
Abstract
Main conclusion
miRNAs are involved in the pollen development during the CMS occurrence in rice.
Abstract
miRNAs are 20–24 nt endogenously expressed small RNAs that play key roles in the regulation of many growth and developmental processes in plants. The knowledge on cytoplasmic male sterility (CMS) regulation by miRNAs in rice is rather limited. In this study, Illumina sequencing was employed to examine the expression profiles of rice anther miRNAs from the CMS line MeixiangA (MxA) and its maintainer line MeixiangB (MxB). A total of 518 known miRNAs and 144 novel miRNAs were identified during rice anther development. Based on the number of sequencing reads, a total of 24 miRNAs were discovered to be differentially expressed between MxA and MxB, and the results were partially validated by qRT-PCR. Among these, 16 miRNAs were decreased and 8 miRNAs were increased in MxA compared with MxB. Target prediction showed that they target genes encoding EF-hand family proteins, F-box domain-containing proteins, MYB transcription factors, PPR-containing proteins and transposons. The expression patterns for targets of osa-miR528, osa-miR5793, osa-miR1432, osa-miR159, osa-miR812d, osa-miR2118c, osa-miR172d and osa-miR5498 were selectively examined, and the results showed that there was a negative correlation on the expression patterns between miRNAs and their targets. These targets have previously been reported to be related with pollen development and male sterility, suggesting that miRNAs might act as regulators of CMS occurrence in rice anthers. Furthermore, miRNA editing events were observed. The U → C and U → A editing phenomenon was validated by molecular cloning and sequencing. These findings contribute to our understanding of the roles of miRNAs during anther development and CMS occurrence in rice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
miRNAs are small, non-coding RNAs of 20–24 nucleotides that play various important roles in post-transcriptional gene regulation (Jones-Rhoades et al. 2006). miRNA genes are transcribed by RNA polymerase II, forming the primary miRNA known as the pri-miRNA, which is subsequently capped, spliced and polyadenylated (Kurihara and Watanabe 2004). In plants, a ribonuclease III-like protein in the nucleus, Dicer-like 1 (DCL1), is responsible for processing pri-miRNA transcripts into an miRNA–miRNA* duplex (Papp et al. 2003). The miRNA duplex is then exported into the cytoplasm by HASTY, the plant orthologue of exportin 5 (Park et al. 2005). The active miRNA strand is then incorporated into the RNA-induced silencing complex (RISC) and guides the RISC to target mRNA transcripts, where they silence the expression of specific mRNA targets by directing either mRNA cleavage or translational repression (Fagard et al. 2000; Jones-Rhoades et al. 2006).
miRNAs have been implicated in a wide range of plant growth and development processes. For example, miR319 controls the expression of TCP factors during Arabidopsis leaf morphogenesis (Palatnik et al. 2003); miR159 regulates the transcript levels of two MYB factors during Arabidopsis seed germination (Reyes and Chua 2007); miR164 regulates NAC1 mRNAs that function in Arabidopsis lateral root initiation (Guo et al. 2005); miR172 controls flowering time by downregulating the APETALA2-like target genes in Arabidopsis (Aukerman and Sakai 2003; Chen 2004); and miR397 improves rice yield by promoting panicle branching and increasing grain size (Zhang et al. 2013). In addition, miRNAs have been confirmed to respond to diverse biotic and abiotic stresses (Sunkar et al. 2006; Ding et al. 2009; Campo et al. 2013).
Cytoplasmic male sterility (CMS) is a widely observed phenomenon in the plant kingdom, and it is characterised by the inability to produce functional pollen grains (Hanson and Bentolila 2004). CMS lines are widely used for the production of hybrid seeds because they eliminate the need for tedious hand emasculation (Bentolila et al. 2002). CMS is generally considered to be caused by mitochondrial genomic rearrangement (Hanson and Bentolila 2004). Numerous CMS-related sterile genes and fertility restorer genes have been cloned from various plants (Cui et al. 1996; Liu et al. 2001; Yi et al. 2002; Klein et al. 2005; Hu et al. 2012). As a class of negative regulators, miRNAs have also been found to be involved in plant reproductive development (Chambers and Shuai 2009; Grant-Downton et al. 2009; Wei et al. 2011). It has been reported that overexpression of miR167 leads to male organ fertility defects in Arabidopsis (Ru et al. 2006). Differential expression patterns of miRNAs between the sterile line and its maintainer line have been reported in maize (Shen et al. 2011), cotton (Wei et al. 2013) and Brassica juncea (Yang et al. 2013), and the differentially expressed miRNAs target many transcription factors associated with cell metabolism and signalling pathways, playing important roles in microspore development and fertility regulation (Shen et al. 2011; Wei et al. 2013; Yang et al. 2013).
To our knowledge, there have been no reports comparing miRNA expression profiles during the CMS occurrence of a sterile line and its maintainer line in rice. To achieve this goal, we identified the miRNAs via a high-throughput sequencing approach from rice anthers at the early uninucleate stage of the sterile line MeixiangA (MxA) and its maintainer line MeixiangB (MxB). MxA is a new CMS line derived from the pollen-free CMS line Yunnan ZidaoA by consecutive backcrossing (Hu et al. 2013; Yan et al. 2014). MxA aborts at the early uninucleate stage, and MxB is its maintainer line. Differential expression patterns of miRNAs were analysed and compared between MxA and MxB. Targets were predicted, and their expression profiles were selectively validated. These results may shed light on the regulatory roles of miRNAs during pollen development as well as the occurrence of CMS in rice.
Materials and methods
Rice materials
The sterile line MeixiangA (MxA) and its maintainer line MeixiangB (MxB) were used in this study. They were grown in a rice paddy at Wuhan University (latitude 30°34″N; longitude 114°17″E) under normal conditions. In general, anthers at the early uninucleate stage with a floret length of ~5 mm were manually collected and stored at −80 °C for RNA isolation. The microspore development was judged by both the floret length as described by Feng et al. (2001) and the auricle distance (AD) between the flag leaf and the penultimate leaf (AD = ~0 mm).
