
Abstract
Main conclusion
Based on findings described herein, we contend that the reduction of vomilenine en route to antiarrhythmic ajmaline in planta might proceed via an alternative, novel sequence of biosynthetic steps.
In the genus Rauvolfia, monoterpenoid indole alkaloids (MIAs) are formed via complex biosynthetic sequences. Despite the wealth of information about the biochemistry and molecular genetics underlying these processes, many reaction steps involving oxygenases and oxidoreductases are still elusive. Here, we describe molecular cloning and characterization of three cinnamyl alcohol dehydrogenase (CAD)-like reductases from Rauvolfia serpentina cell culture and R. tetraphylla roots. Functional analysis of the recombinant proteins, with a set of MIAs as potential substrates, led to identification of one of the enzymes as a CAD, putatively involved in lignin formation. The two remaining reductases comprise isoenzymes derived from orthologous genes of the investigated alternative Rauvolfia species. Their catalytic activity consists of specific conversion of vomilenine to 19,20-dihydrovomilenine, thus proving their exclusive involvement in MIA biosynthesis. The obtained data suggest the existence of a previously unknown bypass in the biosynthetic route to ajmaline further expanding structural diversity within the MIA family of specialized plant metabolites.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Monoterpenoid indole alkaloids (MIAs) are widespread within the Apocynaceae, underscoring the importance of numerous members of the botanical family as sources of high value compounds boasting pharmacological potential. The anticancer agents of Catharanthus roseus, vincristine and vinblastine (Almagro et al. 2015) or the antihypertensive reserpine and the antiarrhythmic ajmaline characteristic of Rauvolfia serpentina (Stöckigt and Zenk 1995) are only the most prominent representatives of the aforementioned class of specialized metabolites, collectively forming several hundred different MIA structures within the two plant genera. Since obtainable amounts of plant secondary metabolites from natural resources are often scarce, unravelling the full potential of a large fraction of this array of conceivably valuable ingredients has been consistently hampered. Recent advances in metabolic engineering now open new exploratory avenues leading to deliberate reconstruction of pathways, ultimate increase in yields, as well as generation of new-to-nature structures (Staniek et al. 2013, 2014). However, in-depth knowledge on the target biosynthetic pathways and cloning of the corresponding genes are prerequisite for successful metabolic engineering applications.
Understanding biosynthetic formation of the complex MIA compounds has posed a challenge for several decades and, despite considerable progress in the area, certain genetic and spatio-temporal factors intrinsic to the metabolic network in question remain unknown. Common to the biosynthesis of all MIAs is the formation of their terpenoid precursor, secologanin. Its subsequent ligation to tryptamine yields the universal intermediate, strictosidine, whence all 2500 MIAs known thus far are derived (Stöckigt and Zenk 1977). This “upstream pathway” was elucidated in detail on enzymatic and genetic level (Geu-Flores et al. 2012; Asada et al. 2013; Salim et al. 2013, 2014; Miettinen et al. 2014) and, with the genetic machinery at hand, heterologous production of the latter plant-specific compound in yeast became feasible (Brown et al. 2015). However, the manifold reactions, spearheaded by deglucosylation of strictosidine, providing the abundance of MIA carbon skeletons, are still elusive. Further, of the various “downstream pathways” leading to the pharmacologically important structures, only a few are known. This lack of information on key enzymes hampers further metabolic engineering approaches aimed at modulation of MIA production. Regarding the number of identified enzymes and genes of the downstream pathways, C. roseus and R. serpentina are, by far, the best studied systems. In the case of Rauvolfia, all nine major biosynthetic steps leading to ajmaline (from the parent precursor, strictosidine) have been elucidated through analysis of purified enzyme fractions. Further, six snakeroot genes exclusively involved in MIA formation have been cloned, including two contributing to side-reactions in ajmaline biosynthesis, greatly expanding our knowledge of this specialized metabolic route. Moreover, structural information on five of the identified enzymes is available (Panjikar et al. 2012), enabling mechanistic studies as well as mutagenesis and design aimed at expansion of their catalytic properties, as recently exemplified for strictosidine synthase (STR1, Bernhardt et al. 2007). Withal, rational design based on structural data facilitated modulation of substrate acceptance of STR1 toward halogenated tryptamine derivatives, affording deliberate modification of MIA structures in vivo (Zhu et al. 2015). Despite the near-complete delineation of the biosynthetic sequence in question, existence of further bypasses to established routes as well as additional enzymes acting on intermediates and generating side-products still cannot be excluded.
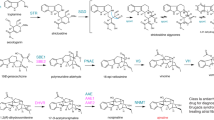
Reduction of vomilenine to 17-O-acetylnorajmaline is an essential step in ajmaline biosynthesis. Previous studies postulated the involvement of two disparate enzymes sequentially reducing the aforesaid intermediate (Stöckigt and Zenk 1995). Later, two NADPH-dependent reductases were purified from cultured cells of R. serpentina and characterized (Gao et al. 2002; von Schumann et al. 2002). Based on the obtained data, the authors posited that an enzyme dubbed vomilenine reductase (VR) was responsible for the initial reduction of the 1,2- double bond within the indolenine ring, forming 2β-(R)-1,2-dihydrovomilenine, with the latter serving as a substrate for 1,2-dihydrovomilenine reductase (DHVR), acting upon the 19,20- double bond to yield 17-O-acetlynorajmaline (Fig. 1). Both enzyme preparations were highly substrate-specific, neither processing the substrate of the other, lending support to the aforementioned reaction sequence as prevailing in planta. However, genes encoding the two enzymes could not be identified. Further, neither of the utilized protein isolates was pure and might have manifested additional enzymatic activities, while ultimate evidence of substrate specificity can only be obtained by testing pure, heterologously produced enzymes.

