metabolism that are known to be involved in the plant response to nematode infection are shown in the map in Fig. 6. Overall, MapMan ontology analysis enabled us to construct a genome-wide outline of the expression of sweetpotato genes that respond to RKN M. incognita infection by identifying pathways involved in the main steps leading to the resistance response. Initially, during nematode infection, recognition by R genes usually triggers effector-triggered immunity, eliciting intermediate enzymes with an active redox state that protect cells from oxidative damage (Holbein et al. 2016). Subsequently, the regulation of various MAPK and TF genes involved in signal transduction pathways promotes the induction of a large set of genes commonly known as defense-related genes, such as PR proteins, heat shock proteins (HSPs), and enzymes involved in the biosynthesis of anti-microbial secondary metabolites. However, our results show that R gene-mediated signaling via MAPK and TF genes responded only weakly to RKN infection in the resistant cultivar Juhwangmi compared with Yulmi (Fig. 6b). Interestingly, genes involved in phytohormone-mediated defense signaling pathways directly or indirectly related to resistance responses, such as JA, SA, ET, and ABA genes that play a role in plant immunity (Santino et al. 2013), were upregulated in Juhwangmi under RKN treatment (Fig. 6b). In addition, some other TF genes and other DEGs identified in the study are also known to be activated by abiotic stress (Fig. 6).
Some aspects of plant immunity include sensing of pathogens or microbe-associated molecular patterns (PAMPs or MAMPs) by cell surface-localized pattern recognition receptors (PRRs) (Zipfel 2008), which leads to induction of pattern-triggered immunity (PTI). This activates downstream immune responses, including induction of an oxidative burst via accumulation of reactive oxygen species (ROS), cell wall reinforcement, activation of CDPKs and MAPKs, and rapid changes in expression of signaling-associated genes (Jones and Dangl 2006; Zipfel 2008). Plants have also evolved resistance mechanisms that recognize specific effectors, which induce effector-triggered immunity (ETI). Plant PRRs are plasma membrane-localized receptor-like kinases (RLKs) or receptor-like proteins (RLPs) that can form multiprotein complexes with PRRs at the cell membrane (Zipfel 2014). The extracellular leucine-rich repeats (LRR)-RLK BRASSINOSTEROID INSENSITIVE-1 (BRI1) is a receptor complex with various LRR containing PRRs for positive PTI regulation (Roux et al. 2011; Sun et al. 2013). The BAK1-dependent signaling pathway is conserved in different PRRs; this involves transphosphorylation of the BAK1-PRR and phosphorylation of the cytoplasmic BOTRYTIS-INDUCED KINASE 1 (BIK1) (Lu et al. 2010; Han et al. 2014). Phosphorylation of BIK1 by BAK1 activates NADPH oxidase D (RbohD), which then activates downstream signaling pathways such as the MAP kinase cascade (Asai et al. 2002; Lu et al. 2010; Kadota et al. 2014; Li et al. 2014). Interestingly, a recent study suggests that nematode infection triggers PTI responses by surface-localized receptors on host cells. Peng and Kaloshian (2014) reported that silencing of tomato BAK1 orthologues SISERK3A or SISERK3B in transgenic plants increased their susceptibility to nematode infection due to defective activation of basal defenses. In Arabidopsis, nematode infection triggers PTI responses via BAK1-dependent and independent pathways. BAK1 mutants show significantly greater susceptibility to RKN than wild-type controls (Teixeira et al. 2016). Nematode-derived elicitors are capable of inducing PTI responses in Arabidopsis in a BAK1-dependent manner (Mendy et al. 2017). Here, we also observed BAK1-dependent responses in the two different sweetpotato cultivars (Fig. 7). The data suggest that nematode-mediated induction of PTI responses by the two sweetpotato cultivars during RKN infection involves BAK1-related signaling via Rbohs, CDPKs, and MAPKs. Taken together, the results indicate that two different sweetpotato cultivars mount BAK1-related PTI responses, suggesting the existence of diverse nematode recognition responses.

Comparison of the relative expression levels of BAK1-dependent response-related genes in two sweetpotato cultivars under control and RKN treatment. DEGs including BAK1, Rbohs, CDPKs, and MAPKs The heatmap, which was constructed using MultiExperiment Viewer v. 4.9 (MEV4.9), displays the normalized transcript levels of each gene
Identifying DEGs in RKN-susceptible and resistant sweetpotato cultivars was of primary importance, since the differential expression of genes involved in hormonal signal transduction during RKN infection might play a key role in the resistance response. The DEGs identified in this study showed comparable expression profiles under both types of analysis, with the qRT-PCR results positively correlated with the RNA-Seq data (Figs. 3, 4). This set of genes exhibited distinctive responses to nematode challenge, including genes related to phytohormone signaling via TFs and PR genes, which participate in plant resistance to RKN in sweetpotato roots.