RNA isolation, small RNA library construction, sequencing and data analysis
Total RNA was extracted from rice anther at the early uninucleate stage (~5 mm) using the pBIOZOL reagent (Bioer Technology, Hangzhou, China) according to the manufacturer’s instructions. RNA quantity was determined with a Qubit Fluorometer. The RNA purity was assayed by two means: 260/280 nm (ratio >2) was detected with a NanoDrop, and the 28S/18S value was detected with an Agilent 2100.
Small RNAs in the range of 18–30 nt were fractionated from 15 % polyacrylamide gel electrophoresis (PAGE). Pairs of Solexa adaptors were ligated to 5′ and 3′ termini of the small RNAs using T4 ligase. The resulting products were reverse transcribed and amplified using PCR to produce a cDNA sequencing library. Solexa sequencing was performed on the HiSeq 2,000 platform (BGI).
After the Illumina sequencing, the raw sequences were passed through a filtering process to generate high quality reads. After trimming out low quality reads, adaptors, contaminating sequences, and sequences shorter than 18 nt, sequences in the range of 18–30 nt were kept for further analysis. All unique sequences were aligned against the rice genome (MSU 7, ftp://ftp.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/) using SOAP (Li et al. 2008) to analyse their expression in the genome. Sequences recognised as rRNA, tRNA, scRNA, snoRNA, and snRNA by searching through the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) and Rfam RNA family databases (Gardner et al. 2009) were discarded. RepeatMasker software (http://www.repeatmasker.org/) was used to filter out sequences originating from repeat regions. The remaining sequences overlapping with intron and exon sequences of mRNA were considered as the degradation fragments of mRNA and excluded from subsequent analysis.
Sequences that could be perfectly mapped to miRNA precursors by a BLAST search in the miRNA database miRBase release 20 (http://mirbase.org/) (Kozomara and Griffiths-Jones 2011) were designated known miRNAs. The remaining unannotated small RNA tags were used to predict novel miRNAs based on the characteristic hairpin structure of miRNA precursors. MIREAP (http://sourceforge.net/projects/mireap/), a miRNA prediction program, was utilised to discover potential novel miRNAs by exploring the secondary structure, the Dicer cleavage site and the minimum free energy (MFE) of the unannotated small RNA tags which could be mapped to the genome. The following parameters were used: (1) minimal miRNA sequence length, 18 nt; (2) maximal miRNA sequence length, 24 nt; (3) maximal free energy allowed for a miRNA precursor, −18 kcal/mol; (4) maximal space between an miRNA and miRNA*, 300; (5) minimal base pairs of miRNA and miRNA*, 16; (6) maximal bulge of miRNA and miRNA*, 4; (7) maximal asymmetry of miRNA/miRNA* duplexes, 4; (8) flank sequence length of miRNA precursors, 20. In addition, the numbers of mature miRNA with predicted hairpins with normalised number of less than 1 TPM (transcript per million) were discarded to decrease background noise. The miRNA precursors’ stem-loop structures were constructed by the Mfold online service (http://mfold.rna.albany.edu/) (Zuker 2003).
Differential expression analysis of miRNAs
To identify miRNAs with differential expression between MxA and MxB, the expression of miRNAs was first quantified in transcripts per million (TPM). Next, fold change and P value were calculated to determine the significance of expression difference. The formulas are listed below: (1) normalised expression (TPM) = count of miRNA/total count of clean sRNAs × 106. (2) Fold change = log2 (normalised expression of miRNA in MxA/normalised expression of miRNA in MxB). The fold change data were used to draw the scatter plot. (3) P value was calculated using the formula as previously described (Audic and Claverie 1997). miRNAs with \(| fold-change|\) >2 and P value <0.01 were recognised to have extremely significant differential expression and were labelled by **; those with \(|fold-change|\) >2 and 0.01 ≤ P value < 0.05 were defined to have significant differential expression and were labelled by *. If one miRNA has no reads in a library, the normalised read count of this miRNA in the library was arbitrarily set to be 0.01 for further calculation (Chen et al. 2012).
Prediction of miRNA targets
The sequences of all known and novel miRNAs were aligned to the rice genome (MSU 7.0) to predict the potential target genes. The rules used for target prediction in this study were based on those proposed by Allen et al. (2005). (1) No more than four mismatches between sRNA and target (G-U bases count as 0.5 mismatches); (2) no more than two adjacent mismatches in the miRNA/target duplex; (3) no adjacent mismatches in positions 2–12 of the miRNA/target duplex (5′ of miRNA); (4) no mismatches in positions 10–11 of miRNA/target duplex; (5) no more than 2.5 mismatches in positions 1–12 of the miRNA/target duplex (5′ of miRNA); (6) minimum free energy (MFE) of the miRNA/target duplex should be ≥75 % of the MFE of the miRNA bound to its perfect complement.
miRNA detection by stem-loop RT-PCR and qRT-PCR
Total RNA was extracted from anthers at the early uninucleate stage (~5 mm) of both MxA and MxB. DNA contamination was removed by DNaseI (Fermentas) incubation at 37 °C for 30 min. miRNAs were detected by the stem-loop RT-PCR method as described previously (Chen et al. 2005). For each miRNA, the DNaseI-treated RNA samples from MxA and MxB were reverse transcribed using a specific stem-loop RT primer with the RevertAid First Strand cDNA Synthesis Kit (Fermentas). The reactions were incubated for 30 min at 16 °C, followed by pulsed RT of 60 cycles at 30 °C for 30 s, 42 °C for 30 s and 50 °C for 1 s, and a final incubation at 70 °C for 5 min to inactivate the reverse transcriptase (Varkonyi-Gasic et al. 2007). The cDNA template for the miRNA target was reverse transcribed using the OligodT20 primer with the RevertAid First Strand cDNA Synthesis Kit (Fermentas).
To compare the expression patterns of miRNA and their targets between MxA and MxB, qRT-PCR was performed using the FastStart Universal SYBR Green Master (Roche) on the StepOnePlus™ Real-Time PCR Platform (Applied Biosystems). The qRT-PCRs were conducted with the following protocol: 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C. Equally expressed miRNA in both libraries, osa-miR167b, was used as internal standard for miRNAs analysis. Actin was used as endogenous control for miRNA targets analysis. After the amplification steps, the melting curve was determined for each primer pair to verify that only one specific product had been amplified. The reactions were performed with three biological replicates, and the error bars represent standard errors. The comparative 2−∆∆Ct method (Livak and Schmittgen 2001) was used to calculate the fold changes in the miRNAs and their targets between MxA and MxB. The primers used are listed in Table S1.
