
Abstract
Main conclusion
A genome-wide survey of Catharanthus roseus receptor-like kinase1-like kinases (CrRLK1Ls) in rice revealed that the pattern of expression by some CrRLK1Ls is controlled by drought or circadian rhythms. This is probably accomplished through the functioning of Gigantea ( OsGI ). Such findings provide a novel angle for using CrRLK1Ls to study the drought-stress response and circadian regulation.
Abstract
The 17 CrRLK1L members of a novel RLK family have been identified in Arabidopsis. Each carries a putative extracellular carbohydrate-binding malectin-like domain. However, their roles in rice, a widely consumed staple food, are not well understood. To investigate the functions of CrRLK1Ls in rice, we utilized phylogenomics data obtained through anatomical and diurnal meta-expression analyses. This information was integrated with a large set of public microarray data within the context of the rice CrRLK1L family phylogenic tree. Chromosomal locations indicated that 3 of 16 genes were tandem-duplicated, suggesting possible functional redundancy within this family. However, integrated diurnal expression showed functional divergence between two of three genes, i.e., peak expression was detected during the day for OsCrRLK1L2, but during the night for OsCrRLK1L3. We found it interesting that OsCrRLK1L2 expression was repressed in osgigantea (osgi) mutants, which suggests that it could function downstream of OsGI. Network analysis associated with OsCrRLK1L2 and OsGI suggested a novel circadian regulation mechanism mediated by OsGI. In addition, two of five OsCrRLK1Ls preferentially expressed in the roots were stimulated by drought, suggesting a potential role for this family in water-use efficiency. This preliminary identification of CrRLK1Ls and study of their expression in rice will facilitate further functional classifications and applications in plant production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protein kinases are essential for regulating growth and development in prokaryotes and eukaryotes. They can be grouped into three distinct classes: serine/threonine, tyrosine, and histidine kinases. In plants, receptor-like kinases (RLKs) comprise a large protein family with potential capacity for cell surface signaling (Cheung and Wu 2011). These transmembrane proteins have putative amino-terminal extracellular and carboxyl-terminal intracellular kinase domains, and their domain organization is strikingly similar to that of animal RLKs, such as the presence of an epidermal growth factor receptor (Shiu and Bleecker 2001).
CrRLK1L, is a novel RLK-type from Catharanthus roseus that has a unique extracellular domain (Schulze-Muth et al. 1996). In contrast to other plant RLKs, this one utilizes an intra- rather than an inter-molecular phosphorylation mechanism for regulation of kinase activity (Schulze-Muth et al. 1996). Studies with Arabidopsis have identified 17 CrRLK1L members that contain a signal peptide, a putative extracellular carbohydrate-binding malectin-like domain, a transmembrane domain, and a kinase domain (Boisson-Dernier et al. 2011; Cheung and Wu 2011; Lindner et al. 2012). In particular, the existence of a malectin-like domain supports the roles this family has in mechanisms associated with cell wall surveillance (Lindner et al. 2012). Functions have been reported for some Arabidopsis CrRLK1Ls in cell elongation, coordination of cell wall integrity, cell wall sensing, and cell-to-cell communication between male and female gametophytes (Lindner et al. 2012). Among these, expression of FERONIA (FER), THESEUS1 (THE1), HERCULES1 (HERK1), and HERCULES2 (HERK2) is stronger in regions within vegetative tissues where cells are elongating, and are also up-regulated by brassinosteroids to modulate growth and development (Guo et al. 2009b). Loss-of-function studies have presented elongation defects in the leaves and stems of fer single mutants, the1/herk1 double mutants, and a the1/herk1/herk2 triple mutant, all indicating that CrRLK1L members have somewhat redundant roles in that process during the vegetative growth phase. FER is also involved in polar growth of root hairs and pollen tube reception through cell-to-cell communications between gametophytes (Escobar-Restrepo et al. 2007; Duan et al. 2010).
Within the CrRLK1L family, ANXUR1 (ANX1) and ANXUR2 (ANX2) are the closest FER homologs in Arabidopsis, and are preferentially expressed in pollen, playing redundant roles in controlling pollen tube integrity (Boisson-Dernier et al. 2009; Miyazaki et al. 2009). In addition, the pollen-expressed NADPH oxidases function downstream of ANX1 and ANX2 to coordinate cell wall integrity in tip-growing cells via ROS production, Ca2+ homeostasis, and exocytosis (Boisson-Dernier et al. 2013). Pollen tube rupture and sperm release appear to be mediated by high levels of ROS at the entrance to the female gametophyte (Duan et al. 2014). The FER signaling pathway is required in calcium responses and for coupling the programmed cell death of pollen tubes and the receptive synergid to control plant sperm delivery (Ngo et al. 2014). In addition to ROS and Ca2+, a secreted peptide—rapid alkalinization factor (RALF)—suppresses cell elongation in primary roots. This is mediated by activating FER through a ligand and receptor pair (Haruta et al. 2014).
The molecular and mechanical properties of cell walls enable them to adapt to extracellular abiotic and biotic stresses through ROS regulation. This possibly occurs when the expression of the CrRLK1L family genes is controlled (Lindner et al. 2012). Interestingly, the CrRLK1Ls, At5g38990 and At5g39020 are up-regulated by elicitor treatments and biotic stresses but down-regulated by drought (Lindner et al. 2012). Unlike in Arabidopsis, none of the CrRLK1Ls has been characterized in Oryza sativa (rice), a model crop plant. We speculate that this manipulation of cell wall elongation and integrity through ROS regulation might be very critical to the maintenance of crop productivity.