Simplified pathway leading from strictosidine via vomilenine to ajmaline. Two routes from vomilenine to 17-O-acetylnorajmaline can be postulated, depending on which of the two double bonds is reduced first: reduction of the indolenine ring in 1,2- position (VR) followed by reduction of the 19,20- double bond (DHVR), as proposed by Gao et al. (2002) and von Schumann et al. (2002). Alternatively, the 19,20- bond of vomilenine is reduced first (VR2, characterized herein) and the 1,2- reduction step (by virtue of a yet unidentified enzyme, ?) follows
Since classical gene identification procedures, like screening of cDNA libraries, have not yielded the corresponding coding sequences thus far, we now wish to make use of the newly available transcriptomic data of the Medicinal Plant Genomics Resource (MPGR, http://medicinalplantgenomics.msu.edu/index.shtml). The large-scale transcriptome and expression profiles (Góngora-Castillo and Buell 2013) have already proved a valid source, facilitating identification of missing-link enzymes in specialized metabolism (Góngora-Castillo et al. 2012a, b). This is especially true of high priority species like C. roseus, boasting a large data set encompassing inputs characteristic of an array of developmental stages and tissues as well as treatment with elicitors. For R. serpentina, only limited transcriptomic resources are available; these comprise eight different organs but no differential expression upon elicitor treatment.
Here, we report on the molecular cloning of a snakeroot gene encoding for a reductase acting exclusively on the 19,20- double bond of vomilenine.
Materials and methods
Plant material and cDNA cloning
Cell suspension cultures of Rauvolfia serpentina (from the collection of Prof. J. Stöckigt, Mainz, Germany) were grown in liquid LS medium (Linsmaier and Skoog 1965) at 25 °C under continuous agitation. After 7 days, plant cells were harvested by filtration and flash-frozen in liquid nitrogen. Roots of greenhouse-grown R. tetraphylla plants from the collection of the Botanical Garden of the Technische Universität Darmstadt, Germany (2 years old) were freed from soil, washed with tap water and flash-frozen in liquid nitrogen. Tissues were ground with a mortar and a pestle in liquid nitrogen to a fine powder and total RNA was extracted with the TriFast reagent (Peqlab, Erlangen, Germany). For cloning, 1 µg RNA samples was reverse transcribed with RevertAid reverse transcriptase (Thermo Fischer Scientific, Waltham, MA USA) and the gene-specific reverse primers. cDNAs were amplified by PCR with the appropriate primer pairs and Pfu DNA polymerase (Thermo Fischer Scientific). Primers were derived from transcriptomic sequence loci 233 and 15454 (http://medicinalplantgenomics.msu.edu/) and contained extensions facilitating addition of restriction sites for cloning into the bacterial expression vector pET32a, NcoI at the 5′-end and XhoI at the 3′-end (DHVR fw, 5′-TTC GCC ATG GCT GGA AAG TCT CCA GAA GAG-3′; DHVR rev, 5′-GAT CCT CGA GTT AGG ACT CAG GTG GAG TTA AGG TG-3′; VR for, 5′-GGC CAT GGC GAA GTC ACC AGA AGT TG-3′; VR rev, 5′-AAG CTC GAG TTA GGC AGA TTT CAA TGT GTT GCC-3′). PCR products were cloned into the pCR Blunt vector (Invitrogen, Life Technologies GmbH, Darmstadt, Germany) and subsequently sequenced.
Sequence alignments were performed with BLAST (NCBI) and against the MPGR database (http://medicinalplantgenomics.msu.edu/).
Expression of cDNAs in bacteria and protein purification
Coding regions of the isolated cDNAs were inserted between NcoI and XhoI sites of the pET32a vector and expressed in E. coli cells, strain T7 SHuffle (New England Biolabs, Frankfurt am Main, Germany), grown in 300 ml of liquid TB medium (Sambrook et al. 1989) amended with 100 µg ml−1 ampicillin at 25 °C and 95 rpm, until OD600 of about 0.2. Cells of the control bacterial culture contained pET32a:MC-SM1. IPTG (0.4 mM) was added to induce protein synthesis. Post-induction, the cultures were maintained overnight (~18 h) at 25 °C and 95 rpm. Bacteria were harvested (6000g, 10 min at 4 °C) and resuspended in 3 ml of LEW buffer (lysis, equilibration, wash; 50 mM NaH2PO4, 300 mM NaCl, pH 8.0) per 1 g of wet cell weight. The suspensions were then supplemented with 6 mM CHAPS, 1 mM AEBSF and 1 mg ml−1 lysozyme, incubated on ice for 30 min, and sonicated (6 × 10 s, in 20 s intervals). Bacterial cell lysates were subsequently treated with DNaseI (5 U, Thermo Fisher Scientific) and, once more, incubated on ice for 20 min. Following centrifugation (6000g, 30 min at 4 °C), supernatants were collected for purification of soluble, N-terminally His-tagged reductases (Protino Ni-TED Resin Kit, Macherey–Nagel, Düren, Germany). Proteins of interest were eluted in a stepwise manner, with LEW of increasing imidazole content (50, 150, and 250 mM). Relevant elution fractions were then desalted and concentrated with the Amicon Ultra-15 Centrifugal Filter Devices (Merck Millipore, Darmstadt, Germany), resulting in recovery of final enzyme preparations in 50 mM NaH2PO4 buffer (pH 5.9). All steps of the protein retrieval procedure were monitored by SDS-PAGE (separating gel buffer, 1.5 M Tris–HCl pH 8.8; stacking gel buffer, 0.5 M Tris–HCl pH 6.8; running buffer, 0.025 M Tris base, 0.2 M glycine, 0.1 % SDS (w/v); 12.5 % acrylamide; PerfectBlue Dual Gel System, Peqlab). Determination of purified protein concentration was performed with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), using bovine serum albumin (BSA) as a standard. Aliquots of the recovered reductase solutions were stored in 25 % (v/v) glycerol at −80 °C.