Results
Overview of small RNA library sequencing
To identify the differentially expressed miRNAs in an early stage of microspore development, small RNA libraries were constructed from the anthers at the early uninucleate stage of the sterile rice line MxA and its maintainer line MxB. In total, 14,576,717 and 14,636,983 raw reads were generated by Illumina sequencing from MxA and MxB, respectively (Table S2). After discarding the low quality reads, 3′ adapter_null, insert_null, 5′ adapter_contaminants, reads shorter than 18 nt and polyA reads, a total of 14,413,675 and 14,490,030 clean reads were obtained for MxA and MxB (Table S2). These reads corresponded to 4,472,864 and 4,793,730 unique small RNA reads in MxA and MxB, respectively (Table 1). The clean small RNA tags were mapped to the rice genome with the SOAP program (Li et al. 2008) to analyse their expression and distribution in the genome. Approximately 67.23 and 66.18 % of the total small RNA sequences corresponding to 58.70 and 58.70 % of the unique small RNAs from MxA and MxB libraries, respectively, were mapped onto the rice genome (MSU 7.0) (Table 1). Various types of non-coding RNAs, including miRNA, rRNA, tRNA, snRNA, and snoRNA, were identified in this study (Table 1). The miRNA category accounted for 4.3 and 3.5 % of the clean reads for MxA and MxB, respectively (Table 1). In all, the annotated total sRNAs accounted for 22.11 and 21.65 %, corresponding to 19.35 and 19.04 % unique sRNAs in MxA and MxB, and a large number of detected sRNAs were not annotated (Table 1).
Regarding the common and specific reads of small RNAs in the two libraries, 74.37 % of the total small RNAs were shared by the two libraries (Fig. 1a), representing a small fraction (17.25 %) of the unique small RNAs (Fig. 1b). These results demonstrated the uniformity and complexity of the small RNAs of the anthers between the sterile line MxA and its maintainer line MxB at the early uninucleate stage.

Summary of common and unique sRNA and miRNA numbers between MxA and MxB. a Total sRNA sequences. b Unique sRNA sequences. c Known miRNA sequences. d First nucleotide bias of the known miRNAs
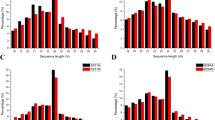
The majority of small RNAs were in length of 21–24 nt for both rice libraries. Small RNAs with a length of 24 nt were the most abundant (~59 % in MxA and ~65 % in MxB), followed by those of 21 nt (~27 % in MxA and ~21 % in MxB) (Fig. S1). The size distribution of small RNAs from the rice MxA and MxB was quite similar to that of Medicago truncatula (Chen et al. 2012), Phaseolus vulgaris (Pelaez et al. 2012), Brassica rapa (Kim et al. 2012) and Arabidopsis thaliana (Rajagopalan et al. 2006), in which the 24 nt class dominates the small RNA transcriptome.
Identification of known miRNAs
To date, there are 592 pre-miRNAs that correspond to 713 mature miRNAs reported for rice in miRBase release 20 (June 2013), and these miRNAs can be classified into more than 100 miRNA families. To identify the known miRNAs in the two rice libraries, unique mature miRNA sequences of rice were downloaded from miRBase, and the known miRNAs were searched through sequence comparison using BLASTN. In the present study, a total of 518 known miRNAs (492 in MxA and 458 in MxB) was identified in the anthers of MxA and MxB at the uninucleate stage (Table S3). Further, most of the known miRNA families deposited in miRBase were found (Table S3). Among these miRNAs, 432 were found in both libraries, accounting for 83.40 % of the total known miRNAs identified, and 60 and 26 were specific to MxA and MxB, respectively (Fig. 1c). These results indicated that a wide range of known miRNAs were involved in the rice microspore development.
In plants, the determination of which strand of the miRNA:miRNA* duplex is incorporated into the RNA-induced silencing complex (RISC) is largely based on the identity of the first nucleotide. In Arabidopsis, there are 10 AGO protein members. AGO1 tends to recruit the miRNAs beginning with a uracil (U), while AGO2 and AGO4 display preferences for miRNAs with a 5′ terminal adenosine (A), and AGO5 would like to enrich for miRNAs initiating with a 5′ cytosine (C) (Mi et al. 2008). In this study, nearly half of the identified known miRNAs showed a bias toward uracil (U) in the first nucleotide, followed by adenosine (A) at approximately 25 % and cytosine/guanine (C/G) at 13 %, respectively (Fig. 1d). It is reported that there are 19 AGO protein members in rice, and some of these share high sequence similarities with the orthologue in Arabidopsis (Wu et al. 2009). This indicated that the identified miRNAs could be recruited by different AGO proteins and enter distinct regulatory pathways.
Generally, the miRNA* strands were thought to be mere by-products of the miRNA biogenesis. In most cases, they were degraded and could not be detected in vivo. However, a few exceptions were found, such as osa-miR169 h, osa-miR1883a, and osa-miR529a, for which the corresponding miRNA* was more abundantly expressed than the annotated miRNAs (Fig. S2a). A similar result was also reported previously during silkworm development (Jagadeeswaran et al. 2010). Recently, Manavella et al. (2013) identified an abundantly expressed miR171a* as an active form, which could silence the expression of SUVH8, a member of the large SET-domain protein family. Additionally, miR171a*-mediated silencing of SUVH8 in specific tissues was important for normal Arabidopsis development (Manavella et al. 2013). Evolutionary analysis indicated that the miRNA* strand might afford potential opportunities for contributing to regulatory networks (Guo and Lu 2010). Thus, we speculated that these osa-miR169h*, osa-miR1883a*, and osa-miR529a* strands might play functional roles during rice anther development.
