
Abstract
Main conclusion
Overexpression of BnaWGR1 causes ROS accumulation and promotes leaf senescence. BnaWGR1 binds to promoters of RbohD and RbohF and regulates their expression.
Manipulation of leaf senescence process affects agricultural traits of crop plants, including biomass, seed yield and stress resistance. Since delayed leaf senescence usually enhances tolerance to multiple stresses, we analyzed the function of specific MAPK–WRKY cascades in abiotic and biotic stress tolerance as well as leaf senescence in oilseed rape (Brassica napus L.), one of the important oil crops. In the present study, we showed that expression of one WRKY gene from oilseed rape, BnaWGR1, induced an accumulation of reactive oxygen species (ROS), cell death and precocious leaf senescence both in Nicotiana benthamiana and transgenic Arabidopsis (Arabidopsis thaliana). BnaWGR1 regulates the transcription of two genes encoding key enzymes implicated in production of ROS, that is, respiratory burst oxidase homolog (Rboh) D and RbohF. A dual-luciferase reporter assay confirmed the transcriptional regulation of RbohD and RbohF by BnaWGR1. In vitro electrophoresis mobility shift assay (EMSA) showed that BnaWGR1 could bind to W-box cis-elements within promoters of RbohD and RbohF. Moreover, RbohD and RbohF were significantly upregulated in transgenic Arabidopsis overexpressing BnaWGR1. In summary, these results suggest that BnaWGR1 could positively regulate leaf senescence through regulating the expression of RbohD and RbohF genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leaf senescence is a fine-tuned active degenerative process through degradation of macromolecules including chlorophylls, proteins and so on, which recycles nutrients released from the old organs to growing young leaves and developing fruits and seeds (Lim and Nam 2005). Leaf senescence is controlled by many factors including age, nutrition status, abiotic and biotic stress conditions, and plant hormones, which interplay in synergistic or antagonistic ways together with calcium signals (Lim et al. 2007). Reactive oxygen species (ROS), including singlet oxygen, superoxide anion, hydrogen peroxide (H2O2) and hydroxyl radical, are both signaling and toxic molecules, depending on the concentration (Mittler et al. 2004). At lower concentrations, ROS are utilized as signaling molecules to regulate development and various physiological responses. At higher concentrations, ROS can cause oxidative damage to proteins, DNA, and lipids (Mittler et al. 2004). Plants have evolved complex arrays of non-enzymatic and enzymatic detoxification mechanisms to detoxify excessive ROS (Apel and Hirt 2004). It is believed that ROS production arising from the disassembly of the chloroplast and the degradation of chlorophyll induces leaf senescence (Rogers and Munne-Bosch 2016). Among the ROS, H2O2 is most studied as it has a relatively longer half-life, facilitating research. It has been shown in Arabidopsis that the hydrogen peroxide level starts to increase at the time points when plants start to bolt and also senescence-associated genes (SAGs) are enhanced by the elevated level of ROS (Ye et al. 2000; Navabpour et al. 2003). It has been reported that there is a causal link between WRKY-mediated leaf senescence and ROS in Arabidopsis (Besseau et al. 2012). However, it remains elusive how the transcriptional activity of a WRKY protein results in modulating cellular ROS levels.
The respiratory burst oxidase homologues (Rbohs) are the most studied ROS-producing enzymes, serving as main molecular ‘hubs’ in ROS-mediated signaling including cell growth, plant development such as root hair, abiotic stress and immune responses (Marino et al. 2012). In Arabidopsis thaliana, there are 10 Rboh genes, which are RbohA through RbohJ (Marino et al. 2012). Of them, AtRbohD and -F are two prominent members and it has been shown that AtRbohD and -F are required in ABA-mediated ROS production and stomatal closure (Kwak et al. 2003), and in ROS burst of pathogen/microbe-associated molecular patterns (PAMP/MAMPs)-triggered immunity (PTI) and effector-triggered immunity (ETI) (Torres et al. 2002). Reports have demonstrated that the activity of Rbohs can be modulated at post-translational level through phosphorylation of the N-terminal cytosolic domain (Baxter et al. 2014). It has also been shown that Rboh genes could be regulated by specific NAC and WRKY transcription factors at the transcriptional level (Lee et al. 2012; Adachi et al. 2015). For instance, phospho-mimicking forms of tobacco WRKY7, -8, -9 and -11 belonging to Group I bind to the W-box element in the RbohB promoter to positively regulate its expression and the activation of RbohB by the MPK-WRKY pathway is involved in R3a/AVR3a-mediated ETI immunity and INF1-triggered immunity (Adachi et al. 2015). However, whether there is any other non-Group I WRKY protein that regulates Rboh transcription remains unknown.
WRKY transcription factors form a large family in either model plant or crops, with 72 members in Arabidopsis (Eulgem et al. 2000). WRKY transcription factors are classified into three major groups: WRKY proteins with two WRKY domains and C2H2 zinc motifs belong to Group I, whereas those with one WRKY domain and C2H2 zinc motifs belong to Group II. WRKY proteins having only one WRKY domain but having C2HC zinc-finger motifs are categorized into Group III. Group II WRKY proteins are further divided into IIa, IIb, IIc, IId, and IIe based on the primary amino acid sequences (Eulgem et al. 2000). WRKYs function in mediating disease resistance and expression of defense-related genes in systemic acquired resistance (SAR) (Zheng et al. 2006; Knoth et al. 2007; Bhattarai et al. 2010; Chen et al. 2010; Birkenbihl et al. 2012), abiotic stress (Bakshi and Oelmuller 2014), and so on. WRKY transcription factors usually recognize and bind to a W-box element, which has a consensus TTGAC(C/T) sequence (Ciolkowski et al. 2008). After binding to the W-box element contained in the promoter region of a downstream target gene, a WRKY transcription factor may activate or repress the transcription of the target gene or both. In Arabidopsis, six WRKY proteins have been shown to positively or negatively regulate leaf senescence, including WRKY6 (Robatzek and Somssich 2001), -22 (Zhou et al. 2011), -53 (Miao et al. 2004), -54 (Besseau et al. 2012), -57 (Jiang et al. 2014) and -70 (Besseau et al. 2012). For instance, AtWRKY6 positively regulated leaf senescence by specifically activating the senescence-induced receptor-like kinase (SIRK) gene (Robatzek and Somssich 2002). AtWRKY53 played a regulatory role in early events of leaf senescence (Hinderhofer and Zentgraf 2001). AtWRKY53 could bind to the promoters of CAT2 and -3 to transactivate their expression, which encode two ROS-scavenging enzymes (Miao et al. 2004). Usually, it is believed that the upregulation of ROS-scavenging enzymes is largely to prevent premature senescence when senescence initiates (Zimmermann et al. 2006). With the proceeding of the senescence, the H2O2 level increases to destroy the cellular integrity leading to the remobilization of the nutrient out of the senescent leaf. It was speculated that the first H2O2 peak might increase the level of WRKY53 (Zimmermann et al. 2006). However, whether AtWRKY53 could directly mediate ROS accumulation during the process of leaf senescence remains unknown.