Enzyme assays and analytics
Putative reductase activity of the isolated proteins was investigated via enzyme assays followed by HPLC analysis. Alkaloids (generously provided by Prof. J. Stöckigt) were dissolved in DMSO and stored at −20 °C; cinnamaldehyde, cinnamyl alcohol, and 1,3-phenylpropanol (Sigma-Aldrich) were dissolved in MeOH and stored at 4 °C. Working solutions of the putative substrates were always prepared freshly; the compounds were diluted in 50 mM NaH2PO4 buffer (pH 5.9) and added to the assays at a final concentration of 0.25 mM. Reaction mixtures were supplemented with either NADPH or NADP+ (2 mM) as cofactors. Proteins were applied at a final concentration of 100 µg ml−1. The assay samples (total volume, 200 µl) were incubated for 1 h at 30 °C. To terminate the reactions, 200 µl of acetonitrile was added. Negative control assays (acetonitrile added prior to incubation) were performed in parallel to ensure detection of specific activity of the investigated enzymes.
After centrifugation (9000g, 10 min), 5 µl of the clear supernatants was injected onto a 150 × 4.6 mm, 5 µm analytical HPLC column (Zorbax 300SB-C18 + Zorbax SB-Aq precolumn, Agilent, Santa Clara, CA, USA). The mobile phase consisted of ddH2O/0.1 % formic acid (A) and acetonitrile (B). The flow rate was set to 1.0 ml min−1 and the following gradient (% B) was applied: 0 min, 10; 12.0 min, 55. After 14 min, the column was flushed with 100 % B for 4 min and re-equilibrated with 10 % B for 3 min. Temperature for the analytical column was set to 30 °C. An ion trap mass spectrometer (Agilent), consisting of the following series 1100 HPLC components: G1379A vacuum degasser, G1312A binary pump, G1313A autosampler, G1316A column thermostat, and G2445D mass selective detector, was applied for the detection of enzyme assay products using electrospray ionisation (ESI) in the positive ion mode. Nitrogen was used as a nebulising (50 psi) and drying gas (300 °C, 12 l min−1). The capillary voltage was 3500 V and the skimmer, octopole 1, octopole 2, and capillary exit voltage values were set to 40, 12, 1.7, and 104 V, respectively.
Kinetic data of the vomilenine reducing enzymes were obtained by applying different substrate concentrations, in a range of 0.025, 0.05, 0.10–1 mM in 0.1 mM increments, into a total sample volume of 100 µl. Final protein concentration in each sample was 100 µg ml−1, while that of NADPH was 2 mM. The samples were preincubated at 30 °C for 10 min. Depletion of the cofactor was detected at 340 nm, measured in intervals of 48 s for 1 h at 30 °C (Epoch 2 microplate reader, BioTek, Bad Friedrichshall, Germany; Immulon 1B 96 well plates, Thermo Fisher Scientific). All kinetic assays were performed in duplicate.
For analysis of LC–MS data, OpenChrom Software was applied. Heat map (Fig. 2) was generated using R with ggplot2, reshape, and grid libraries. Enzyme kinetic parameters (K m and V max) were calculated with GraphPad Prism 6. Chemical structures were drawn by means of ChemDraw Professional 15.

Expression patterns of genes involved in ajmaline biosynthesis as well as selected oxidoreductases according to the R. serpentina transcriptomics data resource (http://medicinalplantgenomics.msu.edu/). SG, strictosidine glucosidase; PNAE, polyneuridine aldehyde esterase; RG, raucaffricine glucosidase; locus_233, putative oxidoreductase (further isolated as clone RR 6 and characterized as RsCAD); locus_15454, putative oxidoreductase (further isolated as clone RR 4 and characterized as RsVR2). Expression abundances reported as log2 fragments per kb transcript per million mapped reads
Results
Previous studies on Rauvolfia reductases (Gao et al. 2002; von Schumann et al. 2002) provided for determination of partial amino acid sequences from endoproteinase LysC digests of purified protein fractions exhibiting either vomilenine reductase (VR) or 1,2-dihydrovomilenine reductase (DHVR) activity (Table 1). To delineate the corresponding cDNA sequences, we utilized the publicly available transcriptomics database of the Medicinal Plant Genomics Resource (medicinalplantgenomics.msu.edu, Góngora-Castillo et al. 2012a, 2012b). We identified locus 233 (rsa_locus_233_iso_7_len_1568_ver_2) as encoding a gene encompassing, upon translation, both peptides derived from the putative VR enzyme fraction, while alignment of the four available DHVR peptide sequences pointed to locus 15454 (rsa_locus_15454_iso_5_len_1727_ver_2) as the corresponding putative gene. We further analyzed differential expression patterns of the two genes in diverse organs of R. serpentina. For both candidate sequences, the highest number of transcripts was detected in roots, either young, for locus 233, or mature, for locus 15454 (Fig. 2). As the observed expression bias was in accord with the Rauvolfia root constituting a hub for accumulation of ajmaline and related alkaloids, we elected to utilize root material as a source of mRNA, alternative to the established R. serpentina cell suspension culture. Moreover, although previously determined in cell suspensions, the culture-inherent, specific reductase activity levels might fluctuate over time and the concomitant decrease in gene expression could hamper cDNA isolation. Due to lack of fresh plant material from R. serpentina, we selected the closely related R. tetraphylla as an available source of root mRNA.