Interestingly, we also observed that the most abundant small RNA tag processed from the miRNA precursor osa-MIR5514 was not its miRNA or miRNA* but another 21-nt sequence (Fig. S2b). From its predicted secondary structure, we speculated that this might be another miRNA or the random degradation product from the miRNA precursor (Fig. S2b). We also examined whether a single miRNA precursor might produce more than two mature miRNAs. The two mature miRNAs osa-miR444c.1 and osa-miR444c.2 were produced from the same osa-MIR444c precursor; meanwhile, the corresponding star sequences osa-miR444c.1* and osa-miR444c.2* were also examined (Fig. S2c). We also note that the abundance of star strands osa-miR444c.1* was considerably higher than that of the mature sequence osa-miR444c.1 (Fig. S2c). In some cases, the same miRNAs could be produced from different precursors, such as osa-miR156a/b/c/d/e/f/j/h/i/j (Fig. S2d). These results demonstrated the complexity and diversity of the miRNA families expressed during rice anther development.
Identification of novel miRNAs
The high-throughput sequencing generated large amounts of information about small RNAs, and the conserved miRNAs could be found by alignment with the known miRNAs deposited in miRBase. However, many novel miRNAs remained to be discovered among the unannotated small RNA tags. Because miRNAs are derived from hairpin-like precursors originating from a single-stranded RNA transcript after sequential processing by Dicer, miRNA precursors should have a characteristic fold-back structure, and this feature can be used to predict novel miRNAs. Considering the potential for sequence errors and to better identify true miRNA candidates, sequence reads with less than 1 TPM were discarded. Existence of a miRNA* sequence is another important rule in miRNA prediction. However, because most miRNA*s are degraded soon after their separation from the corresponding miRNAs, the detected miRNA*s are usually rather low in abundance or even undetectable; in such cases, it is suggested that miRNAs should be detected from multiple independent libraries (Meyers et al. 2008). Thus, the predicted novel miRNAs should be sequenced in both MxA and MxB libraries in this study.
A total of 144 novel miRNAs were available (Table S4). Of these miRNAs, 92 miRNAs were located on the 3′ arm of the miRNA precursor, and 52 miRNAs were located on the 5′ arm; 7 miRNA* sequences were obtained in this study (Table S4). Size distribution analysis indicated that most of the miRNAs displayed a nucleotide length of 21 nt (Fig. S3a). The first nucleotide analysis of these novel miRNAs showed that the bases of C, U, A and G were included at rates of 31.94, 29.17, 21.53 and 17.36 %, respectively (Fig. S3b). The folding structures for the novel miRNA precursors were predicted using the Mfold online service (http://mfold.rna.albany.edu/). Five structures of the novel miRNAs were exemplified and displayed (Fig. 2a). The novel miRNAs were named sequentially, according to their chromosome locations. They are temporarily in the form of osa-mir-number.

Novel miRNAs predicted and validated in this study. a Stem-loop structures of the novel miRNA precursors constructed by the Mfold online service. Identified mature miRNA and miRNA* sequences are shown in red and light blue, respectively. b Expression validation of the novel miRNAs in MxA and MxB by stem-loop RT-PCR amplification. Primers are listed in Table S1-1. The expected size of the PCR product is 60 bp and was observed on the gel
The novel miRNAs had differential expression profiles. Some of them were expressed at relatively low levels, such as osa-mir-4, osa-mir-5, osa-mir-7, osa-mir-12, and osa-mir-13 (Table S4). In contrast, some of the novel miRNAs were abundantly expressed, with more than 1,000 reads, including osa-mir-44, osa-mir-46, osa-mir-47, and osa-mir-50 (Table S4). To validate the expression of these novel miRNAs, stem-loop RT-PCR was performed for five novel miRNAs (osa-mir-46, osa-mir-76, osa-mir-83, osa-mir-101, and osa-mir-116) with relatively high abundance in both rice lines. DNA fragments of approximately 60 bp were amplified, revealing that these five novel miRNAs were all expressed in both MxA and MxB anthers (Fig. 2b). The results indicated that the novel miRNAs might participate in the development of rice anthers.
Observation of miRNA editing events
RNA editing is a post-transcriptional modification of RNA nucleotides from their genome-encoded sequences. miRNA transcripts have been recognised as a major target for RNA editing enzymes, and single-nucleotide changes through editing can impact the biogenesis of mature miRNAs, as well as the target specificity of the specific miRNAs. It has been revealed that miRNA editing events occur during the rice grain-filling stage (Yi et al. 2013). In the present study, a large number of miRNA editing events were found in both rice libraries (Table S5). A previous study showed that 5′ terminal nucleotides were decisive for directing miRNA sorting to Argonaute complexes (Mi et al. 2008). In this study, we observed that the miRNA editing events mainly occurred at nucleotide positions of 11, 4 and 6, with no SNPs at the 5′ terminal nucleotides (Fig. 3a), indicating the conservation of the 5′ terminal nucleotides of miRNAs. We also observed that the miRNA editing patterns were similar between the two rice lines (Fig. 3b). The most dominant nucleotide substitution type was U to C, which accounted for more than 50 % of all of the editing events (Fig. 3b). This was mainly attributed to the considerably higher mutation rate of U to C on the 6th nucleotide of osa-miR166i-3p (47,825 in MxA, 39,935 in MxB) (Table S5). Meanwhile, due to the higher substitution rate of U–A on the 16th nucleotide of osa-miR166 m (22,980 in MxA, 20,969 in MxB), the U to A type was the second most observed editing event, accounting for ~21 % in both rice lines. A previous study reported that the most dominant miRNA editing type was A–U during the rice grain-filling stages (>60 %) (Yi et al. 2013) and that the most common nucleotide substitutions were G–A (24.2 %), A–G (16.4 %), U–C (12.6 %) and C–U (9.7 %) in Arabidopsis (Meng et al. 2010). In the present study, the well-known C to U editing mode was also observed, accounting for 4.2 and 5.1 % in MxA and MxB, respectively (Fig. 3b). However, the cause for the variant editing type remained elusive.