During RKN infection, various TF genes functioned in sweetpotato roots (Fig. S3). Among these, WRKY TFs are involved in SA signaling pathways during pathogen and nematode infection (Grunewald et al. 2008; Rushton et al. 2010; Holbein et al. 2016). ERFs and MYC TFs also regulate ET and JA signaling pathways under biotic stress conditions, including nematode infection (Nahar et al. 2011; Santino et al. 2013; Holbein et al. 2016). Putative SA-dependent WRKY53/WRKY70 genes were not expressed or only slightly induced upon RKN infection in both cultivars, whereas putative ET-dependent ERF1/ERF2 and JA-dependent MYC2/MYC4 genes were more highly upregulated in Juhwangmi than in Yulmi (Fig. 3). PR genes have been implicated in active defense responses and may play a role in restricting pathogen development and spread in plants (Van Loon et al. 2006). These genes are also induced in response to bacterial, fungal, viral, and nematode infection (Holbein et al. 2016).
Analyzing the association of the expression profiles of these DEGs in response to RKN infection with their primary functions in defense might help unravel their putative physiological roles during this incompatible plant–nematode interaction. In general, SA activates the plant defense response to biotrophic and hemi-biotrophic pathogens, induces systemic acquired resistance (SAR), and triggers the expression of SAR-associated PR genes. SA has already been shown to play important physiological and biochemical roles in decreasing susceptibility to RKN, as this phytohormone can induce the accumulation of reactive oxygen species, which occurs during the early stages of incompatible interactions and is a component of the hypersensitive response (HR) (Nandi et al. 2003; Branch et al. 2004; Melillo et al. 2006). During nematode infection in Arabidopsis, PR1, PR2, and PR5, which are markers for SA-dependent SAR, are induced in infected plants (Thomma et al. 1998). In the SA signaling pathway, major TFs, including WRKY53 and WRKY70, regulate the expression of PR1 (Eulgem and Somssich 2007). In the current study, PR genes such as PR1, PR2, and PR5, and WRKY TF genes such as WRKY53 and WRKY70 were slightly responsive to RKN infection in resistant cultivar Juhwangmi (Fig. 3), suggesting that SA signaling pathways likely play a minor role in the resistance mechanism in Juhwangmi plants in response to RKN infection.
JA and ET are important for the induction of nonspecific disease resistance and play an important role in induced resistance, activating genes involved in plant defense in response to pathogen attack and wounding (Thomma et al. 1998). JA is closely associated with the generation of various secondary metabolites, such as flavonoids, and can also trigger the biosynthesis of defensive proteins in roots, which are toxic to nematodes in vitro and have been implicated in plant defense against these nematodes (Soriano et al. 2004). During nematode infection in Arabidopsis, JA-dependent resistance also appears to be effective against necrotrophic pathogens (Staswick et al. 1998; Vijayan et al. 1998), and PR3, PR4, PR6, and PR10, which are markers for the JA-dependent SAR response, are induced in infected plants (Thomma et al. 1998). Among PR6 proteins, patatin from potato and sporamin from sweetpotato exhibits trypsin-inhibiting activity and plays an essential role in the HR and cell death during plant–pathogen interactions, as they mediate the production of JA and defense responses in some plant species (Yeh et al. 1997; Camera et al. 2009). Other proteins involved in mounting resistance responses via the regulation of JA include chitinase (PR3 and PR4) and RNase (PR10), which also play regulatory roles in the JA signaling pathway (Liu and Ekramoddoullah 2006; Hammouch et al. 2011). Major TFs in the JA/ET signaling pathway, including MYCs such as MYC2 and MYC4 and ERFs such as ERF1 and ERF2, regulate the expression of ET- and/or JA-responsive genes (Figs. 3, S2). Our results show that TF genes such as MYC2, MYC4, ERF1, and ERF2 and PR genes such as PR3, PR6, and PR10 were highly responsive to RKN infection in resistant cultivar Juhwangmi (Fig. 3).