Oscillations within the 24-h circadian clock drive diurnal rhythms. Those circadian rhythms have been studied in various organisms including higher plants such as Arabidopsis and rice (McClung 2006). Chen (2013) has reported that the plant circadian clock has three interlocked transcriptional feedback loops. The central loop includes Circadian Clock-Associated 1 (CCA1)/Late Elongated Hypocotyl (LHY) and Timing Of Cab Expression 1 (TOC1)/Pseudo-Response Regulator 1 (PRR1), which reciprocally regulate CCA1/LHY, CCA1 Hiking Expedition (CHE), Early Flowering 3 (ELF3), ELF4, and Phytoclock 1 (PCL1). The morning loop comprises PRR5, PRR7, PRR9, and casein kinase II (CK2), while the evening loop includes Gigantea (GI), Zeitlupe (ZTL), and PRR3. Rice plants are day length-sensitive, and their circadian clock controls key developmental processes such as flowering time (Yano et al. 2000; Izawa et al. 2011; Izawa 2012). A mutation of OsGI, an Arabidopsis GI ortholog, causes late flowering (Izawa et al. 2011). Heading Date 1 (Hd1), an Arabidopsis CONSTANS (CO) homolog, encodes a zinc finger domain protein (Yano et al. 2000) and predominantly regulates the OsGI-dependent photoperiodic pathway under either long or short days (Hayama et al. 2003). The Arabidopsis CCA1/LHY ortholog OsLHY controls flowering time through phosphorylation by CK2 (Ogiso et al. 2010); its expression is significantly decreased by the OsGI mutation (Izawa 2012). In Arabidopsis, an adaptation to salt stress that is mechanistically based on GI protein degradation under saline conditions can retard flowering, thereby indicating crosstalk between the abiotic stress response and circadian regulation (Kim et al. 2013). Drought is a major obstacle for crop production because it profoundly limits leaf and root growth (Farooq et al. 2009). To satisfy increasing demands for food, researchers have been working to develop rice plants with enhanced drought tolerance. In response to water deficits, plants employ mechanisms such as ABA-independent and ABA-dependent pathways (Shinozaki and Yamaguchi-Shinozaki 2007). The BASIC LEUCINE ZIPPER 23 transcription factor (bZIP23) and bZIP46 are involved in drought-induced gene expression through the ABA signaling pathway (Bartels and Sunkar 2005; Fukao and Xiong 2013). Responsive to Abscisic Acid 16 (RAB16) is another drought-responsive gene controlled by ABA (Oono et al. 2014). In rice, Late Embryogenesis Abundant (LEA) 3-1 (OsLEA3-1) is required for conferring tolerance during the response to multiple abiotic stressors, including drought, salt, and cold (Goyal et al. 2005; Xiao et al. 2007). The FER pathway in Arabidopsis negatively regulates the ABA response that is mediated by activation of Arabidopsis ABSCISIC ACID-INSENSITIVE2 phosphatase (Yu et al. 2012). This also suggests that members of the CrRLK1L family modulate plant responses to abiotic stresses by means of the ABA signaling pathway.
In this systematic investigation, we used combined phylogenetics and meta-expression profiling analyses to obtain functional information about all of the CrRLK1L family members in rice. Phylogenomic sequences were compared between rice and Arabidopsis so that we could assign Arabidopsis orthologs with known functions to individual rice CrRLK1Ls. Data were acquired from public microarrays for both meta-anatomical and diurnal expression. These were integrated to develop a phylogenic tree for rice.
Materials and methods
Identification of rice CrRLK1L genes
Rice CrRLK1L genes were obtained from Greenphyl, a phylogenomics database for plant comparative genomics (http://greenphyl.cirad.fr/v2/cgi-bin/index.cgi) (Conte et al. 2008b), and from the Rice Genome Annotation Project database (RGAP; http://rice.plantbiology.msu.edu/). Our list of all 16 CrRLK1L genes in rice (Table S1) included Locus identifier (Locus id), RAP_id (http://www.rapdb.dna.affrc.go.jp/tools/dump), the protein, genomic and cDNA accession number from NCBI, the clone name from Knowledge-based Oryza Molecular biological Encyclopedia (KOME), and gene ontology (GO) terms from the Rice Oligonucleotide Array Database (http://www.ricearray.org/) (Cao et al. 2012; Chandran and Jung 2014). We also compiled the sequences for 17 Arabidopsis CrRLK1L genes (Lindner et al. 2012). Ortholog names were collected from The Arabidopsis Information Resource (TAIR; http://www.Arabidopsis.org/).
Gene mapping onto rice chromosomes
The distribution of 16 CrRLK1Ls on 7 rice chromosomes (Fig. S1) was determined and visualized using the map tool function installed in the Oryzabase database (http://www.shigen.nig.ac.jp/rice/oryzabaseV4/).
Phylogenetic analysis
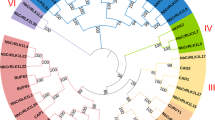
To investigate the phylogenetic relationships among CrRLK1L genes from rice to Arabidopsis thaliana, we first conducted multiple alignments for 16 rice and 17 Arabidopsis protein sequences, using CLUSTAL X software (ver. 2). Those alignments were manually adjusted to retain the conserved RLK motifs. Phylogenetic analysis was then performed with the MEGA5 program based on the Neighbor-Joining (NJ) tree method, complete deletion, and bootstrapping with 500 replicates. These data were used to assign subgroups within the CrRLK1L family (Fig. 1).