Analysis of miRNA editing events. a miRNA editing frequency at each miRNA nucleotide position in both MxA and MxB. b miRNA editing types and their frequency in both MxA and MxB. c Validation of miRNA editing types inferred from high-throughput sequencing by precursor miRNA and mature miRNA cloning and sequencing. Sequencing chromatograms from the two most commonly observed editing types (U → C, U → A) of osa-miR164c and osa-miR166m are shown. The edited positions are highlighted in pink. The upper panel indicates the part of precursor miRNA cloned from genomic DNA, and the lower panel indicates the mature miRNA cloned from cDNA reverse transcribed with stem-loop RT primers. Primers are listed in Table S1-2
To validate the occurrence of the miRNA editing, the two most frequently observed editing types (U → C, U → A) were examined. The precursor miRNA sequence was cloned from the rice genome, and the mature miRNA sequence was cloned from the cDNA reverse transcribed by the stem-loop reverse primers. Our sequencing results confirmed that nucleotide substitutions had truly occurred during the mature miRNA processing (Fig. 3c).
Differential miRNA expression profiles of the sterile line MxA and its maintainer line MxB during anther development
miRNAs with high sequencing frequencies have been shown to play fundamental and essential regulatory roles in maintaining basic biological processes. The read counts for the known miRNA families were checked in this study (Table S6). We found that the sequencing frequencies varied greatly between distinct miRNA families. Some of the miRNA family members, such as osa-miR156, osa-miR164, osa-miR166, osa-miR167, osa-miR168, osa-miR528, osa-miR535, osa-miR5497, osa-miR5791, and osa-miR5792 (Table S6), were abundantly expressed in both rice lines, suggesting their conserved and essential roles for rice anther development. In contrast, other miRNA families, such as osa-miR1442, osa-miR1846, osa-miR1883, osa-miR2093 and osa-miR2120, were expressed at lower levels in both rice lines compared with the above conserved miRNAs, suggesting the functional divergence among these miRNA families. We also detected some miRNAs specifically expressed in one line: for instance, osa-miR5793 was highly expressed in the sterile line MxA (487 reads), but absent from its maintainer line MxB (Table S6).
To decrease the background noise, the miRNAs expressed at low levels (<1 TPM, equal to count reads less than 15 in both libraries) were omitted for further expression analysis in this study. Accordingly, a total of 284 miRNAs were used for differential expression analysis between MxA and MxB (Table S6), and the normalised expression values of these miRNAs from MxA and MxB were used to draw the scatter plot (Fig. 4a). Based on the rules for differentially expressed miRNAs with \(| fold-change|\) >2, most of the miRNAs were equally expressed; however, 24 miRNAs were found to be differentially expressed between the sterile line MxA and its maintainer line MxB (Fig. 4a). Among these differentially expressed miRNAs, 16 miRNAs belonging to 14 miRNA families were downregulated in MxA, whereas 8 miRNAs belonging to 8 miRNA families were upregulated in MxA (Table 2).

Comparison and validation of the differential expression of known miRNAs between the sterile line MxA and its maintainer line MxB. a Scatter plot of expression ratios for known miRNAs in MxA and MxB; data are normalised and log2 transformed. Red points represent miRNAs with fold change >2, indicating upregulated miRNAs in MxA; blue points represent miRNAs with 1/2 ≤ fold change ≤ 2, indicating equally expressed miRNAs in both rice lines; green points represent miRNAs with fold change <1/2, indicating downregulated miRNAs in MxA. Fold change = normalised expression in MxA/normalised expression in MxB. b Comparison of the miRNA expression levels between MxA and MxB by qPCR for osa-miR2118c, osa-miR812d, osa-miR1432, osa-miR528, osa-miR5793. The expression level in MxB was set as 1.0. Primers are listed in Table S1-3. The experiments were performed with three biological replicates, and the error bars represent the standard error
To validate the differential expression data obtained from the high-throughput sequencing (Table 2), we performed a qRT-PCR assay, which allowed us to detect miRNAs at low abundance and acquire more precise data on the relative expression of miRNAs. The expression patterns of five selected miRNAs (osa-miR2118c, osa-miR812d, osa-miR1432, osa-miR528 and osa-miR5793) were examined in both rice lines (Fig. 4b–f). According to the results of the qRT-PCR assay, the expression levels of osa-miR2118c and osa-miR812d were decreased in MxA (Fig. 4b, c), whereas osa-miR1432, osa-miR528 and osa-miR5793 were upregulated in MxA compared with MxB (Fig. 4d–f). The qRT-PCR results were consistent with the high-throughput sequencing, implying that the high-throughput sequencing results were reliable in this study.
miRNA target prediction and functional analysis
Identifying the candidate genes targeted by the miRNAs is crucial to understanding these miRNAs’ biological functions. The transcribed regions of the rice genome (MSU 7) were searched to find sequences complementary to the identified miRNAs by the psRNATarget online service in this study (Dai and Zhao 2011). Among the identified miRNAs, many belong to the same miRNA families and share the same miRNA sequences, such as osa-miR1428b/c/d and osa-miR156b/c/d/e/f/g/h/i/j/k. After removing the redundancies, 328 unique miRNA sequences were obtained, and these unique sequences were used for target prediction. We found 1,802 targets for 328 unique known miRNAs (Table S7) and 843 targets for 144 novel miRNAs (Table S8). In summary, a total of 2,645 targets were predicted for the 472 identified unique miRNAs, with an average of ~5.6 targets per miRNA.
Among the predicted targets, a large proportion of the targets were transposon proteins, retrotransposon proteins, growth-regulating factors, MYB family transcription factors, F-box domain-containing proteins, MADS-box family proteins, and SBP-box gene family members (Tables S7, S8). These proteins are known to play various roles during the plant growth and development processes. To illuminate their functional roles, the predicted targets were subjected to Gene Ontology analysis on the terms of “biological process” and “molecular function” using WEGO (Ye et al. 2006). The results for “biological process” were classified into 13 reference terms, with the most frequent term being “metabolic process”, followed by “cellular process”, “response to stimulus”, “developmental process” and “multicellular organismal process” (Fig. 5). The most frequent “molecular function” terms were “binding”, “catalytic”, “transcription regulator” and “transporter” (Fig. 5).