In summary, we identified changes in the expression of important functional genes in RKN-susceptible and resistant sweetpotato cultivars during nematode parasitism. We identified candidate genes that might trigger changes in specific signaling pathways involved in hormonal regulation and/or defense gene-related resistance to RKN in sweetpotato. The identification of individual or multiple RKN-resistance genes by marker-assisted selection or the introduction of these genes into plants via transgenic expression could offer several benefits for nematode control in an integrated management system and could provide broad-spectrum nematode resistance in a variety of useful crop plants. Further investigation will be required to elucidate the exact functional role of each candidate gene in the regulation of the defense signaling pathways induced in sweetpotato after infection with RKN. Transgenic sweetpotato plants expressing each candidate gene will be generated to analyze their roles in RKN-resistant stress mechanism in plants. We expect that these studies, including those requiring suppression of candidate genes in transgenic plants, will provide valuable information for the development of crops with enhanced resistance to RKN infection.
Author contribution statement
YHK, IHL, DS, and JCJ conceived and designed the experiments. JWY and YWS performed the experiments. KJN analyzed the data. JH and JJL contributed analysis/materials/reagents tools. YHK, IHL, DS, and JCJ wrote the paper.
Abbreviations
- DEG:
-
Differentially expressed genes
- ET:
-
Ethylene
- JA:
-
Jasmonic acid
- PR:
-
Pathogenesis-related
- RKN:
-
Root-knot nematode
- SA:
-
Salicylic acid
- TF:
-
Transcription factor
References
Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15:63–78
Anderson JP, Badruzsaufari E, Schenk PM, Manners JM, Desmond OJ, Ehlert C, Maclean DJ, Ebert PR, Kazan K (2004) Antagonistic interaction between abscisic acid and jasmonate-ethylene signaling pathways modulates defense gene expression and disease resistance in Arabidopsis. Plant Cell 16:3460–3479
Asai T, Tena G, Plotnikova J, Willmann MR, Chiu WL, Gomez-Gomez L, Boller T, Ausubel FM, Sheen J (2002) MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 415:977–983
Boter M, Ruiz-Rivero O, Abdeen A, Prat S (2004) Conserved MYC transcription factors play a key role in jasmonate signaling both in tomato and Arabidopsis. Genes Dev 18:1577–1591
Branch C, Hwang CF, Navarre DA, Williamson VM (2004) Salicylic acid is part of the Mi-1-mediated defense response to root-knot nematode in tomato. Mol Plant Microbe Int 17:351–356
Bridge J, Starr JL (2010) Plant nematodes of agricultural importance a color handbook. Academic Press, San Diego, pp 77–78
Camera SL, Balagué C, Göbel C, Geoffroy P, Legrand M, Feussner I, Roby D, Heitz T (2009) The Arabidopsis patatin-like protein 2 (PLP2) plays an essential role in cell death execution and differentially affects biosynthesis of oxylipins and resistance to pathogens. Mol Plant Microbe Int 22:469–481
Castagnone-Sereno P, Danchin EG, Perfus-Barbeoch L, Abad P (2013) Diversity and evolution of root knot nematodes, genus Meloidogyne: new insights from the genomic era. Annu Rev Phytopathol 51:203–220
Cervantes-Flores JC, Yenchom GC, Pecotam KV, Sosinski B (2008) Detection of quantitative trait loci and inheritance of root-knot nematode resistance in sweetpotato. J Am Soc Hortic Sci 133:844–851
Choi DR, Lee JK, Park BY, Chung MN (2006) Occutrrence of rootknot nematodes in sweet potato fields and resistance screening of sweet potato cultivars. Korean J Appl Entomol 45:211–216
Clark CA, Moyer JW (1998) Compendium of sweetpotato diseases. APS Press, Saint Paul
De Vos M, Van Oosten VR, Van Poecke RM, Van Pelt JA, Pozo MJ, Mueller MJ, Buchala AJ, Metraux JP, Van Loon LC, Dicke M, Pieterse CMJ (2005) Signal signature and transcriptome changes of Arabidopsis during pathogen and insect attack. Mol Plant Microbe In 18:923–937
Diaz JT, Chinn MS, Truong VD (2014) Simultaneous saccharification and fermentation of industrial sweetpotatoes for ethanol production and anthocyanins extraction. Ind Crops Prod 62:53–60