Oilseed rape (Brassica napus L.) is one of the most important oil crops in the world. The regulatory mechanisms and key transcription factors controlling leaf senescence in oilseed rape remain unclear. Dissecting the senescence network in oilseed rape will not only provide approaches to control premature leaf senescence, but also offer alternative strategies of enhancing stress tolerance (Rivero et al. 2007). Previously, we performed a systematic identification, cloning and analysis of WRKY transcription factor genes and their upstream kinase genes in oilseed rape (Yang et al. 2009; Liang et al. 2013). In this study, we report the functional characterization of one of the WRKY-type transcription factor genes BnaWGR1 (WRKY generating ROS 1) from oilseed rape. In addition, we identified that its expression induced ROS accumulation in tobacco and leaf senescence in Arabidopsis, through modulating the expression of RbohD and -F.
Materials and methods
Plant materials and growth conditions
Seeds of oilseed rape (Brassica napus L. spring type, double haploid) were originally supplied by Dr. Michael Deyholos (UBCO, Canada) and those of Arabidopsis thaliana and Nicotiana benthamiana were from Lehle Seeds (Round Rock, TX, USA). Plants were grown in a growth chamber with 22/20 °C day/night temperatures under a light period of 14 h per day and a light intensity of 120 μmol m−2 s−1. Arabidopsis ecotype Col-0 was used as the wild type (WT). Seeds were surface sterilized and plated on half-strength Murashige and Skoog (MS, Caisson Labs, Smithfield, UT, USA) medium (2.15 g L−1 basal salts, 1% sucrose, pH 5.7 and 8 g L−1 Phytoblend) and imbibed for 2 d at 4 °C before germinated in a growth chamber. Seven-day-old seedlings were transferred into a commercial soil mix (Pindstrup, Kongerslev, Denmark).
Promoter cloning and GUS staining
A DNA fragment containing a promoter region of 1.5 kb upstream of the initiation codon was cloned from oilseed rape genomic DNA using primers listed in Table S1. Primers were designed through mining the public bacterial artificial clone (BAC) sequences stored in the NCBI database. After sequencing, the promoter was cloned into the binary vector pCAMBIA1391Z. The resulting construct was introduced into the Agrobacterium tumefaciens strain GV3101 and transformed into wild-type Arabidopsis Col-0 (Clough and Bent 1998). To screen transgenic homozygous lines, T1 seeds were selected on hygromycin (Roche, Mannheim, Germany)-containing 1/2 × MS medium for 7 days before they were transferred to soil to get T2 seeds individually. Then, individual T2 seeds were plated on hygromycin-containing 1/2 × MS medium to evaluate homozygosity. Only homozygous T2 lines resistant to hygromycin were subjected to histochemical staining of GUS as described previously (Jiang and Deyholos 2009).
β-Glucuronidase (GUS) activity assay
Measurement of GUS activity was performed according to Kim et al. (2006). Briefly, tissue was ground in liquid nitrogen and mixed in 200 μL extraction buffer (50 mM sodium phosphate buffer, 10 mM EDTA, 0.1% SDS, 0.1% Triton X-100 and 10 mM β-mercaptoethanol). 15 μg total protein was transferred to 980 μL of 1 mM 4-methylumbelliferyl-β-d-glucuronide (4-MUG) dissolved in the extraction buffer. The reaction was stopped by 0.2 M sodium carbonate buffer (pH11.3) at 5, 10, 20, 40 min, respectively, for fluorescence measurement on a multiple-well plate reader (Spectra Max M2, Molecular Devices, CA, USA) with an excitation wavelength of 365 nm and an emission at 455 nm. The resultant product, 4-methylumbelliferone (4-MU), was quantified against a standard curve.
Subcellular localization assay
To construct the BnaWGR1 fused with GFP expression vectors, the full-length coding sequence of BnaWGR1 was subcloned into a modified pYJGFP vector with C-terminal fusion with GFP and into a p35SNGFP with N-terminal fusion with GFP, respectively, using the primers listed in Supplementary Table S1. The NLS-mCherry expression cassette was constructed by fusing nuclear localization signal (NLS) from pGADT7 vector (Clontech, Mountain View, CA, USA) upstream of mCherry gene in binary vector pYJmCherry using the primers listed in Table S1. These two constructs were individually introduced into A. tumefaciens strain GV3101 and infiltrated into the abaxial surface of 21-day-old N. benthamiana leaves using 1-mL needleless syringes, together with P19, a gene silencing repressor (Liang et al. 2013). After infiltration, N. benthamiana was grown in a growth chamber for 24 h before 50 μM MG132 (Selleck, Houston, TX, USA) was infiltrated. GFP signals were observed 24 h later with a FV1000MPE confocal microscopy (Olympus, Shinjuku-ku, Tokyo, Japan).
Construction of plasmids and generation of transgenic plants
The coding region of BnaWGR1 was cloned to pYWLHA vector through Nco I and Sal I sties under the control of cauliflower mosaic virus (CaMV) 35S promoter. After mobilization into A. tumefaciens strain GV3101 through the freeze–thaw method, pYWLHA-BnaWGR1 was transformed into WT Col-0 through a floral dip protocol (Clough and Bent 1998). Transgenic lines were selected on 1/2 × MS medium containing 25 mg L−1 hygromycin for two generations to harvest homozygous T3 seeds. High-expression lines were screened through qRT-PCR.
Quantitative RT-PCR (qRT-PCR) assay
Leaf tissues of at least three biological replicates were harvested at the specified time points for RNA extraction using Plant RNA kit (Omega bio-tek, Salt Lake City, UT, USA) followed by removal of genomic DNA using DNA-free kit (Ambion, Vilnius, Lithuania). The first-strand cDNA was synthesized from 2.5 μg of RNA using oligo(dT)18 and RNase H-MMLV (TaKaRa, Kusatsu, Shiga, Japan). Relative qRT-PCR was performed as described earlier using tenfold-diluted cDNA and a SYBR Green I kit (CWBIO, Beijing, China) on a CFX96 real-time PCR system (Bio-Rad, Hercules, CA, USA). Each primer pair was examined for amplification specificity and efficiency. The fold changes, normalized to two internal reference genes, were calculated using the formula proposed by Pfaffl (2001). For N. benthamiana, NbL23 (ribosomal large subunit 23) and NbPP2A (type 2A serine/threonine protein phosphatase) were used as reference genes (Liu et al. 2012). As for Arabidopsis, UBIQUITIN CONJUGATING ENZYME 21 (AtUBC21) and POLYUBIQUITIN 10 (AtUBQ10) were used as reference genes (Czechowski et al. 2005).
For absolute qRT-PCR, each gene was cloned into the pJET1.2 vector. Serial tenfold dilutions of plasmids were performed to generate standard curve of each gene by plotting log ratio of the copy number of template DNA against the Ct value (Bustin 2000). The regression equation was generated for each gene to calculate the corresponding template copies using the formula of N (copy numbers) = quantity of plasmid/(base numbers of plasmid × 660 Da) × NA (6.02 × 1023 copies/mol). Primers are listed in Supplementary Table S1.