To obtain full-length cDNAs encoding the two reductases, we designed primer pairs derived from the sequences published at http://medicinalplantgenomics.msu.edu/. Both oligo-pairs provided for amplification of cDNA clones from the R. serpentina cell suspension culture, whereas for R. tetraphylla roots, only the DHVR primer combination yielded a valid DNA complement. Sequencing and alignment studies revealed that all isolated sequences were highly homologous, but in no case identical, to those of the original transcriptome loci. Clone RR 4 from R. serpentina cell culture and clone RR 2 from R. tetraphylla roots showed high homology to locus 15454 (94.2 and 93.5 %, respectively) and to each other (93.9 %). For clone RR 6 from R. serpentina cell culture, 91.1 % identity could be assigned to locus 233. Further, presence of all previously determined peptide sequences was confirmed within the corresponding cDNA translates; RR 6 contained the VR-derived peptides, while RR 4 and RR 2 encompassed amino acid chains characteristic of the putative DHVR (Fig. 3). The following GenBank accession numbers were assigned: RR 6, KT369739; RR 4, KT369740; RR 2, KT369741. Upon subsequent homology search against the database of protein sequences of assigned and verified function (NCBI), all candidate enzymes showed high homology (around 75 % identity) to sinapyl alcohol dehydrogenase (SAD) from Populus tremuloides (Li et al. 2001). Sequence RR 6 presented similar homology to cinnamyl alcohol dehydrogenases (CAD) from various origins, while RR 4 and RR 2 conformed to CAD peptide chains to a lesser extent (around 60 % identity). The comparison demonstrated that the identified Rauvolfia proteins exhibit features of medium chain reductase/dehydrogenase (MDR)/zinc-dependent alcohol dehydrogenase-like family and, therefore, comprise potential reductase activity. To determine subcellular enzyme localization, the retrieved amino acid sequences were subjected to a series of sorting predictions in silico (TargetP and SignalP at http://www.cbs.dtu.dk/services/; seqNLS at http://mleg.cse.sc.edu/seqNLS/). For all three queries, neither N-terminal targeting sequences nor nuclear localization signals could be detected, spurring the assumption that the investigated reducing catalysts are cytosolic.

Alignment of amino acid sequences retrieved in course of the present study (RR 6, RR 4, and RR 2) with AtCAD4 (AEE76241.1), AtCAD5 (AEE86345.1) and PtrSAD (AAK58693.1). Amino acids identical in all sequences are shaded in grey. Sequence motifs previously identified by Edmann degradation (Gao et al. 2002; von Schumann et al. 2002) are indicated in bold and underlined
Since the applied RT-PCR method afforded no further retrieval of cDNA sequences from the investigated Rauvolfia RNA pools, the three aforementioned amplicons were subjected to heterologous expression in E. coli. All sequences were cloned into the pET32a vector as thioredoxin/His-tag fusions. Expression in T7 SHuffle cells resulted in inducible accumulation of recombinant proteins, while purification via immobilized metal ion affinity chromatography (IMAC) and subsequent desalting yielded protein preparations of 2.1 mg ml−1 for RR 6, 5.2 mg ml−1 for RR 4, and 1 mg ml−1 for RR 2. As illustrated in Fig. 4, the heterologously generated Rauvolfia reductase isolates were of sufficient purity (>90 %). In parallel, to serve as a control in all subsequent experiments, we expressed another plant gene via the pET32a system, resulting in a thioredoxin/His-tag fusion of Momordica cochinchinensis trypsin inhibitor I (MCoTI-II, Avrutina et al. 2005) mutant, with no detectable reductase activity. The protein (termed MC-SM1), produced in the same cells and purified accordingly was retrieved at 5.5 mg ml−1.

Recombinant reductases (RR 6, RR 4, and RR 2) produced in E. coli T7 SHuffle. 12.5 µl of the IMAC-purified protein solutions was subjected to SDS-PAGE. As control, recombinant MCoTI-II (MC-SM1), with no reductase activity, was produced and purified accordingly. Numbers to the left indicate molecular mass of marker proteins (M)
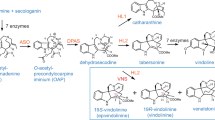
The obtained enzymatic proteins were characterized concerning their substrate and cofactor specificity as well as the products of their catalytic activity. In parallel, appropriate control experiments were performed and the assay samples were analyzed using HPLC and LC–MS. With vomilenine as a substrate, no enzymatic activity was observed in case of RR 6. In contrast, both RR 4 and RR 2 converted the MIA intermediate (R t, 8.17; [MH]+ m/z, 351) to a new product characterized by a slightly higher retention time (R t, 8.42) and an [MH]+ m/z of 353, corresponding to dihydrovomilenine (Fig. 5). The initial LC–MS results, coupled with the fact that the reaction was clearly NADPH-dependent, corroborated that enzymes RR 4 and RR 2 are reductases acting on vomilenine and, therefore, belong to the biosynthetic pathway leading to ajmaline. Since vomilenine reduction could take place at the 1,2- double bond of the indolenine ring as well as at the 19,20- double bond, further unambiguous identification of the product was required. Comparing the various authentic samples of vomilenine derivatives showed that the newly formed product exhibited the exact same R t value and UV spectrum as 20α-(S)-19,20-dihydrovomilenine, indicating that the indolenine ring was not affected by the reduction. Finally, the daughter ion spectrum, upon fragmentation of the product of the investigated enzymatic reaction, was identical to the one obtained for the authentic 19,20-dihydrovomilenine (Fig. 5), clearly corroborating its identity.