Dombrecht B, Xue GP, Sprague SJ, Kirkegaard JA, Ross JJ, Reid JB, Fitt GP, Sewelam N, Schenk PM, Manners JM, Kazan K (2007) MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 19:2225–2245
Eulgem T, Somssich IE (2007) Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10:366–371
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. https://doi.org/10.1093/bioinformatics/bts565
Fujita M, Fujita Y, Noutoshi Y, Takahashi F, Narusaka Y, Yamaguchi-Shinozaki K, Shinozaki K (2006) Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Curr Opin Plant Biol 9:436–442
Grace MH, Yousef GG, Gustafson SJ, Truong VD, Yencho GC, Lila MA (2014) Phytochemical changes in phenolics, anthocyanins, ascorbic acid, and carotenoids associated with sweetpotato storage and impacts on bioactive properties. Food Chem 145:717–724
Grunewald W, Karimi M, Wieczorek K, Van de Cappelle E, Wischnitzki E, Grundler F, Inzé D, Beeckman T, Gheysen G (2008) A role for AtWRKY23 in feeding site establishment of plant-parasitic nematodes. Plant Physiol 148:358–368
Ha J, Won JC, Jung YH, Yang JW, Lee HU, Nam KJ, Park SC, Jeong JC, Lee SW, Lee DW, Chung JS, Lee JJ, Kim YH (2017) Comparative proteomic analysis of the response of fibrous roots of nematode-resistant and -sensitive sweetpotato cultivars to root-knot nematode Meloidogyne incognita. Acta Physiol Plant 39:262. https://doi.org/10.1007/s11738-017-2560-0
Hamamouch N, Li C, Seo PJ, Park CM, Davis EL (2011) Expression of Arabidopsis pathogenesis-related genes during nematode infection. Mol Plant Pathol 12:355–364
Han Z, Sun Y, Chai J (2014) Structural insight into the activation of plant receptor kinases. Curr Opin Plant Biol 20:55–63
Holbein J, Grundler FMW, Siddique S (2016) Plant basal resistance to nematodes: an update. J Exp Bot 67:2049–2061
Jones JD, Dangl JL (2006) The plant immune system. Nature 444:323–329
Kadota Y, Sklenar J, Derbyshire P, Stransfeld L, Asai S, Ntoukakis V, Jones JD, Shirasu K, Menke F, Jones A, Zipfel C (2014) Direct regulation of the NADPH oxidase RBOHD by the PRR-associated kinase BIK1 during plant immunity. Mol Cell 54:43–55
Kazan K, Manners JM (2008) Jasmonate signaling: toward an integrated view. Plant Physiol 146:1459–1468
Kistner MH, Daiber KC, Bester C (1993) The effect of root-knot nematodes (Meloidogyne spp.) and dry land conditions on the production of sweetpotato. JS Afr Soc Hortic Sci 3:108–110
Kreuze J (2002) Molecular studies on the sweetpotato virus disease and its two causal agents. In: Acta Universitatis Agriculturae Sueciae Agraria 335. Department of Plant Biology, Sveriges lantbrukuniversitet, Uppsala
Lee JJ, Park KW, Kwak YS, Ahn JY, Jung YH, Lee BH, Jeong JC, Lee HS, Kwak SS (2012) Comparative proteomic study between tuberous roots of light orange- and purple-fleshed sweetpotato cultivars. Plant Sci 193–194:120–129
Li L, Li M, Yu L, Zhou Z, Liang X, Liu Z, Cai G, Gao L, Zhang X, Wang Y, Chen S, Zhou JM (2014) The FLS2-associated kinase BIK1 directly phosphorylates the NADPH oxidase RbohD to control plant immunity. Cell Host Microbe 15:329–338
Liu JJ, Ekramoddoullah AKM (2006) The family 10 of plant pathogenesis-related proteins: their structure, regulation, and function in response to biotic and abiotic stresses. Physiol Mol Plant Pathol 68:3–13
Lorenzo O, Chico JM, Sanchez-Serrano JJ, Solano R (2004) JASMONATE-INSENSITIVE1 encodes a MYC transcription factor essential to discriminate between different jasmonate-regulated defense responses in Arabidopsis. Plant Cell 16:1938–1950
Lu DP, Wu SJ, Gao XQ, Zhang YL, Shan LB, He P (2010) A receptor-like cytoplasmic kinase, BIK1, associates with a flagellin receptor complex to initiate plant innate immunity. Proc Natl Acad Sci USA 107:496–501
Melillo MT, Leonetti P, Bongiovanni M, Castagnone-Sereno P, Bleve-Zacheo T (2006) Modulation of reactive oxygen species activities and H2O2 accumulation during compatible and incompatible tomato-root knot nematode interactions. New Phytol 170:501–512