Transient expression and physiological assays
Plasmids of pYWLHA-BnaWGR1 contained in GV3101 were used for transient expression in true leaves of 30-day-old N. benthamiana plants. Agrobacteria harboring P19, a silencing suppressor, and BnaWGR1 plasmids, respectively, were mixed at a ratio of 1:1 before infiltration. Leaf discs were immersed in deionized water for 30 min at 25 °C. Electric conductivity was measured using a DDS-307 conductivity meter (Leici, Shanghai, China) (Oh and Martin 2011).
3,3′-Diaminobenzidine (DAB, MP Biomedicals, Solon, OH, USA) staining was performed according to Daudi et al. (2012). For nitroblue tetrazolium (NBT, Amresco, Solon, OH, USA) staining, leaves were cut from plants and placed in 9-cm Petri dishes containing 10 mM NaN3 (Sigma-Aldrich, St. Louis, MO, USA) in 10 mM potassium phosphate buffer (pH 7.8) before vacuumed for 30 min. Then, leaves were transferred into 0.1% NBT solution and 10 mM potassium phosphate buffer (pH7.8) and vacuumed for 30 min again. After incubation and shaking for 2 h at room temperature, leaves were decolorized in AGE solution (acetic acid:glycerol:ethanol, 1:1:3, by vol.) and boiled for 30 min before photographed (Jambunathan 2010). The content of H2O2 was quantified as described previously (Li et al. 2015). Measurement of chlorophyll was performed as described based on the method reported previously (Niu et al. 2016). In brief, 0.1 g tissue was immersed in 5 mL absolute ethanol overnight followed by measuring the absorbance at 665, 649 and 470 nm on a spectrophotometer (Thermo Scientific, Waltham, MA, USA). Malondialdehyde (MDA) was extracted from 0.1 g leaf tissue by 0.1% trichloroacetic acid (TCA, Alfa Aesar, Tianjing, China) followed by centrifugation. One milliliter of supernatant was then transferred and mixed with 2 mL of 20% TCA and 2 mL of 0.5% TBA before boiling. After cooling down, the absorbance was measured at 532 and 600 nm, respectively (Jambunathan 2010).
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL, Promega, Madison, WI, USA) assay was performed as described (Li et al. 2015).
Dual-luciferase reporter assay
Promoter regions plus 5′untranslated region (UTR) were amplified from genomic DNA using the primers listed in Table S1. After confirmed through sequencing, they were cloned upstream of firefly luciferase (LUC) gene in the pGreenII0800-LUC vector as reporter plasmids (Hellens et al. 2005). An expression cassette harboring an endogenous control Renilla luciferase (REN) gene driven by 35S promoter was also located in the T-DNA region of pGreenII0800-LUC vector. pYWLHA-BnaWGR1 and pYJHA-GFP were used as effector plasmid and control plasmid, respectively. The dual-LUC reporter assay was performed on 30-day-old N. benthamiana leaves using a dual-LUC kit (Promega) as described previously (Niu et al. 2016).
Senescence assay
Seeds of different genotypes were harvested from plants grown under the same conditions and at the same time. Seven-day-old seedlings of different genotypes were transferred from 1/2 × MS medium to soil and grown as described previously. For age-dependent leaf senescence comparison, age-matched true leaves from 38- and 43-day-old plants were used for analyzing the percentage of each group, including green, green/yellow, fully yellow, and brown/dry. The 5th, 6th and 7th true leaves of 45-day-old plants were used for chlorophyll content measurement. A percentage of yellow leaves was analyzed from 6th to 10th true leaves when the plants were 59 days old.
Prokaryotic expression, purification and electrophoretic mobility shift assay (EMSA)
BnaWGR1 was subcloned to pGEX-4T-1 vector (Amersham, Fairfield, CT, USA) through the restriction sites of BamH I and Xho I for prokaryotic expression. Glutathione S-transferase (GST)-fused BnaWGR1 was expressed and purified from E.coli. Briefly, pGEX-4T-1-BnaWGR1 was transformed into BL21 (DE3) Codonplus strain (Stratagene, Santa Clara, CA, USA) and fusion protein was induced by isopropyl thio-β-d-galactoside (IPTG) at 25 °C for 6 h in 500 mL of 2 × YT medium. Cell pellets were harvested for lysing and purification was performed using GST-bind resin (Novagen, Madison, WI, USA). GST proteins expressed from the empty vector pGEX-4T-1 were also purified and used as a negative control.
Different DNA fragments containing W-box elements were PCR amplified using primers listed in Table S1. Purified fragments were labeled with biotin using the biotin 3′ end DNA labeling kit (Pierce, Waltham, MA, USA). To perform the DNA–protein binding reaction, 2000 ng purified BnaWGR1-GST was incubated with 20 fmol 3′-biotin-labeled DNA and 50 ng Poly (dI·dC) in a total volume of 20 μL for 20 min at room temperature. Binding reactions were stopped by adding loading buffer and resolved on a 4–6% native polyacrylamide gel run in 0.5 × Tris–borate–EDTA (TBE) buffer. The DNA–protein complex was electrophoretically transferred to Hybond N+ nylon membrane (Amersham) followed by cross-linking at 120 mJ cm−2 for 45–60 s. To detect biotin-labeled DNA, the membrane was stabilized with streptavidin–horseradish peroxidase conjugate in a blocking buffer with 1–300 dilution after blocking. After equilibrating with substrate equilibration buffer, the membrane was subjected to mixture of luminol/enhancer solution and stable peroxide solution in a 1–1 ratio according to the manufacturer’s manual of LightShift Chemiluminescent EMSA Kit (Pierce). The membrane was then exposed on a ChemiDoc XRS+ system (Bio-Rad).
Statistical analysis
Statistical analysis was performed using SPSS Statistics 17.0, and differences were analyzed with one-way ANOVA followed by Duncan’s multiple comparison test. Student’s t test was performed in Microsoft Excel 2003. Graphs were edited in Photoshop CS (Adobe) or plotted by Sigmaplot 12.3 (Systat Software, Inc., San Jose, CA, USA).
Results
Expression pattern of BnaWGR1 and subcellular localization
Previously, we cloned over 53 cDNA sequences of WRKY genes from oilseed rape and investigated their response towards different abiotic and biotic stresses (Yang et al. 2009; Liang et al. 2013). A further microarray profiling of transcriptomic change during leaf senescence in oilseed rape indicated that quite a few WRKY genes were highly expressed in senescent leaves, one of which is BnaWGR1. The translated sequence of BnaWGR1 contains one DNA-binding WRKY domain, which harbors a C2HC-type zinc-finger motif at the C terminus. Thus, BnaWGR1 belongs to Group III in the WRKY family. When compared to the 72 Arabidopsis WRKYs and about 100 rice WRKYs, BnaWGR1 shows the highest homology to At4g23810 (AtWRKY53) and Os08g29660 (OsWRKY69), with an identity of 71.6 and 28.2%, respectively, at the amino acid levels.