Comparative phylogenetic analysis of rice and Arabidopsis CrRLK1L proteins. Unrooted phylogenetic tree was constructed using MEGA5 program and Neighbor-Joining method. Five colors indicate distinct CrRLK1L subgroups (I–V) in rice and Arabidopsis
Meta-expression analysis
Anatomical expression data for each CrRLK1L gene were collected using 1,150 Affymetrix arrays (GPL2025) and 143 Agilent 44 K arrays (GSE21494). Raw data were downloaded from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database at the National Center for Biotechnology Information (NCBI) (Barrett et al. 2011). The Affymetrix microarray data were then normalized by the Mas 5.0 method implemented in the R package affy, and normalized values were converted to a log2 scale. For anatomical expression analysis, we organized the Affymetrix data into 18 groups of tissues/organs. For the Agilent arrays, we used the median Cy3 signal intensities in the Agilent 44 K microarray data sets (GSE21494) at log2 median intensities, then normalized the log2 intensities using a quantile normalization method (Bolstad et al. 2003; Cao et al. 2012). Data were then assigned to the 18 groups. In addition, we downloaded and analyzed diurnal expression data which were generated using leaves representing four developmental stages from wild-type (WT) japonica rice ‘Norin 8’ and the osgi mutant using Agilent array (GSE18685) from GEO. These were sampled each week between June 6 and June 27, in 2008. For transcriptome analysis (Itoh and Izawa 2011), field-grown plants were used from each developmental stage. Two leaves were harvested every 2 h for 2 days (13 time points over 24 h, beginning at 07:00 on 12 August). Heat maps were generated from the log-transformed data and average calculations using Multi Experiment Viewer (http://www.tm4.org/mev/). They were integrated into the phylogenetic tree context.
Plant materials and growing conditions
Wild-type (WT) seeds of Oryza sativa L. ssp. Japonica, genotype Dongjin (Rural Development Administration of Korea) were germinated in a Murashige and Skoog (1962) agar medium for 7 days. The resultant seedlings were grown at 28 °C and 60 % humidity under a 12-h photoperiod. For evaluating anatomical expression, 7-day-old seedlings were transferred to water and incubated for 1 day under continuous light. Afterward, their roots and shoots were harvested, frozen in liquid nitrogen, and stored at −80 °C. For the flag leaves, tissues were collected after 3 months of growth in the field. To induce drought stress in a growth chamber (28 °C, 12-h photoperiod), we removed the water from the container having 30-day-old WT seedlings grown in watered plastic seed starting trays with 32 cells and examined them at 3 and 4 days after treatment (DAT). Non-stressed samples were maintained without water removal under the same environmental conditions. Roots were harvested simultaneously from both settings for RNA extraction. For examining the circadian influence, 10-day-old WT and osgi mutant seedlings harboring a T-photoperiod). Beginning at 30 DAT, their leaves were sampled every 4 h for 2 days, then frozen in liquid nitrogen and stored at −80 °C.
RNA isolation, RT-PCR, and real-time RT-PCR analyses
Total RNA was extracted with an RNAiso Plus Kit (Takara, Kyoto, Japan). For semi-quantitative reverse transcription-PCR (RT-PCR), 4 µg of total RNA was reverse-transcribed to cDNA using SuperScript II Reverse Transcriptase, according to the manufacturer’s instructions. The same amount of cDNA was utilized for each PCR procedure. Two internal control genes—rice UBIQUITIN 5 (OsUBI5, LOC_Os01g22490) and rice ELONGATION FACTOR1 (OsEF1, LOC_Os03g08010)—were used to balance the transcripts among samples.
For real-time RT-PCR analysis, primers were designed to amplify products between 150 and 200 bp (Wong and Medrano 2005). Each primer was searched using the NCBI database BLAST tool to check specificity by gene. The 2X SYBR green PCR premix Ex Taq (Takara) was used, and PCRs were run on a Rotor-Gene 6000 (Qiagen, Hilden, Germany). OsUbi5 served as an internal control for normalization (Jung et al. 2013). Quantitative RT-PCR and the 2−∆Ct comparative threshold cycle method were used to quantify gene expression (Wong and Medrano 2005), and PCRs were performed three times. All primers are listed in Table S2.
Results
CrRLK1L gene identification and chromosomal location in rice
We used the GreenphylDB database and obtained 25 models representing 16 loci annotated as “the CrRLK1L homolog”. All 16 were verified by the RGAP database and a representative gene model was selected for each locus. These were denoted as OsCrRLK1L1 through OsCrRLK1L16. Because all OsCrRLK1Ls were annotated as expressed protein, we first tried to find orthologs in Arabidopsis from TAIR. We also identified GO terms to provide functional classification information about the category of ‘cellular component’ for each family member as previously preceded (Botstein et al. 2000; Nguyen et al. 2013). An annotation overview of all rice CrRLK1L proteins is provided in Fig. S2.
Family members were distributed on 7 of 12 rice chromosomes, with none found on chromosomes 2, 8, 9, 11, and 12. Five were located on chromosome 5, four on chromosome 3, two each on chromosomes 1 and 6, and one each on chromosomes 4, 7, and 10. We thought it is noteworthy that a tandem array of three OsCrRLK1Ls occurred on chromosome 5, namely OsCrRLK1L2, -3, and -12. We compared the these protein sequences using blastp suite-2sequences tool in NCBI BLAST and identified that OsCrRLK1L2 has 67 % identity under 100 % query coverage with OsCrRLK1L3, and 69 % with OsCrRLK1L12. Therefore, we suspected that these were duplicated and might have redundant functions (Fig. S1).
Comparative phylogenetic analysis of CrRLK1L family between rice and Arabidopsis
Relying on RGAP and TAIR sequence information, we performed a phylogenetic analysis to examine the evolutionary relationship between CrRLK1L family genes in rice and Arabidopsis. A phylogenetic tree was constructed by aligning the full-length protein sequences for the 16 rice members and 17 Arabidopsis members. These genes were divided among Subgroups I through V. Subgroups IV and V contained only Arabidopsis CrRLK1L proteins (Fig. 1), evidence of unique evolutionary subgroups in that species. The organization for rice included eight members in Subgroup I (OsCrRLK1L1, -4, -6, -7, -10, -13, -14, and -16), two members in Group II (OsCrRLK1L9 and -15), and six members in Group III (OsCrRLK1L2, -3, -5, -8, -11, and -12). Arabidopsis orthologs were assigned to 13 of those rice members (Fig. S2), five with known functions: Arabidopsis HERK2, an ortholog of OsCrRLK1L9 and -15; Arabidopsis ANX1 and ANX2, both orthologs of OsCrRLK1L11; Arabidopsis FER, an ortholog of both OsCrRLK1L5 and -8; and Arabidopsis ERULUS, an ortholog of OsCrRLK1L1. We expected that those rice members would have functions similar to their Arabidopsis orthologs. However, it was more difficult to predict roles for the others due to the lack of functionally characterized Arabidopsis orthologs.