Gene ontology analysis for the targets of the identified known and novel miRNAs based on the terms of biological process and molecular function
Expression profiles of miRNA targets examined by qRT-PCR
To examine the correlation between the targets and the corresponding miRNAs, the expression levels of 10 selected targets were examined by qRT-PCR analysis. The high-throughput sequencing results indicate that osa-miR1432, osa-miR159 and osa-miR528 were upregulated in MxA (Table 2). osa-miR1432 targets a metal cation transporter (LOC_Os05g07210.1), osa-miR159 targets MYB family transcription factors (LOC_Os01g59660.1, LOC_Os06g40330.1), and osa-miR528 targets F-box-domain- and LRR-containing proteins (LOC_Os06g06050.1) (Table S9). qRT-PCR analysis showed that the expression profiles of the above miRNA targets were downregulated in MxA (Fig. 6). osa-miR5793 was detected to be highly expressed in MxA than in MxB by high-throughput sequencing (Table 2) and qPCR (Fig. 4b) analysis. osa-miR5793 was predicted to target two genes encoding receptor-like protein kinase (LOC_Os02g11930.1) and dehydrin (LOC_Os11g26780.1) (Table S9). The expression of dehydrin was undetectable in both rice lines, and the receptor-like protein kinase was sharply reduced in MxA compared with MxB, based on the qPCR analysis (Fig. 6). In contrast, osa-miR172d, osa-miR2118c/q, osa-miR5498 and osa-miR812d were downregulated in MxA (Table 2). osa-miR172d targets phytochrome B (LOC_Os03g19590.1), osa-miR2118c/q targets an NBS-LRR disease-resistance protein (LOC_Os01g05600.1), osa-miR5498 targets retrotransposon proteins (LOC_Os10g09960.1), and osa-miR812d targets proteins containing helix-loop-helix DNA-binding domains (LOC_Os02g23823.2) and CAMK (a family of calcium/calmodulin-dependent protein kinase) (LOC_Os03g22050.4) (Table S9). The expression levels of these targets were all upregulated in MxA, except for CAMK (Fig. 6). Some negative correlations were found between the expression levels of the target genes and their corresponding miRNAs in the anthers of the sterile line MxA and its maintainer line MxB, implying that miRNA-mediated mRNA silencing occurred during anther development in both the sterile line MxA and its maintainer line MxB.

Quantitative real-time PCR analysis of the relative expression of miRNA targets in the sterile line MxA and its maintainer line MxB. F-box (LOC_Os06g06050.1), MCT (LOC_Os05g07210.1), RLPK (LOC_Os02g11930.1), CAMK (LOC_Os03g22050.1), HLH (LOC_Os02g23823.2), NBS (LOC_Os01g05600.1), MYB1 (LOC_Os01g59660.1), MYB2 (LOC_Os06g40330.1), PhytoB (LOC_Os03g19590.1), Retro (LOC_Os10g09960.1). Actin was used as endogenous control. Primers for these genes are listed in Table S1-4. Corresponding miRNA for each target is included in brackets. The expression level in MxB is set as 1.0. The experiments were performed with three biological replicates, and the error bars represent the standard errors
Discussion
Studies have shown that miRNAs regulate anther development in plants. However, knowledge of the relationship between the miRNA biogenesis and the CMS occurrence is very limited. Recently, the rapid advances in high-throughput sequencing have provided an effective way to identify and estimate the expression profiles of miRNAs in plant tissues at different developmental stages or from different tissues. To explore the roles of miRNAs during the occurrence of CMS, anthers at the early uninucleate stage were used to examine the expression profiles of the miRNAs in the sterile line MxA and its maintainer line MxB using high-throughput sequencing. To the best of our knowledge, this is the first report of a comprehensive comparison of the miRNA expression profiles between a sterile line and its maintainer line in rice.
In the present study, a total of 24 conserved miRNAs were identified as differentially expressed between the sterile line MxA and its maintainer line MxB (Table 2). Among them, 8 are upregulated and 16 are downregulated in MxA compared with MxB (Table 2). Targets of these differentially expressed miRNAs include MYB family proteins, PPR-containing proteins, kinases, laccase proteins, and disease-resistance proteins (Table S9). This work demonstrates that miRNAs are involved in many biological processes, including carbohydrate metabolic pathways, signal transduction, reproductive processes, death and response to stimulus.
Kinases are major regulatory components that control various pathways. This naturally leads to the presumption of their involvement in pollen development. In this study, we identified a miRNA specifically expressed in the sterile line MxA, osa-miR5793 (Table 2), which is predicted to target two transcripts encoding receptor-like protein kinase (LOC_Os02g11930.1) and dehydrin (LOC_Os11g26780.1) (Table S9). It has been reported that the receptor-like kinase genes were highly expressed and upregulated in the fertile buds in Brassica rapa (Dong et al. 2013). Moreover, mutation of the receptor-like protein kinase 2 could lead to male sterility in Arabidopsis (Mizuno et al. 2007). These results demonstrate that the receptor-like kinase is a key regulator during anther development. In the present study, qPCR analysis showed that the expression of receptor-like protein kinase (LOC_Os02g11930.1) was downregulated in MxA (Fig. 6), implying that osa-miR5793 might be involved in the occurrence of CMS via silencing the receptor-like kinase and further affecting the tapetum function in the sterile line MxA.
miR156 targets SBP-box genes, including the SPL family, a group of transcription factor genes with important regulatory functions throughout the growth and development stages in plants. It is reported that miR156 regulates the timing of flower formation via SPL3, activating the expression of LEAFY, FRUITFULL, and APETALA1 (Yamaguchi et al. 2009). Another study reported that overexpression of miR156 in an spl8 mutant background resulted in sterility in Arabidopsis (Xing et al. 2010). In this study, the entire miR156 miRNA family members (osa-miR156a-k) were identified as upregulated in MxA compared with MxB (Table S6), indicating that miR156 may participate in fertility regulation in the sterile line MxA.
miR159 is a highly conserved miRNA family, essential for plant development and fertility (Jones-Rhoades et al. 2006). Previous studies have indicated that miR159 is widely expressed in various organs, predominantly in floral organs, in plants (Achard et al. 2004; Axtell and Bartel 2005; Tsuji et al. 2006). They target a class of MYB transcription factors, which play important roles in flower development. Loss-of-function mutations in OsGAMYB resulted in defects in floral organ development, especially the anthers and pollen (Kaneko et al. 2004). Consistent with this result, overexpression of miR159 leads to male sterility in Arabidopsis (Achard et al. 2004) and shrunken anthers in rice (Tsuji et al. 2006). In this study, three members of osa-miR159 (osa-miR159a.1, osa-miR159b and osa-miR159f) were found to be upregulated in MxA (Table S6). Bioinformatics analysis indicates that osa-miR159 targets four MYB family proteins (Table S9). The expression levels of two MYB proteins (LOC_Os01g59660.1, LOC_Os06g40330.1) were examined by qPCR. These targets were downregulated in the sterile line MxA (Fig. 6), showing that the increased abundance of osa-miR159 partially silenced the expression of the MYB proteins, hampering normal anther development in the sterile line MxA.