Next, we examined the subcellular localization of BnaWGR1 by both N- and C-terminal fusion with GFP using Agrobacterium-mediated infiltration in leaves of N. benthamiana. We observed water-soaking symptoms on leaves infiltrated with BnaWGR1–GFP and GFP–BnaWGR1 plasmids, suggesting ROS accumulation and hypersensitive response-like cell death. We failed to detect any green signal in N. benthamiana leaves via agroinfiltration at the first try. We used a well-known 26S proteosome inhibitor, MG132, and co-infiltrated it into leaf cells 1.5 days after initial agroinfiltration and observed the GFP signal 12 h later. As a result, we successfully observed green signals in nuclei (Fig. 1a), which is consistent with the role of a transcription factor. Our results suggested that there was a rapid turnover of expressed BnaWGR1 in leaf cells, which was likely attributed to proteosomal degradation. It is known that protein levels of some specific TFs are tightly controlled in plant cells and a recent study on a tomato NAC TF gene also indicates this (Huang et al. 2013). In contrast, the GFP alone was localized to both cytoplasm and nuclei (Fig. 1a).

Assays of subcellular localization and promoter-GUS transgenic lines of BnaWGR1. a Subcellular localization assay of GFP and BnaWGR1–GFP in leaf cells of N. benthamiana. A plasmid harboring NLS-mCherry was co-transformed as a nuclear marker. GFP, bright and merge represent the GFP, bright and merged fields, respectively. Bars = 50 μm. b Histochemical staining of ProBnaWGR1-GUS transgenic plants. Two independent transgenic lines (17-2 and 19-5) were examined. The left to right panels indicate 7-day-old seedling, flowers, rosette leaves and siliques of 6-week-old mature plants
To examine the tissue specificity of BnaWGR1 expression, the promoter region together with 5′UTR was cloned and fused with β-glucuronidase (GUS) reporter gene. We examined the GUS expression driven by promoter of BnaWGR1 in two representative transgenic T2 lines. In 7-day-old seedlings, moderate GUS staining in vascular system of the cotyledons and in hypocotyls was observed (Fig. 1b). In 6-week-old mature plants, strong GUS expression in the sepals and rosette leaves was detected. In young siliques, moderate to strong GUS expression was detected in the two transgenic lines (Fig. 1b). At the age of 36 and 43 days, GUS activity of 9th–11th leaves was significantly higher than GUS activity of 9th–11th leaves at the age of 30 days (Fig. S1).
Expression of BnaWGR1 induces ROS accumulation and cell death
To further study the function of BnaWGR1, we cloned it into a binary vector, in which BnaWGR1 was under the control of CaMV 35S promoter. The transient expression of BnaWGR1 in leaves of N. benthamiana began to induce H2O2 accumulation and cell death from 3 days post-infiltration (dpi) compared to the GFP expression control (Fig. 2a). As time went on, the symptom of cell death became more severe.

Expression of BnaWGR1 induces accumulation of ROS and cell death. a Symptom of leaves of N. benthamiana after expression of BnaWGR1 and GFP at 2–9 days post-infiltration (dpi). DAB staining shows H2O2 accumulation. b NBT staining of elicited O2− in leaves expressing BnaWGR1 and GFP at 3–7 h post-infiltration (hpi). c Quantitative assay of H2O2 contents at three time points. d TUNEL assay of nuclear fragmentation in leaf tissues. DAPI staining was used to show nuclei. Scale bars = 50 μm. e Quantitative assay of malondialdehyde (MDA) contents in BnaWGR1- and GFP-expressing leaf tissues. f Measurement of electrolyte leakages in leaf tissues expressing BnaWGR1 and GFP. Data in c, e and f were averages of three biological replicates with error bars representing standard errors. Asterisks indicate significant difference between BnaWGR1- and GFP-expressing tissues (P < 0.05)
We also performed NBT staining to monitor the change in the content of superoxide anion (O2−), which is highly mobile, instable and could be converted to H2O2. Leaves expressing BnaWGR1 showed obvious accumulation of O2−, while GFP expression did not have such an effect (Fig. 2b). The content of H2O2 in the GFP- and BnaWGR1-expressing leaves was significantly higher in the BnaWGR1-expressing tissues compared to GFP control beginning from 3 dpi (Fig. 2c).
As there was an active genomic DNA degradation during cell death, we employed TUNEL assay to test this. The green signals of fluorescein isothiocyanate (FITC) were observed in nuclei of the BnaWGR1-expressing leaf tissue, while there was no signal of FITC signal in the control of GUS-expressing tissue (Fig. 2d), indicative of fragmentation of genomic DNA induced by BnaWGR1 expression. Moreover, malondialdehyde (MDA), from lipid peroxidation due to the attacked cell membrane by intracellular ROS, increased significantly at 5, 7 and 9 dpi in leaf tissues expressing BnaWGR1 (Fig. 2e). In addition, ion leakage increased significantly in BnaWGR1-expressing tissues compared to GFP at the three time points examined (Fig. 2f), which further supported that BnaWGR1 positively modulated ROS accumulation and cell death.
WGR1 regulates developmental leaf senescence
To further characterize the function of BnaWGR1, we first performed absolute quantitative RT-PCR to investigate the expression of BnaWGR1 in leaves of four different developmental stages of oilseed rape plants, which were represented by young leaves (YL), mature leaves (ML), early senescent (ES) and late senescent (LS) leaves (Fig. 3a). The four stages of oilseed rape leaves were also validated by monitoring transcript levels of three marker genes, which are BnaSAG12, BnaSEN1 and -4. The standard curve of each gene was generated by log ratio of the copy number of template DNA plotted against the Ct values (Fig. S2). As we expected, BnaSAG12 was specifically induced at LS, and BnaSEN1 and -4 were induced at ML, ES and LS (Fig. 3b). BnaWGR1 was significantly induced at ES and reduced to normal level at LS (Fig. 3b).

Expression analysis of BnaWGR1 during leaf senescence in oilseed rape. a The fourth true leaves of oilseed rape at four different developmental stages. YL young leaves of 21-day-old seedlings, ML fully expanded mature leaves of 28-day-old seedlings, ES early senescent leaves of 35-day-old seedlings, LS late senescent leaves of 44-day-old seedlings, with > 50% leaf area yellowing. b Absolute qRT-PCR analysis of transcript levels of BnaSAG12, BnaSEN1, BnaSEN4 and BnaWGR1 in wild-type leaves at different developmental stages. Data represent means of three biological replicates ± standard errors (SE). Different letters indicate significant difference (P < 0.05)
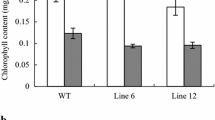
Furthermore, we expressed BnaWGR1 under the control of CaMV35S promoter in wild-type Arabidopsis. Among over 30 transgenic T3 lines, two BnaWGR1-overexpressing lines, which are OX#24 and OX#26, were identified by qRT-PCR (Fig. S3). A categorization of four groups of leaves according to the leaf colors including green, green/yellow, fully yellow, and brown/dry was used. The percentage of each category was recorded from 16 individually grown plants for statistical analysis. Using this categorization, BnaWGR1-OX#24 and #26 exhibited accelerated leaf senescence compared to 38- and 43-day-old WT plants (Fig. 4a and b). The percentages of green/yellow leaves of OX#24 and OX#26 were significantly higher than that of WT (Fig. 4a), whereas the percentages of green leaves of OX#24 and OX#26 were lower than that of WT (Fig. 4a, b). At 45 and 49 days post-stratification, earlier leaf senescence occurred in the two overexpression lines compared to WT as evidenced by rosette leaves (Fig. 4c). The content of chlorophyll between overexpression lines and WT was also compared in the age-matched rosette leaves. In 5th and 6th rosette leaves, the two overexpression plants had a lower chlorophyll content than WT at the age of 45 days (Fig. 4d). Similarly, the chlorophyll content in the 7th rosette leaves of BnaWGR1-overexpression plants was also lower than that of WT (Fig. 4e). A simple comparison of phenotypes of the 6th through 10th rosette leaves of 59-day-old soil-grown plants also supported the conclusion (Fig. 4f). We repeated the assay independently and the results were very similar, that is, OX#24 and OX#26 showed early leaf senescence and lower chlorophyll content, compared to WT (Fig. S4). These data indicate that BnaWGR1 positively regulates leaf senescence.