Analysis of products of enzymatic conversions. HPLC chromatograms of vomilenine incubated with inactivated enzyme (a), with RR 4 (b), or RR 2 (c). Boxes show mass spectra of indicated peaks and, in c, a daughter ion spectrum of the product. UV absorption spectra of products of enzymatic conversion as well as authentic standards (d). Structure of the product, 19,20-dihydrovomilenine (e). Daughter ion spectrum of authentic 19,20-dihydrovomilenine (f)
Conversion of vomilenine was observed solely in the presence of NADPH. If NADP+ or no cofactor were applied, the substrate was not processed, indicating explicit dependence of the investigated catalytic reaction on NADPH as well as specific reductase, and not oxidoreductase, activity. Since absorption of the investigated alkaloids, vomilenine and its product, 19,20-dihydrovomilenine, occurs at 260–280 nm, giving rise to similar maxima in the respective UV spectra, the catalytic activity of enzymes RR 2 and RR 4 was analyzed indirectly, through detection of NADPH depletion at 340 nm. RR 2 was characterized by a K m value of 13.56 µmol (±3.07) and a V max rate of 0.90 µmol l−1 min−1 (±0.02). In case of RR 4, the respective values of 43.16 µmol (±23.21) and 0.56 µmol l−1 min−1 (±0.05) were estimated.
To further examine substrate specificity of the three isolated enzymes, a number of Rauvolfia alkaloids were tested. No activity against the closely related structures 2β-(R)-1,2-dihydrovomilenine, vinorine, raucaffricine, or perakine (Fig. 6) was observed. Additionally, application of 19,20-dihydrovomilenine as a substrate (and NADP+ as a cofactor) for RR 2 and RR 4 did not yield any product, suggesting that the reductases do not catalyze its reverse oxidation. Since protein sequences of all investigated Rauvolfia enzymes exhibited high homology to NADPH-dependent reductases acting on cinnamaldehyde derivatives, we further tested the aldehyde as well as cinnamyl alcohol as substrates. Only RR 6 converted cinnamaldehyde to the corresponding alcohol, but did not reduce its double bond. Neither RR 4 nor RR 2 showed any activity against the simple phenylpropanoid compounds.

Structures of substrates tested with the three Rauvolfia reductases. −, no detectable activity; +*, conversion to 19,20-dihydrovomilenine; +**, conversion to cinnamyl alcohol
Finally, we assayed the ability of the reductases to form alkaloids comprising a heteroyohimbine structure from appropriate precursors. In a recent publication (Stavrinides et al. 2015), a reductase from C. roseus was described capable of producing tetrahydroalstonine (and was hence dubbed tetrahydroalstonine synthase, THAS) from strictosidine aglycon. Since heteroyohimbine alkaloids are present in Rauvolfia and similar enzymatic reduction steps can be surmised, we incubated the purified enzymes with cathenamine, the direct precursor of THA. No product formation could be detected in this setting.
As we were able to demonstrate specific activity for all three enzymes, we assigned relevant names to the so far arbitrarily termed reductases. RR 6 was, therefore, dubbed R. serpentina cinnamyl alcohol dehydrogenase (RsCAD). Since the remaining enzyme process exclusively vomilenine but catalyze a reaction alternative to the previously described vomilenine reductase (von Schumann et al. 2002), we suggest the designations R. serpentina vomilenine reductase 2 (RsVR2) for RR 4 and R. tetraphylla vomilenine reductase 2 (RtVR2) for RR 2.
Discussion
Previous studies have proposed a (by now, universally acknowledged, http://www.kegg.jp) reaction sequence from vomilenine via 1,2-dihydrovomilenine to acetylnorajmaline (Gao et al. 2002; von Schumann et al. 2002). The postulated biosynthetic progression en route to antiarrhythmic ajmaline was based on activity testing of purified enzyme fractions from R. serpentina cell suspension cultures with authentic substrates. While one of the investigated protein isolates presented vomilenine reductase activity exclusively targeting the indolenine double bond (1,2-) of the MIA intermediate, another, acting on 1,2-dihydrovomilenine alone, further reduced the 19,20- double bond showing no measurable activity against vomilenine. In this study, we identified a new reductase, targeting solely the 19,20- double bond of vomilenine, hence representing a novel, yet unclassified enzyme. We, therefore, dubbed it vomilenine reductase 2 (VR2). Our findings further suggest that both possible reduction sequences of vomilenine might take place in planta, although a corresponding 19,20-dihydrovomilenine reductase has yet to be identified (Fig. 1). Withal, no genes correlating with the reductases described by Gao et al. (2002) and von Schumann et al. (2002) have been annotated thus far and supplementary studies with recombinant enzymes are needed to unequivocally define the diverse reduction steps of the pathway segment in question.
Although biochemical experiments provided clear evidence that VR2 acts on an intermediate of the ajmaline biosynthetic pathway, its physiological relevance cannot be unambiguously delimited based upon the current data. Yet, regarding the pathway as a solitary sequence of reactions might be misleading. In fact, Rauvolfia yields an assorted array of diverse alkaloids. While ajmaline constitutes a predominant and vital component of the myriad, it is by no means a sole metabolic target of the plant, as numerous snakeroot compounds, i.e., side-products, like sarpagine (Stöckigt et al. 1981), raucaffricine (Warzecha et al. 1999), or raucaffrinoline (Rosenthal et al. 2006) do not emerge en route to the antiarrhythmic. This heterogeneity is generated by a highly branched or, indeed, a matrix pathway that could be considered “diversity-oriented” (Fischbach and Clardy 2007). Hence, the herein delineated activity of VR2 lends itself to speculation on its biodiversity-contributing role in providing an alternative processing route for vomilenine.