The predicted targets of osa-miR397b include laccases, F-box domain-containing proteins and PPR-containing proteins (Table S9). Recently, it has been clearly reported that overexpression of osa-miR397 increases rice grain yield by downregulating the expression of laccase (Zhang et al. 2013). Another previous study has demonstrated that the laccase genes are expressed during pollen development in loblolly pine, indicating that laccases are involved in plant pollen development (Sato et al. 2001). It has been suggested that the high expression level of osa-miR397 could downregulate peroxidase and laccase, leading to the failure to remove oxyradicals, thus contributing to pollen abortion in the CMS line (Shen et al. 2011). F-box proteins are involved in the regulation of various developmental processes in plants, including photomorphogenesis, circadian clock regulation, self-incompatibility, and floral meristem and floral organ identity determination (Jain et al. 2007). In the present study, osa-miR528 was identified as increased in MxA compared with MxB (Table 2; Fig. 4). A negative correlation was discovered between the levels of osa-miR528 and its target F-box protein (LOC_Os06g06050.1) (Fig. 6), indicating that osa-miR528 might be involved in the regulation of the abortion process in MxA. ARF (Auxin Response Factor) proteins can bind to auxin response promoter elements and mediate gene expression responses to the plant hormone auxin. It has previously been reported that ARF6 and ARF8 regulate flower maturation (Nagpal et al. 2005). Further, it was shown that ARF6 and ARF8 are targeted by miR167. Loss of miR167 regulation in mARF6 and mARF8 expression caused arrested ovule development and anther indehiscence in Arabidopsis (Wu et al. 2006). There are 10 members of the miR167 family (osa-miR167a–osa-miR167j) deposited in miRBase (http://www.mirbase.org/). In this study, 4 osa-miR167 star sequences (−3p) were found at low expression levels and slightly downregulated in MxA (Table S6). However, all 10 of the osa-miR167 family members (−5p) were highly expressed in both rice lines, and their expression levels were higher in the sterile line MxA (Table S6). Thus, we speculate that the higher expression of osa-miR167 may partially silence the expression of ARF6 and ARF8, finally resulting in abnormal pollen development in the sterile line MxA.
Ca2+ is a crucial regulator of growth and development in plants. osa-miR1432 is predicted to target genes of the EF-hand family (LOC_Os03g59770.1, LOC_Os03g59790.1) and metal cation transporter (LOC_Os05g07210.1). The EF-hand domain is a conserved helix–loop–helix structure that can bind a single Ca2+ molecule, and the EF-hand proteins are thought to function as Ca2+ sensors whose conformational changes upon Ca2+ binding play critical roles in Ca2+-mediated signalling (DeFalco et al. 2010). It has been reported that metal cation transporters are essential for mediating metal transfer between cells and organs and for maintaining intracellular metal homeostasis (Thomine et al. 2000). In this study, osa-miR1432 was found to be upregulated in MxA (Table 2). qPCR analysis indicates that the expression of a metal cation transporter (LOC_Os05g07210.1) on the mRNA level is sharply reduced in MxA (Fig. 6), but the expression of the EF-hand protein mRNAs is not detected in anthers of either rice line. Further, another miRNA, miR812d, which is predicted to target a calcium/calmodulin-dependent protein kinase (LOC_Os03g22050.4), was found to be downregulated in MxA (Table 2). The mRNA expression level of calcium/calmodulin-dependent protein kinase (LOC_Os03g22050.4) was also found to be downregulated in MxA (Fig. 6). These results implied that abnormal expression of osa-miR1432 and osa-miR812 might disrupt the balance of Ca2+ in the anther cells and impair the Ca2+-mediated signalling pathway during the rice anther development in MxA. However, the details of this relationship require further study.
It is interesting to note that a majority of transposons and retrotransposons found in this study are predicted to be targets of osa-miR530-3p and osa-miR5498. However, their functional roles during the rice reproductive development have not been reported, and this requires further study.
Conclusion
In the present study, we employed high-throughput sequencing to identify the miRNA expression profiles of the sterile line MxA and its maintainer line MxB. A total of 518 known and 144 novel miRNAs were identified, and some of these miRNAs were validated by stem-loop RT-PCR analysis. miRNA editing events were also observed and validated during the rice anther development. Target prediction analysis indicates that some of the miRNAs are involved in anther development and male sterility. The characterisation and comparative expression profiling of the miRNA transcriptome lay a solid foundation for unravelling the complex miRNA-mediated regulatory networks in the development of rice anthers. Further functional studies on the differentially expressed miRNAs will provide a better understanding of the miRNA-mediated regulation mechanisms during the CMS occurrence in the ZD-CMS rice line.
Author contribution
JY and YD conceived and designed research. JY and HZ conducted experiments. YZ contributed new reagents or analytical tools. JY and YD analyzed data and wrote the manuscript. All authors read and approved the manuscript.