Overexpression of BnaWGR1 accelerates age-dependent leaf senescence. a, b The percentages of green, green/yellow, yellow and brown/dry leaves of 16 independent plants of 38-day-old (a) and 43-day-old (b) genotypes were calculated and compared. c Photographs of a 45- and 49-day-old WT and two overexpression lines. Rosette leaves excised from age-matched plants were arranged in rows from bottom to top. d, e Quantitative assay of total chlorophyll in the 5th and 6th true leaves (d) and 7th true leaves (e) of three 45-day-old genotypes from three independent plants. f Percentages of yellow leaves from the 6th to 10th true leaves of three 59-day-old genotypes. 16 independent plants for each genotype were calculated. In a, b and d–f, different letters represent significant differences from ANOVA test. FW fresh weight. Values were averages ± SE (n = 3), and significant differences are indicated by different letters based on one-way ANOVA test (P < 0.05)
BnaWGR1 induces the transcription of ROS and cell death-related genes
To better understand how BnaWGR1 modulated ROS accumulation and cell death, we determined to screen the putative downstream components of the BnaWGR1 signaling pathway through examining the transcript levels of genes coding for Rboh and metacaspase (MC) both in tobacco and transgenic Arabidopsis plants. qRT-PCR assay of leaf tissues of tobacco plants transiently expressing BnaWGR1 and GFP showed that RbohA and MC5 were significantly induced in BnaWGR1-expressing tissues at 2 dpi and 3 dpi compared to 0 dpi (Fig. 5a). RbohB was also induced at 2 dpi in BnaWGR1-expressing tissues (Fig. 5a). However, MC8 was not significantly changed at all (Fig. 5a). Together, these data confirmed that BnaWGR1 mediated ROS accumulation and cell death, possibly through affecting the expression of genes directly implicated in homeostasis of ROS and cell death.

Screening of putative target genes of BnaWGR1 through qRT-PCR and dual-LUC assays. a Identification of transcriptional changes of ROS- and cell death-related genes through qRT-PCR at 0, 2 and 3 dpi. Each value represents the mean ± SE of four biological replicates. Asterisks denote significant differences (compared to the level at 0 dpi) by Student t test (P < 0.05). b, c Expression analysis of AtRbohD and AtRbohF in the two independent BnaWGR1-expression transgenic lines at the indicated different leaf ages of 43-day-old plants by qRT-PCR. Data are means of four independent biological replicates ± SE. Significant differences are indicated by different letters based on one-way ANOVA test (P < 0.05). d Schematic diagrams of reporter and effector plasmids used in the dual-luciferase (LUC) assay. Promoters of RbohD and RbohF driving firefly LUC reporter gene were used as reporters while the CaMV 35S promoter-controlled Renilla luciferase (REN) was used for internal normalization. The effector construct consists of either BnaWGR1 or GFP driven by the CaMV 35S promoter. Black rectangles depict terminator sequences. e, f The dual-LUC assay results of transcriptional regulation of AtRbohD (e) and AtRbohF (f) mediated by BnaWGR1 in leaves of N. benthamiana. The relative LUC activity was calculated as the ratio of LUC/REN. Data are means of three independent biological replicates ± SE. Asterisks denote significant differences by Student’s t test (P < 0.05)
Next, the expression levels of RbohD and -F were examined in the rosette leaves of 43-day-old transgenic Arabidopsis plants expressing BnaWGR1. qRT-PCR assay demonstrated a significant increase of AtRbohD and AtRbohF in BnaWGR1-overexpressing lines compared to WT, while expression of AtMC5 and AtMC8 showed a significant increase only in one of the two overexpression lines (Fig. 5b, c, Fig. S5). In parallel, the expression level of a senescence marker gene SAG12 was also significantly higher in transgenic plants compared to WT (Fig. S5).
We further utilized a dual-luciferase (LUC) system to investigate whether BnaWGR1 directly activates the transcription of RbohD and RbohF, which are orthologs of RbohB and RbohA from tobacco, respectively. A 2.1 and 2.8 kb of promoter and 5′UTR fragments of AtRbohD and AtRbohF, respectively, were cloned into the reporter vector, in which a CaMV 35S promoter-controlled Renilla luciferase (REN) was used as the internal normalization gene (Fig. 5d). The effector was BnaWGR1 driven by CaMV 35S while that of GFP was used as the control (Fig. 5d). The different combinations of effector and reporter constructs were co-expressed in a transient system. The relative LUC activity represented by LUC/REN ratio was monitored at three time points. It showed that transient expression of BnaWGR1 significantly induced the transcription of LUC driven by the promoters of AtRbohD and -F (Fig. 5e, f). The data suggest that BnaWGR1 could possibly directly regulate the transcription of AtRbohD and AtRbohF.
BnaWGR1 directly binds to promoters of RbohD and RbohF
To test if BnaWGR1 could specifically bind to the W-box element, EMSA was performed. A recombinant BnaWGR1 fused with GST tag (BnaWGR1-GST) was purified from a prokaryotic system and the tandem repeat W-box probe was labeled with biotin (Fig. 6a). A thick band of protein–DNA complex with reduced mobility was observed when recombinant BnaWGR1-GST was incubated with 4xW-box probes, but not with a mutated 4xW-box, in which TTGACC was mutated to TTGAAC (Fig. 6b). Furthermore, a competition experiment using unlabeled 4xW-box probes confirmed the binding specificity of BnaWGR1 to W-box, but not to the mutated W-box sequence (Fig. 6b).

BnaWGR1 binds to W-box elements. a Schematic diagram of quadruple W-box and the mutated W-box (mW-box) sequences. b EMSA assay of binding of recombinant BnaWGR1-GST to canonical W-box sequence, but not to mW-box sequence. The BnaWGR1 fused to GST tag and GST protein (negative control) were used to detect the binding. Biotin-labeled probes were incubated with recombinant BnaWGR1–GST or GST proteins. 10- and 20-fold molar excess of unlabelled W-box or mW-box fragments were used as competitive probes
An analysis of the promoters of AtRbohD and AtRbohF indicates that there are five and two canonical W-box elements, respectively (Fig. S6). Hence, we speculated that BnaWGR1 might directly bind to some of these W-boxes during the transcriptional regulation of AtRbohD and F. Hence, we designed five probes, namely P1 through P5 for the AtRbohD (Fig. 7a). It is noted that two W-box elements are clustered in the P1 fragment. The results showed that shifted bands (retarded binding complex) for some probes were clearly detected (Fig. 7b). Moreover, weaker retarded bands for P1, P2 and P5 fragments were observed when unlabeled competitive probes were added (Fig. 7c). The results indicate that BnaWGR1 can recognize and bind to the W-box cis-elements in AtRbohD promoter.