Integration of anatomical expression patterns with the CrRLK1L family phylogenic tree
In the next sections, the phylogenetic tree with this classification was integrated to the transcriptome data to represent expression levels of the OsCrRLK1Ls. For our anatomical work, the Affymetrix array data were organized into 18 types of tissues/organs, while the Agilent 44 K array data fell into 13 classifications. The expression data heat map was integrated into the phylogenic tree context (Fig. 2). For the Affymetrix meta-expression database, we analyzed all CrRLK1L expression data. However, in the case of Agilent 44 K, expression data were produced for all genes except OsCrRLK1L1 and -13 because probes for those genes had not been designed for that particular array.

Anatomical expression patterns integrated with CrRLK1L phylogenetic tree. Data for 18 organs/tissues were classified from 1,150 Affymetrix arrays and 209 Agilent 44 K arrays. Yellow high expression; blue low expression. RT-PCR was performed to confirm patterns for 16 OsCrRLK1Ls in leaves, flag leaves, and roots (red box). OsUbi5 and OsEF1α were used as internal controls for reactions. Gray dots on right-hand side, high expression in roots; black dots, high expression in leaves and flag leaves, based on RT-PCR results
The genes in the OsCRLK1L family were considered ubiquitously expressed, vegetative organ-preferential, or floral organ-preferential (Fig. 2). In the first category, OsCrRLK1L8 was detected across all tissue types examined, suggesting a housekeeping role.
Another eight genes showed higher expression in vegetative organs than in reproductive tissues. Of these, OsCrRLK1L4 and -7; OsCrRLK1L6 and -16; and OsCrRLK1L2, -3, and -12 were clustered together in the phylogenic tree. The eighth gene—OsCrRLK1L1—was the outlier in that second category. Both OsCrRLK1L4 and -7 were more highly expressed in the leaves than in the roots. However, the latter gene also demonstrated strong expression in callus tissue and embryos, indicating that it has a unique function. Whereas OsCrRLK1L6 showed higher expression in the roots than in the leaves, the opposite was true for OsCrRLK1L16, which possibly meant functional divergence. Levels of expression in the roots and leaves were similar between OsCrRLK1L3 and -12, while OsCrRLK1L2 tended to be leaf-preferential, suggesting a unique feature when compared with its paralogs. In addition to being expressed in the roots, transcripts in the stigma/ovary and seed were higher for OsCrRLK1L3 and -12 than for OsCrRLK1L2. Expression patterns also differed, however, between OsCrRLK1L3 and -12. Because transcript levels of OsCrRLK1L12 are generally very low in indica rice samples, our functional characterization might explain the evolutionary difference between japonica and indica cultivars. Predominant expression was most obvious in OsCrRLK1L2, suggesting major role among those three clustered members. The root-preferential expression of OsCrRLK1L1 implied that it has a unique role in the development of that organ.
Transcripts of six OsCrRLK1L genes were more abundant in reproductive organs than in vegetative organs. Both OsCrRLK1L5 and -14 showed fairly high expression in all floral organs analyzed, although that of the latter was mild in the endosperm. OsCrRLK1L11 and -13 were preferentially expressed in anthers and pollen. Those two pairs of genes were also separately located in the phylogenic tree, indicating that each might have had unique functions during evolution of that species. In contrast, OsCrRLK1L9 and -15 were clustered together in the tree, and showed similar patterns overall, although expression was higher in the pistils and embryos for OsCrRLK1L15 than for OsCrRLK1L9. This implied functional redundancy between the two in most anatomical functions except those pertaining to the pistils and embryos.
As the sixteenth gene in this family, OsCrRLK1L10 had higher expression in the roots, developing seeds, and endosperm than in other organs. This pattern closely overlapped that of OsCrRLK1L14, although its levels were inconsistent in the roots and seeds, suggesting antagonistic regulation between them in these organs. Expression by OsCrRLK1L10 was also higher in the endosperm when compared with OsCrRLK1L14, suggesting a unique role in endosperm development.
Expression patterns for the 16 OsCrRLK1Ls were analyzed in the vegetative organs via RT-PCR. Transcript levels for OsCrRLK1L5, -6, -10, -14, and -15 were higher in roots than in leaves and flag leaves, while OsCrRLK1L2, -3, -4, -7, -12, and -16 showed stronger expression in the leaves and flag leaves. For most genes, expression patterns were similar to those revealed by the meta-anatomical expression data (Fig. 2). Preferential expression by OsCrRLK1L1, -8, -9, and -13 was unclear for individual tissues or organs and no transcripts were detected for OsCrRLK1L11 (Fig. 2). Overall, our findings demonstrate that 11 of these genes can be used to study functions related to early root or leaf development in seedlings.