Alignment with sequences of enzymes of verified function showed that the hereby isolated VR2 translates exhibit high similarity to cinnamyl alcohol dehydrogenases (CADs) of different origins. CADs are involved in monolignol biosynthesis and are, therefore, essential in lignification processes and wood development (Lewis and Yamamoto 1990). Previous research on various species has shown that CADs are encoded by a multigene family, with numerous isoenzymes frequently present in a given representative of the plant kingdom (Kim et al. 2004; Guo et al. 2010; Chao et al. 2014). Functional characterization has revealed variability of CADs in terms of substrate acceptance, with enzymes of gymnosperms showing specificity towards coniferaldehyde or a P. tremuloides CAD selectively targeting sinapaldehyde (SAD, Li et al. 2001). While most investigated CADs exhibit high promiscuity among phenylpropenyl aldehyde derivatives, some CAD-like enzymes show no activity whatsoever (Chao et al. 2014). Hence, divergent roles of the dehydrogenase family members, played in defense or in response to stress, have been postulated (Kim et al. 2004). With the identification of the CAD-like VR2, we further demonstrate that their catalytic function might veer from lignification to alkaloid biosynthesis.
In contrast to the RsCAD, exhibiting detectable activity against cinnamaldehyde, the two isolated VR2 isoenzymes exclusively converted vomilenine. It seems likely that VR2 has evolved in course of a gene duplication process within the CAD family, and a change in function recruited this paralog from the more basic involvement in lignification towards a defense-related capacity within MIA biosynthesis. Studies in genera like Arabidopsis (Kim et al. 2004) or Populus (Chao et al. 2014) have indicated that among the identified CAD enzymes, a few do not convert phenylpropenyl aldehydes and, therefore, might have acquired different functions as well.
Not only are several oxidoreductases involved in the R. serpentina-characteristic ajmaline biosynthetic pathway; the same is, in fact, true of other MIA metabolic routes in distinct plant species. Furthermore, the recently discovered tetrahydroalstonine synthase (THAS, Stavrinides et al. 2015) and 8-hydroxygeraniol dehydrogenase (Miettinen et al. 2014), an enzyme involved in the upstream MIA pathway leading to the formation of the monoterpenoid precursor, secologanin, from C. roseus exhibit significant homology to CADs. Since both aforementioned intermediates can be found in Rauvolfia, the existence of orthologous snakeroot genes seems likely, yet still requires corroboration. Other oxidoreductases inherent to the ajmaline pathway, although catalyzing similar reaction sequences, proved to be derived from alternative gene families. Perakine, for example, an alkaloid with a reducible carbonyl group and not a substrate of VR2, is reduced by perakine reductase (PR, Sun et al. 2008). The latter belongs to aldo–keto reductases (AKR); again, an enzyme family with members involved in multiple metabolic processes (Sengupta et al. 2015). The enumerated considerations and examples make evident that to predict a candidate gene/enzyme involved in the proposed reaction sequence is almost impossible. Although other reductases, like the hydroxycinnamoyl-CoA double bond reductases implicated in dihydrochalcone formation, might appear more legitimate candidates to tackle the alkene moiety within vomilenine (Youn et al. 2006; Ibdah et al. 2014), the newly acquired function of VR2, seemingly deriving from the CAD enzyme family, comprises the NADPH-dependent reduction of the aforementioned alkene component with very high specificity. Proximity of the hydroxyl group at position 21 of the parent scaffold seems, therefore, mandatory, since neither the structurally related vinorine (no OH-group at C-21) nor raucaffricine (vomilenine-21-D-glucopyranoside) were processed as VR2 substrates. The decisive insight into the mechanistics of the investigated reaction might come from future studies on crystal structures of the retrieved isoenzymes (Panjikar et al. 2012).
Furthermore, we identified the new VR2 relying on peptide sequences obtained from a protein fraction exhibiting 1,2-dihydrovomilenine reductase (DHVR) activity. This is surprising, as the DHVR fraction did not show any VR2-specific conversion (Gao et al. 2002). The most plausible explanation is our incidental identification of a highly homologous protein, with similar sequence stretches, but of different function. In fact, none of the enzymes retrieved from the transcriptomics database exhibited sequences corresponding exactly to those obtained in course of Edmann degradation of the active enzyme fractions from snakeroot cell suspension cultures. Even the two isoenzymes recovered from different Rauvolfia species show slight sequence variations, despite identical activity. This observation raises two questions. First, is the coverage of the available transcriptome data incomplete, suggesting that not all RNA species in a given tissue are represented in the resource or that contig assembly might have generated deficient outcomes (Góngora-Castillo et al. 2012a, b)? Second, was the plant material used for RNA isolation indeed derived from R. serpentina? Rauvolfia is a genus of 60–70 species (Cullen et al. 2011) which poses difficulties in distinguishing between its representatives, especially when not all identification hallmarks, like morphological traits of flowers and fruits, can be taken into account. In case of the investigated cell suspension culture, propagated for decades now, no such disambiguation possibilities exist, and its identity can only be verified through analysis of the spectrum of generated compounds and genes heretofore isolated from this resource. While taxonomic classification of the utilized R. tetraphylla plant was achieved through examination of its vegetative and generative features, no retrospective identification of the species used for generation of the transcriptomic data could be performed. What is more, differences within the R. serpentina species, resulting in genetic variance of MIA biosynthetic genes, cannot be excluded. This quandary can only be solved through application of genetic markers affording unequivocal taxonomic identification (e.g., described in Eurlings et al. 2013).
Author contribution statement
MG and MB conducted experiments and analyzed data. HW and AS conceived and designed research, analyzed data, and wrote manuscript. JV contributed analytical tools. All authors read and approved the manuscript.