BnaWGR1 directly binds to the promoter region of AtRbohD and AtRbohF. a Schematic diagram of the promoter region of AtRbohD and W-box elements. Black rectangles indicate W-box sequences. The five fragments (P1 through P5) used for EMSA were indicated. Numerals represent the sites relative to the transcription start site (TSS), which is referred to be + 1. b EMSA analysis of binding of recombinant BnaWGR1-GST to W-box, mutated W-box and different fragments of AtRbohD promoter. c A competitive binding assay of BnaWGR1-GST to P1, P2 and P5 fragments of AtRbohD promoter. Unlabeled probes were added at a molar excess of 100- or 200-fold. d The schematic diagram of the promoter region of AtRbohF and W-box elements. Black rectangles indicate W-box sequences. The three fragments (P1 through P3) used for EMSA were indicated. Numerals represent the sites relative to the transcription start site (TSS), which is referred to be + 1. e EMSA analysis of binding of recombinant BnaWGR1–GST and GST to different fragments of AtRbohF promoter. f A competitive binding assay of BnaWGR1–GST to P1 and P3 fragments of AtRbohF promoter. Unlabeled probes were added at a molar excess of 50- or 100-fold
Similarly, the binding of BnaWGR1 to promoter of AtRbohF was tested through EMSA. Three fragments (P1 through P3) were amplified and labeled with biotin (Fig. 7d). The results showed that recombinant BnaWGR1-GST bound to P1 and P3, but not to P2 (Fig. 7e). In parallel, BnaWGR1-GST also bound to the quadruple W-box sequence (Fig. 7f). A further competition experiment showed that unlabelled probes successfully diminished the binding of BnaWGR1 to P1 and P3 fragments, which suggests the specificity of binding (Fig. 7f).
Moreover, promoter regions of BnaRbohD and BnaRbohF from oilseed rape were cloned and multiple W-box elements were also identified in the sequences (Fig. 8a, b, Fig. S5). Three and two fragments harboring W-box elements were prepared and subjected to binding assay with recombinant BnaWGR1-GST proteins. BnaWGR1 bound to all the fragments, although with different affinities (Fig. 8c). We further performed a dual-luciferase (LUC) assay to examine the transactivation of BnaRbohD and F by BnaWGR1. A 1.2 kb of promoter and 5′UTR fragments of BnaRbohD and -F, respectively, were cloned into the reporter vector (Fig. 8d). The relative LUC activity revealed that transient expression of BnaWGR1 significantly induced the transcription of LUC driven by the promoters of BnaRbohD and -F at the three time points, compared to the control (Fig. 8e, f). Taken together, these data reveal that BnaWGR1 regulates the expression of RbohD and RbohF genes of Arabidopsis through directly binding to their promoters and also enhances the promoter activities of RbohD and RbohF orthologs in oilseed rape.

BnaWGR1 directly binds to the promoter region of BnaRbohD and BnaRbohF. a, b The schematic diagrams of the promoter regions of BnaRbohD (a) and BnaRbohF (b). Black rectangles indicate W-box and W-box-like sequences. The different fragments used in EMSA were indicated. c EMSA analysis of binding of recombinant BnaWGR1–GST to different fragments of promoters. The canonical tandem repeat W-box probe was used as a positive control. d Schematic diagrams of reporter and effector plasmids used in the dual-luciferase (LUC) assay. Promoters of BnaRbohD and BnaRbohF driving firefly luciferase (LUC) reporter gene were used as reporters while the CaMV 35S promoter-controlled Renilla luciferase (REN) was used for internal normalization. The effector construct consists of either BnaWGR1 or GFP driven by the CaMV 35S promoter. Black rectangles depict terminator sequences. e, f The dual-LUC assay of transcriptional regulation of BnaRbohD (e) and BnaRbohF (f) mediated by BnaWGR1 in leaves of N. benthamiana. The relative LUC activity was calculated as the ratio of LUC/REN. Data are means of three independent biological replicates ± SE. Asterisks denote significant differences by Student’s t test (*P < 0.05; **P < 0.01; ***P < 0.001)
Discussion
Leaf senescence is a developmental programmed cell death process providing plants with phenotypic plasticity to help them to adapt to adverse environmental conditions (Lim et al. 2007). Leaf senescence also determines crop plant yields and, therefore, a better understanding of the essential genes regulating this process is important. In this study, we characterized the function and molecular mechanism of one of a WRKY-type transcription factor gene in regulating leaf senescence of the oil crop, oilseed rape.
Expression of BnaWGR1 promotes leaf senescence
Previously, a transcriptomic profiling of early senescent leaves of oilseed rape identified BnaWGR1 as a WRKY-type gene highly induced in early senescent leaves (unpublished data). Further, a transient expression of BnaWGR1 in leaves of N. benthamiana indicated significant accumulation of ROS and hypersensitive response-like cell death (Fig. 2). A transcriptional profiling of BnaWGR1 expression at the four different stages of leaf development revealed high expression levels only in the early senescent leaves (Fig. 3), suggesting a potential role of BnaWGR1 in process of leaf senescence. This observation was complemented with the GUS activity assay when senescence is beginning (Fig. S1). A stronger GUS activity of 9th–11th leaves of the transgenic lines harboring the ProBnaWGR1-GUS construct was observed at the age of 36 and 43 days (Fig. S1). In Arabidopsis, some TF genes are preferentially expressed in early senescencing leaves, while others in late senescencing leaves (Gepstein et al. 2003; Balazadeh et al. 2008). We, therefore, propose that BnaWGR1 may play an important role when leaf senescence procedure proceeds. Furthermore, one of the transgenic Arabidopsis expressing BnaWGR1 showed a significantly precocious leaf senescence compared to WT and the other transgenic line also showed the trend of earlier senescence than WT (Fig. 4, Fig. S4). This result indicates that BnaWGR1 can positively regulate leaf senescence. It should be noted that the expression level of BnaWGR1 might be post-translationally regulated or tightly controlled as a signal of GFP fused BnaWGR1 was only detected when the 26S proteosome inhibitor MG132 was used (Fig. 1a).