Circadian regulation associated with the rice CrRLK1L family
The circadian clock regulates physiological events such as flowering time, hypocotyl elongation, and leaf movement (Sugiyama et al. 2001). We analyzed the publicly available microarray data related to circadian rhythms for 17 Arabidopsis CrRLK1Ls, using Affymetrix Arabidopsis ATH1 genome array data sets (GSE3416) that contained information about gene expression from three biological replicates of a diurnal time series spanning 24 h (sampling every 4 h). Based on those data, THE1 (At5g54380) showed a clear pattern of diurnal expression while the other 16 did not, suggesting that, in the Arabidopsis family, THE1 was likely to be under the control of the circadian clock (Fig. S3). To elucidate the role of OsCrRLK1Ls in circadian-guided physiological events, we used expression data from leaves of the ‘Norin 8’ WT and the osgi mutant collected through four developmental stages in the Agilent 44 K microarray from NCBI GEO. Microarray and RT-PCR analyses indicated that 6 of the 16 genes (OsCrRLK1L2, -3, -4, -7, -12, and -16) demonstrated leaf-preferential expression during the early seedling stages. All of them except OsCrRLK1L4 also showed diurnal expression in the meta-analysis. Transcript levels were reduced in the osgi mutant (Fig. 3). Meta-expression analysis of the two marker genes, OsLHY and HD1, indicated that, as expected, expression of OsLHY peaked during the day, but then declined at the onset of darkness before increasing as daylight returned. By contrast, HD1 showed peak expression under darkness; its regulatory patterns were antagonistic to those of OsLHY. Patterns for OsCrRLK1L3, -7, and -16 were similar to that of HD1 whereas OsCrRLK1L2 and -12 resembled the pattern of OsLHY. Diurnal expression was most obvious for OsCrRLK12 based on both microarray and quantitative real-time RT-PCR analyses, and its expression was significantly reduced in the osgi mutant (Fig. 3). Therefore, these results suggested that OsCrRLK1L2 functions downstream of OsGI. Data were rather inconclusive for the others, leading us to recommend that further examination is required to confirm these changes in gene expression as presented by the meta-expression data.

Circadian regulation associated with 6 CrRLK1Ls in rice. a Heat map was generated using Agilent 44 K microarray data sets (GSE18685) for regulation in WT and osgi mutant confirmed by RT-PCR. Yellow high expression, blue low expression. For RNA expression analysis after 30 days, leaves of WT and GIGANTEA (GI) mutant were harvested every 4 h for 2 days. Gray bar daytime, black bar night time. OsUbi5 and OsEFα1 were used as internal controls. Red box indicates genes that were differentially expressed between WT and mutant. b Expression of OsLHY, Hd1, and OsCrRLK1L2 in WT and OsGI. OsUbi5 was used as internal control for real-time PCR. Error bars represent standard error of three replicates
Effect of hd1 mutant on OsCrRLK1L2 expression
To investigate further how the circadian clock regulates the expression of OsCrRLK1L2, we used the mutant (hd1) of the circadian clock-controlling gene, HD1, which is part of a short day-dependent flowering pathway (Izawa et al. 2003). Expression in the mutant was compared with that of the field-grown WT as well as that of plants exposed to short days (12-h light/12-h dark cycle). Transcripts of OsCrRLK1L2 were up-regulated in the mutant under both environmental scenarios (Fig. 4), indicating that HD1 represses OsCrRLK1L2 expression. This finding suggested a possible molecular link between OsGI-mediating circadian rhythms or flowering pathway and OsCrRLK1L2 (Fig. 4).

Quantitative RT-PCR analyses for OsCrRLK1L2 in WT and hd1 mutant. a Expression in field-grown plants. b Expression under short days (12-h photoperiod). OsUbi1 was used as internal control
Drought-stress response associated with CrRLK1L family
Anatomical roles were revealed via meta-expression analysis and RT-PCR for OsCrRLK1L5, -6, -10, -14, and -15, all of which were preferentially expressed in the roots during the early stage of seedling development (Fig. 2). As we were also interested in understanding their levels of activity under drought stress, we investigated their responses by quantitating their expression. After 3 days of stress, leaf morphology was severely damaged, as manifested by rolling, drying, and wilting when compared with leaves from well-watered control plants (Fig. S4). The expression patterns of several well-characterized drought stress-responsive genes were monitored, including bZIP23 (LOC_Os02g52780), LEA3-1 (LOC_Os05g46480), and RAB16 (LOC_Os11g26790). As expected, all three were strongly up-regulated in the roots at 3 and 4 days after stress treatment (DAT) (Fig. 5). Moreover, of the five root-preferential genes, a milder response was found with OsCrRLK1L10 and -14, which were induced by 3.1-fold and 2.0-fold at 4 DAT, respectively (Fig. 5). These results suggested that OsCrRLK1L10 and -14 have roles in both root development and the response to drought stress.

Quantitative RT-PCR analyses for two OsCrRLK1Ls isolated in drought-stressed roots. Relative transcript levels were normalized with respect to internal control OsUbi5. Error bars represent standard error of three replicates. C control, Dr drought-stressed (water withheld). 3D and 4D indicate sampling at 3 and 4 DAT. Numbers at top of each bar are fold-changes for treatment over control value; plus indicates upregulation
Discussion
Members of the Arabidopsis CrRLK1L family have diverse functions in regulating cell wall integrity (Hématy et al. 2007; Boisson-Dernier et al. 2013), cell-to-cell communications (Escobar-Restrepo et al. 2007; Ngo et al. 2014), and responses to biotic and abiotic stresses (Kessler et al. 2010; Yu et al. 2012). However, their biological and genetic roles are still completely unknown in crop plants such as rice. All Arabidopsis and rice CrRLK1L family have one or two extracellular carbohydrate-binding malectin-like domains and a kinase domain (Fig. S5), indicating that all rice CrRLK1L family members in this study retain the structural features of the CrRLK1L family. Our comparative phylogenetic analysis indicated that genes in Subgroups IV and V are specific to the Arabidopsis family, suggesting unique evolutionary roles. Of the known Arabidopsis CrRLK1Ls, HERK1 and THE1 do not have assigned orthologs in rice. Likewise, OsCrRLK1L2, -3, and -12 from rice do not have assigned orthologs in Arabidopsis (Fig. S2), again implying unique roles. However, absolute functional conservation between rice and Arabidopsis CrRLK1L families must still be determined in future experiments.