Abbreviations
- MIAs:
-
Monoterpenoid indole alkaloids
- CAD:
-
Cinnamyl alcohol dehydrogenase
- VR:
-
Vomilenine reductase
- DHVR:
-
Dihydrovomilenine reductase
References
Almagro L, Fernández-Pérez F, Pedreño MA (2015) Indole alkaloids from Catharanthus roseus: bioproduction and their effect on human health. Molecules 20:2973–3000. doi:10.3390/molecules20022973
Asada K, Salim V, Masada-Atsumi S, Edmunds E, Nagatoshi M, Terasaka K, Mizukami H, De Luca V (2013) A 7-deoxyloganetic acid glucosyltransferase contributes a key step in secologanin biosynthesis in Madagascar periwinkle. Plant Cell 25:4123–4134. doi:10.1105/tpc.113.115154
Avrutina O, Schmoldt HU, Gabrijelcic-Geiger D, Le Nguyen D, Sommerhoff CP, Diederichsen U, Kolmar H (2005) Trypsin inhibition by macrocyclic and open-chain variants of the squash inhibitor MCoTI-II. Biol Chem 386:1301–1306. doi:10.1515/BC.2005.148
Bernhardt P, McCoy E, O’Connor SE (2007) Rapid identification of enzyme variants for reengineered alkaloid biosynthesis in periwinkle. Chem Biol 14:888–897. doi:10.1016/j.chembiol.2007.07.008
Brown S, Clastre M, Courdavault V, O’Connor SE (2015) De novo production of the plant-derived alkaloid strictosidine in yeast. Proc Natl Acad Sci USA 112:3205–3210. doi:10.1073/pnas.1423555112
Chao N, Liu SX, Liu BM, Li N, Jiang XN, Gai Y (2014) Molecular cloning and functional analysis of nine cinnamyl alcohol dehydrogenase family members in Populus tomentosa. Planta 240:1097–1112. doi:10.1007/s00425-014-2128-9
Cullen JK, Knees SG, Cubey HS (2011) The European garden flora. Angiospermae-Dicotyledons, vol IV. Cambridge University Press, Cambridge
Eurlings MC, Lens F, Pakusza C, Peelen T, Wieringa JJ, Gravendeel B (2013) Forensic identification of Indian snakeroot (Rauvolfia serpentina Benth. ex Kurz) using DNA barcoding. J Forensic Sci 58:822–830. doi:10.1111/1556-4029.12072
Fischbach MA, Clardy J (2007) One pathway, many products. Nat Chem Biol 3:353–355. doi:10.1038/nchembio0707-353
Gao S, von Schumann G, Stöckigt J (2002) A newly-detected reductase from Rauvolfia closes a gap in the biosynthesis of the antiarrhythmic alkaloid ajmaline. Planta Med 68:906–911. doi:10.1055/s-2002-34935
Geu-Flores F, Sherden NH, Courdavault V, Burlat V, Glenn WS, Wu C, Nims E, Cui Y, O’Connor SE (2012) An alternative route to cyclic terpenes by reductive cyclization in iridoid biosynthesis. Nature 492:138–142. doi:10.1038/nature11692
Góngora-Castillo E, Buell CR (2013) Bioinformatics challenges in de novo transcriptome assembly using short read sequences in the absence of a reference genome sequence. Nat Prod Rep 30:490–500. doi:10.1039/c3np20099j
Góngora-Castillo E, Childs KL, Fedewa G, Hamilton JP, Liscombe DK, Magallanes-Lundback M, Mandadi KK, Nims E, Runguphan W, Vaillancourt B, Varbanova-Herde M, DellaPenna D, McKnight TD, O’Connor SE, Buell CR (2012a) Development of transcriptomic resources for interrogating the biosynthesis of monoterpene indole alkaloids in medicinal plant species. PLoS One 7(12):e52506. doi:10.1371/journal.pone.0052506
Góngora-Castillo E, Fedewa G, Yeo Y, Chappell J, DellaPenna D, Buell CR (2012b) Chapter Seven—Genomic approaches for interrogating the biochemistry of medicinal plant species. Methods Enzymol 517:139–159. doi:10.1016/B978-0-12-404634-4.00007-3
Guo DM, Ran JH, Wang XQ (2010) Evolution of the cinnamyl/sinapyl alcohol dehydrogenase (CAD/SAD) gene family: the emergence of real lignin is associated with the origin of bona fide CAD. J Mol Evol 71:202–218. doi:10.1007/s00239-010-9378-3
Ibdah M, Berim A, Martens S, Valderrama AL, Palmieri L, Lewinsohn E, Gang DR (2014) Identification and cloning of an NADPH-dependent hydroxycinnamoyl-CoA double bond reductase involved in dihydrochalcone formation in Malus × domestica Borkh. Phytochemistry 107:24–31. doi:10.1016/j.phytochem.2014.07.027
Kim SJ, Kim MR, Bedgar DL, Moinuddin SG, Cardenas CL, Davin LB, Kang C, Lewis NG (2004) Functional reclassification of the putative cinnamyl alcohol dehydrogenase multigene family in Arabidopsis. Proc Natl Acad Sci USA 101:1455–1460. doi:10.1073/pnas.0307987100
Lewis NG, Yamamoto E (1990) Lignin: occurrence, biogenesis and biodegradation. Annu Rev Plant Physiol Plant Mol Biol 41:455–496. doi:10.1146/annurev.pp.41.060190.002323
Li L, Cheng XF, Leshkevich J, Umezawa T, Harding SA, Chiang VL (2001) The last step of syringyl monolignol biosynthesis in angiosperms is regulated by a novel gene encoding sinapyl alcohol dehydrogenase. Plant Cell 13:1567–1586
Linsmaier EM, Skoog F (1965) Organic growth factor requirements of tobacco tissue cultures. Physiol Plant 18:100–127