BnaWGR1 positively regulates the expression of RbohD and RbohF
For a regulator mastering the early events of senescence, it is interesting to investigate the downstream targets. For example, three abscisic acid (ABA)-responsive element (ABRE)-binding transcription factors, ABF2, ABF3, and ABF4 act as positive regulators of chlorophyll catabolism genes to trigger ABA-dependent chlorophyll degradation and leaf senescence (Gao et al. 2016). AtNAP promotes chlorophyll degradation and leaf senescence via enhancing ABA synthesis mediated by abscisic aldehyde oxidase3 (AAO3) (Yang et al. 2014). OsNAP, an ortholog of AtNAP, also acts as a positive regulator of onset of senescence by modulating ABA biosynthesis and directly targeting SAGs in rice (Liang et al. 2014). As for AtWRKY53, it could bind to the promoters of a few transcription factor genes including WRKY6, -62, senescence-associated and defense-related genes (Miao et al. 2004). Surprisingly, AtWRKY53 can bind to promoters and transactivate the expression of CAT2 and -3, which encode two ROS-scavenging enzymes decreasing the content of H2O2 (Miao et al. 2004). We observed that the expression of BnaWGR1 induced significant ROS accumulation (Fig. 2). It implies that BnaWGR1 may disturb ROS homeostasis by either repressing genes encoding ROS-scavenging enzymes or activating genes encoding ROS-producing enzymes. Using a combination of methods, we identified two possible new direct targets of BnaWGR1, which are ROS-producing genes encoding RbohD and -F to be activated by BnaWGR1 through dual-LUC and EMSA assays.
ROS are highly reactive molecules being able to damage cellular components but are also proposed as signaling elements at lower concentrations (Apel and Hirt 2004). Plant homologs of mammalian NADPH oxidase (NOX), also known as Rbohs, act as a group of plasma membrane-localized enzymes producing H2O2 that mediate the switch of plants from growth to defense or adapt to environmental changes (Marino et al. 2012). Previously, it has been demonstrated that NbRbohA and NbRbohB, orthologs of AtRbohF and AtRbohD, respectively, are the two main members responsible for ROS production in N. benthamiana (Yoshioka et al. 2003). AtRbohD and -F are two famous enzymes mediating ABA promotion of ROS production, ABA-induced cytosolic Ca2+ increase, ABA-activated plasma membrane Ca2+-permeable channels in guard cells, and ABA-induced stomatal closing, as well as plant immunity (Torres et al. 2002; Kadota et al. 2014; Adachi et al. 2015). The diverse functions of Rbohs suggest they may serve as important molecular “hubs” in ROS signaling in plants (Marino et al. 2012). However, the role of RbohD and RbohF in leaf senescence has not been reported yet. Here, we provide supporting evidence that BnaWGR1 may regulate the expression of RbohD and -F to accelerate leaf senescence through increasing ROS levels.
In conclusion, this study reveals that the oilseed rape WGR1 transcription factor acts as a possible positive regulator of leaf senescence potentially through promoting the expression of two central ROS-producing enzyme genes, RbohD and –F, by directing binding to their promoters to advance ROS production. This is a finding not reported for any WRKY transcription factor gene in plants. It could be possible that other sources of ROS within cells may be present and hence make a global analysis of ROS levels and interactions more challenging. Collectively, the current work uncovers the potential role of an oilseed rape BnaWGR1 in leaf senescence and will be helpful for a better understanding and manipulation of leaf senescence in oilseed rape.
Author contribution statement
BY and YQJ conceived and designed research. LY, CY and YZ conducted experiments. XC and YW contributed materials. BY and YQJ analyzed data. BY and YQJ wrote the manuscript. All authors read and approved the manuscript.
Abbreviations
- CaMV:
-
Cauliflower mosaic virus
- EMSA:
-
Electrophoretic mobility shift assay
- GUS:
-
β-Glucuronidase
- NBT:
-
Nitroblue tetrazolium
- ROS:
-
Reactive oxygen species
- SAG:
-
Senescence-associated gene
References
Adachi H, Nakano T, Miyagawa N, Ishihama N, Yoshioka M, Katou Y, Yaeno T, Shirasu K, Yoshioka H (2015) WRKY transcription factors phosphorylated by MAPK regulate a plant immune NADPH oxidase in Nicotiana benthamiana. Plant Cell 27:2645–2663
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399
Bakshi M, Oelmuller R (2014) WRKY transcription factors: Jack of many trades in plants. Plant Signal Behav 9:e27700
Balazadeh S, Riano-Pachon DM, Mueller-Roeber B (2008) Transcription factors regulating leaf senescence in Arabidopsis thaliana. Plant Biol (Stuttg) 10(Suppl 1):63–75
Baxter A, Mittler R, Suzuki N (2014) ROS as key players in plant stress signalling. J Exp Bot 65:1229–1240
Besseau S, Li J, Palva ET (2012) WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J Exp Bot 63:2667–2679
Bhattarai KK, Atamian HS, Kaloshian I, Eulgem T (2010) WRKY72-type transcription factors contribute to basal immunity in tomato and Arabidopsis as well as gene-for-gene resistance mediated by the tomato R gene Mi-1. Plant J 63:229–240
Birkenbihl RP, Diezel C, Somssich IE (2012) Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol 159:266–285
Bustin SA (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169–193
Chen L, Zhang L, Yu D (2010) Wounding-induced WRKY8 is involved in basal defense in Arabidopsis. Mol Plant Microbe Interact 23:558–565
Ciolkowski I, Wanke D, Birkenbihl RP, Somssich IE (2008) Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol Biol 68:81–92
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17
Daudi A, Cheng Z, O’Brien JA, Mammarella N, Khan S, Ausubel FM, Bolwell GP (2012) The apoplastic oxidative burst peroxidase in Arabidopsis is a major component of pattern-triggered immunity. Plant Cell 24:275–287
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Gao S, Gao J, Zhu X, Song Y, Li Z, Ren G, Zhou X, Kuai B (2016) ABF2, ABF3, and ABF4 promote ABA-mediated chlorophyll degradation and leaf senescence by transcriptional activation of chlorophyll catabolic genes and senescence-associated genes in Arabidopsis. Mol Plant 9:1272–1285
Gepstein S, Sabehi G, Carp MJ, Hajouj T, Nesher MF, Yariv I, Dor C, Bassani M (2003) Large-scale identification of leaf senescence-associated genes. Plant J 36:629–642
Hellens RP, Allan AC, Friel EN, Bolitho K, Grafton K, Templeton MD, Karunairetnam S, Gleave AP, Laing WA (2005) Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 1:13
Hinderhofer K, Zentgraf U (2001) Identification of a transcription factor specifically expressed at the onset of leaf senescence. Planta 213:469–473
Huang W, Miao M, Kud J, Niu X, Ouyang B, Zhang J, Ye Z, Kuhl JC, Liu Y, Xiao F (2013) SlNAC1, a stress-related transcription factor, is fine-tuned on both the transcriptional and the post-translational level. New Phytol 197:1214–1224