Chromosomal mapping of rice members revealed an array of tandem-duplicated genes on chr 5, including OsCrRLK1L2/LOC_Os05g25370, OsCrRLK1L3/LOC_Os05g25450, and OsCrRLK1L12/LOC_Os05g25350. This suggested functional redundancy among them. When their anatomical expression was integrated into our phylogenetic tree, OsCrRLK1L2, -3, and -12 in Subgroup III showed conserved expression in the leaves and flag leaves, possibly because of redundancy in those organs. However, we found it interesting that the diurnal expression data integrated to the phylogenetic tree suggested functional divergence between OsCrRLK1L2 and -3 because expression by the former was increased at sunrise. Furthermore, expression of OsCrRLK1L3 was stimulated when OsCrRLK1L2 expression was repressed. This implied that the two genes function antagonistically by phases in the circadian rhythm. Genes with redundant functions are a potentially important source of evolutionary modifications (Krakauer and Nowak 1999). Several examples exist among CrRLK1Ls in Arabidopsis, such as between THE1 and either HERK1 or HERK2 with regard to cell expansion (Guo et al. 2009a); or between ANX1 and ANX2 during pollen tube growth (Boisson-Dernier et al. 2009; Miyazaki et al. 2009). Thus, future genetic evaluations of functional redundancy or divergence are necessary for rice CrRLK1L family members.
Identification of orthologs is a common method used for genome-wide assignment of conserved functions between different plant species (Conte et al. 2008a). Therefore, orthologs found in Arabidopsis are very useful for predicting functions by rice CrRLK1L family members. Our orthology search showed that six of 16 OsCrRLK1Ls have Arabidopsis orthologs with known functions. Namely, OsCrRLK1L9 and -15 is an ortholog of HERK2; OsCrRLK1L11, ANX1 and ANX2; OsCrRLK1L5 and -8, FER; and OsCrRLK1L5, ERULUS. If expression patterns of orthologous pairs are conserved, we would expect to have higher certainty of their functional orthology. For example, OsCrRLK1L11 with anther- and pollen-preferential expression is a functional ortholog of tissue-specific ANX1 and ANX2, which control pollen tube growth and integrity (Boisson-Dernier et al. 2009; Miyazaki et al. 2009). Likewise, FER regulates pollen tube reception (Rotman et al. 2003) as well as root hair growth and integrity (Duan et al. 2010). The rice orthologs OsCrRLK1L5 and -8 showed high levels of expression in the ovary and root, both of which are locations for FER transcripts in Arabidopsis.
Some OsCrRLK1Ls may have roles in regulating circadian rhythms, as proposed by meta-expression analysis. We also showed that expression of OsCrRLK1L2 was regulated positively by OsGI but negatively by HD1. Previous studies have indicated that OsGI controls HD1 in a manner similar to the way that CO is regulated by GI in Arabidopsis during the daytime (Fowler et al. 1999). In addition, expression of Early Heading Daye 1 (EHD1), which is involved in the short day-promoted flowering pathway in rice, is induced by HD1 under short days (Doi et al. 2004) as well as by blue light in an OsGI-dependent manner (Izawa 2012). The downstream pathway of EHD1 includes HD3/FT-LIKE genes in rice and three Mads Box genes (OsMADS1, OsMADS14, and OsMADS15) (Doi et al. 2004). During the day, OsGI controls OsLHY expression, but OsLHY does not control OsGI (Izawa 2012). Moreover, the oslhy mutant is associated with late flowering (Ogiso et al. 2010). Therefore, our observations for OsCrRLK1L2 suggest it plays a novel role in regulating the flowering time signaling pathway or circadian rhythms. Future genetic analysis using gain-of-function or loss-of-function for OsCrRLK1L2 will clarify our model illustrated in Fig. 6.

Functional network construction associated with OsCrRLK1L2 and simplified model for regulating flowering time and circadian rhythms. Circadian clock includes gene series downstream of OsGI: OsLHY, HD1, EHDd1, OsMADS1, OsMADS14, OsMADS15, and HD3a/FT-LIKE. Brown boxes genes associated with daytime, blue boxes genes associated with night time, brown arrows day signaling pathway, blue arrow night signaling pathway, red arrow short day signaling pathway, T line negative regulation. Dotted arrows indicate pathways less characterized; solid arrows show well-characterized pathways. Question marks represent unconfirmed data
In conclusion, basing phylogenomics and meta-expression profiling analyses on a large set of microarray data are a useful means for obtaining functional annotation of rice CrRLK1L family members. Integrating the meta-expression data within the context of a phylogenetic tree provides more precise estimates of the functional redundancy among closely linked rice CrRLK1Ls. Moreover, the involvement of OsCrRLK1L2 in the downstream pathway of OsGI hints at the existence of a novel pathway for flowering or circadian rhythm-mediated regulation.