Miettinen K, Dong L, Navrot N, Schneider T, Burlat V, Pollier J, Woittiez L, van der Krol S, Lugan R, Ilc T, Verpoorte R, Oksman-Caldentey KM, Martinoia E, Bouwmeester H, Goossens A, Memelink J, Werck-Reichhart D (2014) The seco-iridoid pathway from Catharanthus roseus. Nat Commun 5:3606. doi:10.1038/ncomms4606
Panjikar S, Stöckigt J, O’Connor SE, Warzecha H (2012) The impact of structural biology on alkaloid biosynthesis research. Nat Prod Rep 29:1176–1200. doi:10.1039/C2np20057k
Rosenthal C, Mueller U, Panjikar S, Sun L, Ruppert M, Zhao Y, Stöckigt J (2006) Expression, purification, crystallization and preliminary X-ray analysis of perakine reductase, a new member of the aldo–keto reductase enzyme superfamily from higher plants. Acta Crystallogr Sect F Struct Biol Cryst Commun 62:1286–1289. doi:10.1107/S174430910605041X
Salim V, Yu F, Altarejos J, De Luca V (2013) Virus-induced gene silencing identifies Catharanthus roseus 7-deoxyloganic acid-7-hydroxylase, a step in iridoid and monoterpene indole alkaloid biosynthesis. Plant J 76:754–765. doi:10.1111/tpj.12330
Salim V, Wiens B, Masada-Atsumi S, Yu F, De Luca V (2014) 7-Deoxyloganetic acid synthase catalyzes a key 3 step oxidation to form 7-deoxyloganetic acid in Catharanthus roseus iridoid biosynthesis. Phytochemistry 101:23–31. doi:10.1016/j.phytochem.2014.02.009
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sengupta D, Naik D, Reddy AR (2015) Plant aldo–keto reductases (AKRs) as multi-tasking soldiers involved in diverse plant metabolic processes and stress defense: a structure–function update. J Plant Physiol 179:40–55. doi:10.1016/j.jplph.2015.03.004
Staniek A, Bouwmeester H, Fraser PD, Kayser O, Martens S, Tissier A, van der Krol S, Wessjohann L, Warzecha H (2013) Natural products—modifying metabolite pathways in plants. Biotechnol J 8:1159–1171. doi:10.1002/biot.201300224
Staniek A, Bouwmeester H, Fraser PD, Kayser O, Martens S, Tissier A, van der Krol S, Wessjohann L, Warzecha H (2014) Natural products—learning chemistry from plants. Biotechnol J 9:326–336. doi:10.1002/biot.201300059
Stavrinides A, Tatsis EC, Foureau E, Caputi L, Kellner F, Courdavault V, O’Connor SE (2015) Unlocking the diversity of alkaloids in Catharanthus roseus: nuclear localization suggests metabolic channeling in secondary metabolism. Chem Biol 22:336–341. doi:10.1016/j.chembiol.2015.02.006
Stöckigt J, Zenk MH (1977) Strictosidine (isovincoside): the key intermediate in the biosynthesis of monoterpenoid indole alkaloids. J Chem Soc Chem Commun 18:646–648. doi:10.1039/C39770000646
Stöckigt J, Zenk MH (1995) Biosynthesis in Rauvolfia serpentina—modern aspects of an old medicinal plant. In: Cordell GA (ed) The alkaloids. Chemistry and pharmacology. Academic Press, San Diego, pp 115–172
Stöckigt J, Pfitzner A, Firl J (1981) Indole alkaloids from cell suspension cultures of Rauwolfia serpentina benth. Plant Cell Rep 1:36–39. doi:10.1007/BF00267656
Sun L, Ruppert M, Sheludko Y, Warzecha H, Zhao Y, Stöckigt J (2008) Purification, cloning, functional expression and characterization of perakine reductase: the first example from the AKR enzyme family, extending the alkaloidal network of the plant Rauvolfia. Plant Mol Biol 67:455–467. doi:10.1007/s11103-008-9331-7
von Schumann G, Gao S, Stöckigt J (2002) Vomilenine reductase—a novel enzyme catalyzing a crucial step in the biosynthesis of the therapeutically applied antiarrhythmic alkaloid ajmaline. Bioorg Med Chem 10:1913–1918
Warzecha H, Obitz P, Stöckigt J (1999) Purification, partial amino acid sequence and structure of the product of raucaffricine-O-β-d-glucosidase from plant cell cultures of Rauwolfia serpentina. Phytochemistry 50:1099–1109
Youn B, Kim SJ, Moinuddin SG, Lee C, Bedgar DL, Harper AR, Davin LB, Lewis NG, Kang C (2006) Mechanistic and structural studies of apoform, binary, and ternary complexes of the Arabidopsis alkenal double bond reductase At5g16970. J Biol Chem 281:40076–40088. doi:10.1074/jbc.M605900200
Zhu H, Kerčmar P, Wu F, Rajendran C, Sun L, Wang M, Stöckigt J (2015) Using strictosidine synthase to prepare novel alkaloids. Curr Med Chem 22:1880–1888
Acknowledgments
We are indebted to Markus Krischke for his help with LC–MS analysis. The support of COST Action FA1006 is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
M. Geissler and M. Burghard contributed equally to the work.
Rights and permissions
About this article

Cite this article
Geissler, M., Burghard, M., Volk, J. et al. A novel cinnamyl alcohol dehydrogenase (CAD)-like reductase contributes to the structural diversity of monoterpenoid indole alkaloids in Rauvolfia . Planta 243, 813–824 (2016). https://doi.org/10.1007/s00425-015-2446-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-015-2446-6