Jambunathan N (2010) Determination and detection of reactive oxygen species (ROS), lipid peroxidation, and electrolyte leakage in plants. Methods Mol Biol 639:292
Jiang Y, Deyholos MK (2009) Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol 69:91–105
Jiang Y, Liang G, Yang S, Yu D (2014) Arabidopsis WRKY57 functions as a node of convergence for jasmonic acid- and auxin-mediated signaling in jasmonic acid-induced leaf senescence. Plant Cell 26:230–245
Kadota Y, Sklenar J, Derbyshire P, Stransfeld L, Asai S, Ntoukakis V, Jones JD, Shirasu K, Menke F, Jones A, Zipfel C (2014) Direct regulation of the NADPH oxidase RBOHD by the PRR-associated kinase BIK1 during plant immunity. Mol Cell 54:43–55
Kim K, Franceschi V, Davin L, Lewis N (2006) β-Glucuronidase as reporter gene. Advantages and limitations. In: Salinas J, Sanchez-Serrano JJ (eds) Arabidopsis protocols. Humana Press Inc, Totowa, pp 263–273
Knoth C, Ringler J, Dangl JL, Eulgem T (2007) Arabidopsis WRKY70 is required for full RPP4-mediated disease resistance and basal defense against Hyaloperonospora parasitica. Mol Plant Microbe Interact 20:120–128
Kwak JM, Mori IC, Pei ZM, Leonhardt N, Torres MA, Dangl JL, Bloom RE, Bodde S, Jones JD, Schroeder JI (2003) NADPH oxidase AtrbohD and AtrbohF genes function in ROS-dependent ABA signaling in Arabidopsis. EMBO J 22:2623–2633
Lee S, Seo PJ, Lee HJ, Park CM (2012) A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J 70:831–844
Li L, Ye C, Zhao R, Li X, Liu WZ, Wu F, Yan J, Jiang YQ, Yang B (2015) Mitogen-activated protein kinase kinase kinase (MAPKKK) 4 from rapeseed (Brassica napus L.) is a novel member inducing ROS accumulation and cell death. Biochem Biophys Res Commun 467:792–797
Liang W, Yang B, Yu B, Zhou Z, Li C, Sun Y, Zhang Y, Jia M, Wu F, Zhang H, Wang B, Deyholos M, Jiang Y-Q (2013) Identification and analysis of MKK and MPK gene families in canola (Brassica napus L.). BMC Genom 14:392
Liang C, Wang Y, Zhu Y, Tang J, Hu B, Liu L, Ou S, Wu H, Sun X, Chu J, Chu C (2014) OsNAP connects abscisic acid and leaf senescence by fine-tuning abscisic acid biosynthesis and directly targeting senescence-associated genes in rice. Proc Natl Acad Sci USA 111:10013–10018
Lim PO, Nam HG (2005) The molecular and genetic control of leaf senescence and longevity in Arabidopsis. Curr Top Dev Biol 67:49–83
Lim PO, Kim HJ, Nam HG (2007) Leaf senescence. Annu Rev Plant Biol 58:115–136
Liu D, Shi L, Han C, Yu J, Li D, Zhang Y (2012) Validation of reference genes for gene expression studies in virus-infected Nicotiana benthamiana using quantitative real-time PCR. PLoS ONE 7:e46451
Marino D, Dunand C, Puppo A, Pauly N (2012) A burst of plant NADPH oxidases. Trends Plant Sci 17:9–15
Miao Y, Laun T, Zimmermann P, Zentgraf U (2004) Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol Biol 55:853–867
Mittler R, Vanderauwera S, Gollery M, Van Breusegem F (2004) Reactive oxygen gene network of plants. Trends Plant Sci 9:490–498
Navabpour S, Morris K, Allen R, Harrison E, Mackerness SAH, Buchanan-Wollaston V (2003) Expression of senescence-enhanced genes in response to oxidative stress. J Exp Bot 54:2285–2292
Niu F, Wang C, Yan J, Guo X, Wu F, Yang B, Deyholos MK, Jiang YQ (2016) Functional characterization of NAC55 transcription factor from oilseed rape (Brassica napus L.) as a novel transcriptional activator modulating reactive oxygen species accumulation and cell death. Plant Mol Biol 92:89–104
Oh CS, Martin GB (2011) Tomato 14-3-3 protein TFT7 interacts with a MAP kinase kinase to regulate immunity-associated programmed cell death mediated by diverse disease resistance proteins. J Biol Chem 286:14129–14136
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Rivero RM, Kojima M, Gepstein A, Sakakibara H, Mittler R, Gepstein S, Blumwald E (2007) Delayed leaf senescence induces extreme drought tolerance in a flowering plant. Proc Natl Acad Sci USA 104:19631–19636
Robatzek S, Somssich IE (2001) A new member of the Arabidopsis WRKY transcription factor family, AtWRKY6, is associated with both senescence- and defence-related processes. Plant J 28:123–133
Robatzek S, Somssich IE (2002) Targets of AtWRKY6 regulation during plant senescence and pathogen defense. Genes Dev 16:1139–1149
Rogers H, Munne-Bosch S (2016) Production and scavenging of reactive oxygen species and redox signaling during leaf and flower senescence: similar but different. Plant Physiol 171:1560–1568
Torres MA, Dangl JL, Jones JD (2002) Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc Natl Acad Sci USA 99:517–522
Yang B, Jiang Y, Rahman MH, Deyholos MK, Kav NN (2009) Identification and expression analysis of WRKY transcription factor genes in canola (Brassica napus L.) in response to fungal pathogens and hormone treatments. BMC Plant Biol 9:68
Yang J, Worley E, Udvardi M (2014) A NAP-AAO3 regulatory module promotes chlorophyll degradation via ABA biosynthesis in Arabidopsis leaves. Plant Cell 26:4862–4874
Ye Z, Rodriguez R, Tran A, Hoang H, de los Santos D, Brown S, Vellanoweth RL (2000) The developmental transition to flowering represses ascorbate peroxidase activity and induces enzymatic lipid peroxidation in leaf tissue in Arabidopsis thaliana. Plant Sci 158:115–127
Yoshioka H, Numata N, Nakajima K, Katou S, Kawakita K, Rowland O, Jones JD, Doke N (2003) Nicotiana benthamiana gp91phox homologs NbrbohA and NbrbohB participate in H2O2 accumulation and resistance to Phytophthora infestans. Plant Cell 15:706–718
Zheng Z, Qamar SA, Chen Z, Mengiste T (2006) Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J 48:592–605
Zhou X, Jiang Y, Yu D (2011) WRKY22 transcription factor mediates dark-induced leaf senescence in Arabidopsis. Mol Cells 31:303–313
Zimmermann P, Heinlein C, Orendi G, Zentgraf U (2006) Senescence-specific regulation of catalases in Arabidopsis thaliana (L.) Heynh. Plant Cell Environ 29:1049–1060
Acknowledgements
This work was supported by the National Natural Science Foundation of China [No. 31301648 to BY and 31270293 to Y-QJ]. Natural Science Foundation of Shaanxi Province [no. 2014JQ3102 to BY]. The Fundamental Research Funds for the Central Universities of China [no. 2109021137, nos. 2452015413, 2452016009, to BY]. We thank Profs. Jian-Ye Chen (South China Agricultural University) and Benke Kuai (Fudan University) for technical assistance.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, L., Ye, C., Zhao, Y. et al. An oilseed rape WRKY-type transcription factor regulates ROS accumulation and leaf senescence in Nicotiana benthamiana and Arabidopsis through modulating transcription of RbohD and RbohF. Planta 247, 1323–1338 (2018). https://doi.org/10.1007/s00425-018-2868-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-018-2868-z