Abbreviations
- DAT:
-
Days after treatment
- GO:
-
Gene ontology
- RGAP:
-
Rice Genome Annotation Project
- RLK:
-
Receptor-like kinase
- TAIR:
-
The Arabidopsis Information Resource
References
Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM (2011) NCBI GEO: archive for functional genomics data sets—10 years on. Nucleic Acids Res 39:D1005–D1010
Bartels D, Sunkar R (2005) Drought and salt tolerance in plants. Crit Rev Plant Sci 24:23–58
Boisson-Dernier A, Kessler SA, Grossniklaus U (2011) The walls have ears: the role of plant CrRLK1Ls in sensing and transducing extracellular signals. J Exp Bot 62:1581–1591
Boisson-Dernier A, Lituiev DS, Nestorova A, Franck CM, Thirugnanarajah S, Grossniklaus U (2013) ANXUR receptor-like kinases coordinate cell wall integrity with growth at the pollen tube tip via NADPH oxidases. PLoS Biol 11:e1001719
Boisson-Dernier A, Roy S, Kritsas K, Grobei MA, Jaciubek M, Schroeder JI, Grossniklaus U (2009) Disruption of the pollen-expressed FERONIA homologs ANXUR1 and ANXUR2 triggers pollen tube discharge. Development 136:3279–3288
Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193
Botstein D, Cherry J, Ashburner M, Ball C, Blake J, Butler H, Davis A, Dolinski K, Dwight S, Eppig J (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29
Cao P, Jung K-H, Choi D, Hwang D, Zhu J, Ronald PC (2012) The rice oligonucleotide array database: an atlas of rice gene expression. Rice 5:1–9
Chandran AKN, Jung KH (2014) Resources for systems biology in rice. J Plant Biol 57:80–92
Chen ZJ (2013) Genomic and epigenetic insights into the molecular bases of heterosis. Nat Rev Genet 14:471–482
Cheung AY, Wu HM (2011) THESEUS 1, FERONIA and relatives: a family of cell wall-sensing receptor kinases? Curr Opin Plant Biol 14:632–641
Conte MG, Gaillard S, Droc G, Perin C (2008a) Phylogenomics of plant genomes: a methodology for genome-wide searches for orthologs in plants. BMC Genom 9:183
Conte MG, Gaillard S, Lanau N, Rouard M, Périn C (2008b) GreenPhylDB: a database for plant comparative genomics. Nuc Acids Res 36:D991–D998
Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, Shimatani Z, Yano M, Yoshimura A (2004) Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev 18:926–936
Duan Q, Kita D, Li C, Cheung AY, Wu HM (2010) FERONIA receptor-like kinase regulates RHO GTPase signaling of root hair development. Proc Nat Acad Sci USA 107:17821–17826
Duan Q, Kita D, Johnson EA, Aggarwal M, Gates L, Wu H-M, Cheung AY (2014) Reactive oxygen species mediate pollen tube rupture to release sperm for fertilization in Arabidopsis. Nat Commun 5:3129
Escobar-Restrepo JM, Huck N, Kessler S, Gagliardini V, Gheyselinck J, Yang WC, Grossniklaus U (2007) The FERONIA receptor-like kinase mediates male-female interactions during pollen tube reception. Science 317:656–660
Farooq M, Wahid A, Lee D-J, Ito O, Siddique KH (2009) Advances in drought resistance of rice. Crit Rev Plant Sci 28:199–217
Fowler S, Lee K, Onouchi H, Samach A, Richardson K, Morris B, Coupland G, Putterill J (1999) GIGANTEA: a circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. EMBO J 18:4679–4688
Fukao T, Xiong L (2013) Genetic mechanisms conferring adaptation to submergence and drought in rice: simple or complex? Currt Opin Plant Biol 16:196–204
Goyal K, Walton L, Tunnacliffe A (2005) LEA proteins prevent protein aggregation due to water stress. Biochem J 388:151–157
Guo H, Li L, Ye H, Yu X, Algreen A, Yin Y (2009a) Three related receptor-like kinases are required for optimal cell elongation in Arabidopsis thaliana. Proc Nat Acad Sci USA 106:7648–7653
Guo H, Ye H, Li L, Yin Y (2009b) A family of receptor-like kinases are regulated by BES1 and involved in plant growth in Arabidopsis thaliana. Plant Sign Behav 4:784–786
Haruta M, Sabat G, Stecker K, Minkoff BB, Sussman MR (2014) A peptide hormone and its receptor protein kinase regulate plant cell expansion. Science 343:408–411
Hayama R, Yokoi S, Tamaki S, Yano M, Shimamoto K (2003) Adaptation of photoperiodic control pathways produces short-day flowering in rice. Nature 422:719–722
Hématy K, Sado P-E, van Tuinen A, Rochange S, Desnos T, Balzergue S, Pelletier S, Renou J-P, Höfte H (2007) A receptor-like kinase mediates the response of Arabidopsis cells to the inhibition of cellulose synthesis. Curr Biol 17:922–931
Itoh H, Izawa T (2011) A study of phytohormone biosynthetic gene expression using a circadian clock-related mutant in rice. Plant Sign Behav 6:1932–1936
Izawa T (2012) Physiological significance of the plant circadian clock in natural field conditions. Plant Cell Environ 35:1729–1741
Izawa T, Takahashi Y, Yano M (2003) Comparative biology comes into bloom: genomic and genetic comparison of flowering pathways in rice and Arabidopsis. Curr Opin Plant Biol 6:113–120
Izawa T, Mihara M, Suzuki Y, Gupta M, Itoh H, Nagano AJ, Motoyama R, Sawada Y, Yano M, Hirai MY, Makino A, Nagamura Y (2011) Os-GIGANTEA confers robust diurnal rhythms on the global transcriptome of rice in the field. Plant Cell 23:1741–1755
Jung KH, Gho HJ, Nguyen MX, Kim SR, An G (2013) Genome-wide expression analysis of HSP70 family genes in rice and identification of a cytosolic HSP70 gene highly induced under heat stress. Funct Integr Gen 13:391–402
Kessler SA, Shimosato-Asano H, Keinath NF, Wuest SE, Ingram G, Panstruga R, Grossniklaus U (2010) Conserved molecular components for pollen tube reception and fungal invasion. Science 330:968–971