Mendy B, Wangombe MW, Radakovic ZS, Holbein J, Ilyas M, Chopra D, Holton N, Zipfel C, Grundler FMW, Siddique S (2017) Arabidopsis leucine-rich repeat receptor-like kinase NILR1 is required for induction of innate immunity to parasitic nematodes. Plos Pathol 13:e1006284
Nahar K, Kyndt T, De Vleesschauwer D, Hofte M, Gheysen G (2011) The jasmonate pathway is a key player in systemically induced defense against root knot nematodes in rice. Plant Physiol 157:305–306
Nandi B, Kundu K, Banerjee N, Babu SPS (2003) Salicylic acid-induced suppression of Meloidogyne incognita infestation of okra and cowpea. Nematology 5:747–752
Palomares-Rius JE, Kikuchi T (2013) Omics fields of study related to plant-parasitic nematodes. J Integr Omics 3:1–10
Park SC, Kim YH, Ji CY, Park S, Jeong JC, Lee HS, Kwak SS (2012) Stable internal reference genes for the normalization of real-time PCR in different sweetpotato cultivars subjected to abiotic stress conditions. PLoS ONE 7:e51502
Peng HC, Kaloshian I (2014) The tomato leucine-rich repeat receptor-like kinases SISERK3A and SISERK3B have overlapping functions in bacterial and nematode innate immunity. PLoS ONE 9:e93302
Roux M, Schwessinger B, Albrecht C, Chinchilla D, Jones A, Holton N, Malinovsky FG, Tor M, de Vries S, Zipfel C (2011) The Arabidopsis leucine-rich repeat receptor-like kinases BAK1/SERK3 and BKK1/SERK4 are required for innate immunity to hemibiotrophic and biotrophic pathogens. Plant Cell 23:2440–2455
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15:247–258
Santino A, Taurino M, De Domenico S, Bonsegna S, Poltronieri P, Pastor V, Flors V (2013) Jasmonate signaling in plant development and defense response to multiple (a)biotic stresses. Plant Cell Rep 32:1085–1098
Soriano I, Riley I, Potter M, Bowers W (2004) Phytoecdysteroids: a novel defense against plant-parasitic nematodes. J Chem Ecol 30:1885–1899
Staswick PE, Yuen GY, Lehman CC (1998) Jasmonate signaling mutants of Arabidopsis are susceptible to the soil fungus Pythium irregulare. Plant J 15:747–754
Sun YD, Li L, Macho AP, Han ZF, Hu ZH, Zipfel C, Zhou JM, Chai JJ (2013) Structural basis for flg22-induced activation of the Arabidopsis FLS2-BAK1 immune complex. Science 342:624–628
Teixeira MA, Wei L, Kaloshian I (2016) Root-knot nematodes induce pattern- triggered immunity in Arabidopsis thaliana roots. New Phytol 211:279–287
Thomma BPHJ, Eggermont K, Penninckx IAMA, Mauch-Mani B, Vogelsang R, Cammue BPA, Broekaert WF (1998) Separate jasmonate-dependent and salicylate-dependent defense-response pathways in Arabidopsis are essential for resistance to distinct microbial pathogens. Proc Natl Acad Sci USA 95(15107–15):111
Van Loon LC, Rep M, Pieterse CMJ (2006) Significance of inducible defense-related proteins in infected plants. Ann Rev Phytopathol 44:135–162
Viaene N, Smol N, Bert W (2012) General techniques in nematology. Academia Press, Gent. Belgium, pp 58–59
Vijayan P, Shockey J, Levesque CA, Cook RJ, Browse J (1998) A role for jasmonate in pathogen defense of Arabidopsis. Proc Natl Acad Sci USA 95:7209–7214
Williamson VM, Kumar A (2006) Nematode resistance in plants: the battle underground. Trends Genet 22:396–403
Woolfe JA (1992) Sweetpotato: an untapped food resource. Cambridge University Press, Cambridge
Yeh KW, Lin MI, Tuan SJ, Chen YM, Lin CY, Kao SS (1997) Sweetpotato (Ipomoea batatas) trypsin inhibitors expressed in transgenic tobacco plants confer resistance against Spodoptera litura. Plant Cell Rep 16:696–699
Zipfel C (2008) Pattern-recognition receptors in plant innate immunity. Curr Opin Immunol 20:10–16
Zipfel C (2014) Plant pattern-recognition receptors. Trends Immunol 35:345–351
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (2018R1A1A1A05018446).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, I.H., Shim, D., Jeong, J.C. et al. Transcriptome analysis of root-knot nematode (Meloidogyne incognita)-resistant and susceptible sweetpotato cultivars. Planta 249, 431–444 (2019). https://doi.org/10.1007/s00425-018-3001-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-018-3001-z