Kim WY, Ali Z, Park HJ, Park SJ, Cha JY, Perez-Hormaeche J, Quintero FJ, Shin G, Kim MR, Qiang Z, Ning L, Park HC, Lee SY, Bressan RA, Pardo JM, Bohnert HJ, Yun DJ (2013) Release of SOS2 kinase from sequestration with GIGANTEA determines salt tolerance in Arabidopsis. Nat Commun 4:1352
Krakauer DC, Nowak MA (1999) Evolutionary preservation of redundant duplicated genes. Semin Cell Develop Biol 10:555–559
Lindner H, Müller LM, Boisson-Dernier A, Grossniklaus U (2012) CrRLK1L receptor-like kinases: not just another brick in the wall. Curr Opin Plant Biol 6:659–669
McClung CR (2006) Plant circadian rhythms. Plant Cell 18:792–803
Miyazaki S, Murata T, Sakurai-Ozato N, Kubo M, Demura T, Fukuda H, Hasebe M (2009) ANXUR1 and 2, sister genes to FERONIA/SIRENE, are male factors for coordinated fertilization. Curr Biol 19:1327–1331
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Ngo QA, Vogler H, Lituiev DS, Nestorova A, Grossniklaus U (2014) A calcium dialog mediated by the FERONIA signal transduction pathway controls plant sperm delivery. Develop Cell 29:491–500
Nguyen MX, Moon S, Jung KH (2013) Genome-wide expression analysis of rice aquaporin genes and development of a functional gene network mediated by aquaporin expression in roots. Planta 238:669–681
Ogiso E, Takahashi Y, Sasaki T, Yano M, Izawa T (2010) The role of casein kinase II in flowering time regulation has diversified during evolution. Plant Physiol 152:808–820
Oono Y, Yazawa T, Kawahara Y, Kanamori H, Kobayashi F, Sasaki H, Mori S, Wu J, Handa H, Itoh T (2014) Genome-wide transcriptome analysis reveals that cadmium stress signaling controls the expression of genes in drought stress signal pathways in rice. PLoS One 9:e96946
Rotman N, Rozier F, Boavida L, Dumas C, Berger F, Faure J-E (2003) Female control of male gamete delivery during fertilization in Arabidopsis thaliana. Curr Biol 13:432–436
Schulze-Muth P, Irmler S, Schröder G, Schröder J (1996) Novel type of receptor-like protein kinase from a higher plant (Catharanthus roseus). cDNA, gene, intramolecular autophosphorylation, and identification of a threonine important for auto- and substrate phosphorylation. J Biol Chem 271:26684–26689
Shinozaki K, Yamaguchi-Shinozaki K (2007) Gene networks involved in drought stress response and tolerance. J Exp Bot 58:221–227
Shiu S-H, Bleecker AB (2001) Plant receptor-like kinase gene family: diversity, function, and signaling. Sci STKE 2001:re22
Sugiyama N, Izawa T, Oikawa T, Shimamoto K (2001) Light regulation of circadian clock-controlled gene expression in rice. Plant J 26:607–615
Wong ML, Medrano JF (2005) Real-time PCR for mRNA quantitation. Biotechniques 39:75
Xiao B, Huang Y, Tang N, Xiong L (2007) Over-expression of a LEA gene in rice improves drought resistance under the field conditions. Theor App Gene 115:35–46
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12:2473–2483
Yu F, Qian L, Nibau C, Duan Q, Kita D, Levasseur K, Li X, Lu C, Li H, Hou C (2012) FERONIA receptor kinase pathway suppresses abscisic acid signaling in Arabidopsis by activating ABI2 phosphatase. Proc Nat Acad Sci USA 109:14693–14698
Acknowledgments
This work was supported by the Cooperative Research Program for Agriculture Science and Technology Development (Project title: Systematic Identification of Key Genes in Rice for Increasing Yield Using Integrating Omics Technology; Project No. PJ00911002 and Project title: Construction of rice signalom network for regulating hormone biosynthesis and metabolism and the identification of the key regulator for enhancing crop yield, SSAC, Project No.PJ00951405).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
425_2014_2203_MOESM1_ESM.jpg
Supplementary material 1 Fig. S1 CrRLK1L mapping on rice chromosomes. Red box indicates tandem-duplicated genes. Chromosome numbers are shown at top of each bar. (JPEG 862 kb)
425_2014_2203_MOESM2_ESM.jpg
Supplementary material 2 Fig. S2 CrRLK1L rice family phylogenomics with GO and ortholog information in Arabidopsis. Each Locus id and gene name is defined from RGAP. Pale-orange box, Group I; pale-green box,Group II; pale-yellow box, Group III. (JPEG 1373 kb)
425_2014_2203_MOESM3_ESM.jpg
Supplementary material 3 Fig. S3 Circadian expression of 17 Arabidopsis CrRLK1L members. Heat map was generated using Affymetrix Arabidopsis ATH1 genome array data sets (GSE3416) that contain expression information from 3 biological replicates of diurnal time series (4-h intervals for 24 h). Yellow, high expression; blue, low expression. (JPEG 1212 kb)
425_2014_2203_MOESM4_ESM.jpg
Supplementary material 4 Fig. S4 Effect of drought stress on morphology of WT rice. Plants were initially well-watered for 30 d, then exposed to either water-deficit or control (well-watered) conditions for 3 or 4 d. Length of white bar = 5 cm. (JPEG 3229 kb)
425_2014_2203_MOESM5_ESM.jpg
Supplementary material 5 Fig. S5 Primary structures of Arabidopsis and rice CrRLK1L family members. Proteins contain extracellular carbohydrate-binding malectin-like domain(s) (red box) and transmembrane domain (yellow box), plus protein kinase domain (blue box) that shares catalytic functions found in serine/threonine-protein kinases, tyrosine-protein kinases, and dual-specificity protein kinases. (JPEG 1772 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Nguyen, QN., Lee, YS., Cho, LH. et al. Genome-wide identification and analysis of Catharanthus roseus RLK1-like kinases in rice. Planta 241, 603–613 (2015). https://doi.org/10.1007/s00425-014-2203-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-014-2203-2