Abbreviations
- CMS:
-
Cytoplasmic male sterility
- TPM:
-
Transcript per million
- DCL1:
-
Dicer-like 1
References
Achard P, Herr A, Baulcombe DC, Harberd NP (2004) Modulation of floral development by a gibberellin-regulated microRNA. Development 131:3357–3365
Allen E, Xie Z, Gustafson AM, Carrington JC (2005) microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121:207–221
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7:986–995
Aukerman MJ, Sakai H (2003) Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15:2730–2741
Axtell MJ, Bartel DP (2005) Antiquity of microRNAs and their targets in land plants. Plant Cell 17:1658–1673
Bentolila S, Alfonso AA, Hanson MR (2002) A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proc Natl Acad Sci USA 99:10887–10892
Campo S, Peris-Peris C, Sire C, Moreno AB, Donaire L, Zytnicki M, Notredame C, Llave C, San Segundo B (2013) Identification of a novel microRNA (miRNA) from rice that targets an alternatively spliced transcript of the Nramp6 (Natural resistance-associated macrophage protein 6) gene involved in pathogen resistance. New Phytol 199:212–227
Chambers C, Shuai B (2009) Profiling microRNA expression in Arabidopsis pollen using microRNA array and real-time PCR. BMC Plant Biol 9:87. doi:10.1186/1471-2229-9-87
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179
Chen L, Wang T, Zhao M, Tian Q, Zhang WH (2012) Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 235:375–386
Cui X, Wise RP, Schnable PS (1996) The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 272:1334–1336
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
DeFalco T, Bender K, Snedden W (2010) Breaking the code: Ca2+ sensors in plant signalling. Biochem J 425:27–40
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y (2009) Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot 103:29–38
Dong X, Feng H, Xu M, Lee J, Kim YK, Lim YP, Piao Z, Park YD, Ma H, Hur Y (2013) Comprehensive analysis of genic male sterility-related genes in Brassica rapa using a newly developed Br 300 K oligomeric chip. PLoS ONE 8:e72178
Fagard M, Boutet S, Morel JB, Bellini C, Vaucheret H (2000) AGO1, QDE-2, and RDE-1 are related proteins required for post-transcriptional gene silencing in plants, quelling in fungi, and RNA interference in animals. Proc Natl Acad Sci USA 97:11650–11654
Feng J, Lu Y, Liu X, Xu X (2001) Pollen development and its stages in rice (Oryza sativa L.). Chin J Rice Sci 15:21–28
Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A (2009) Rfam: updates to the RNA families database. Nucleic Acids Res 37:D136–D140
Grant-Downton R, Le Trionnaire G, Schmid R, Rodriguez-Enriquez J, Hafidh S, Mehdi S, Twell D, Dickinson H (2009) MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. BMC Genomics 10:643
Guo L, Lu Z (2010) The fate of miRNA* strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? PLoS ONE 5:e11387
Guo HS, Xie Q, Fei JF, Chua NH (2005) MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 17:1376–1386
Hanson MR, Bentolila S (2004) Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 16 Suppl:S154–S169
Hu J, Wang K, Huang W, Liu G, Gao Y, Wang J, Huang Q, Ji Y, Qin X, Wan L, Zhu R, Li S, Yang D, Zhu Y (2012) The rice pentatricopeptide repeat protein RF5 restores fertility in hong-lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 24:109–122
Hu J, Yi R, Zhang H, Ding Y (2013) Nucleo-cytoplasmic interactions affect RNA editing of cox2, atp6 and atp9 in alloplasmic male-sterile rice (Oryza sativa L.) lines. Mitochondrion 13:87–95
Jagadeeswaran G, Zheng Y, Sumathipala N, Jiang H, Arrese EL, Soulages JL, Zhang W, Sunkar R (2010) Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics 11:52
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, Kapoor S, Tyagi AK, Khurana JP (2007) F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143:1467–1483
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kaneko M, Inukai Y, Ueguchi-Tanaka M, Itoh H, Izawa T, Kobayashi Y, Hattori T, Miyao A, Hirochika H, Ashikari M, Matsuoka M (2004) Loss-of-function mutations of the rice GAMYB gene impair alpha-amylase expression in aleurone and flower development. Plant Cell 16:33–44
Kim B, Yu HJ, Park SG, Shin JY, Oh M, Kim N, Mun JH (2012) Identification and profiling of novel microRNAs in the Brassica rapa genome based on small RNA deep sequencing. BMC Plant Biol 12:218
Klein RR, Klein PE, Mullet JE, Minx P, Rooney WL, Schertz KF (2005) Fertility restorer locus Rf1 [corrected] of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the colinear region of rice chromosome 12. Theor Appl Genet 111:994–1012
Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39:D152–D157
Kurihara Y, Watanabe Y (2004) Arabidopsis microRNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA 101:12753–12758
Li R, Li Y, Kristiansen K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24:713–714
Liu F, Cui X, Horner HT, Weiner H, Schnable PS (2001) Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 13:1063–1078
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408
Manavella PA, Koenig D, Rubio-Somoza I, Burbano HA, Becker C, Weigel D (2013) Tissue-specific silencing of Arabidopsis SU(VAR)3-9 HOMOLOG8 by miR171a. Plant Physiol 161:805–812
Meng Y, Chen D, Jin Y, Mao C, Wu P, Chen M (2010) RNA editing of nuclear transcripts in Arabidopsis thaliana. BMC Genomics 11(Suppl 4):S12
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant MicroRNAs. Plant Cell 20:3186–3190
Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, Chen S, Hannon GJ, Qi Y (2008) Sorting of small RNAs into Arabidopsis Argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133:116–127
Mizuno S, Osakabe Y, Maruyama K, Ito T, Osakabe K, Sato T, Shinozaki K, Yamaguchi-Shinozaki K (2007) Receptor-like protein kinase 2 (RPK 2) is a novel factor controlling anther development in Arabidopsis thaliana. Plant J 50:751–766
Nagpal P, Ellis CM, Weber H, Ploense SE, Barkawi LS, Guilfoyle TJ, Hagen G, Alonso JM, Cohen JD, Farmer EE, Ecker JR, Reed JW (2005) Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 132:4107–4118
Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D (2003) Control of leaf morphogenesis by microRNAs. Nature 425:257–263
Papp I, Mette MF, Aufsatz W, Daxinger L, Schauer SE, Ray A, van der Winden J, Matzke M, Matzke AJ (2003) Evidence for nuclear processing of plant microRNA and short interfering RNA precursors. Plant Physiol 132:1382–1390
Park MY, Wu G, Gonzalez-Sulser A, Vaucheret H, Poethig RS (2005) Nuclear processing and export of microRNAs in Arabidopsis. Proc Natl Acad Sci USA 102:3691–3696
Pelaez P, Trejo MS, Iniguez LP, Estrada-Navarrete G, Covarrubias AA, Reyes JL, Sanchez F (2012) Identification and characterization of microRNAs in Phaseolus vulgaris by high-throughput sequencing. BMC Genomics 13:83
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Reyes JL, Chua NH (2007) ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J 49:592–606
Ru P, Xu L, Ma H, Huang H (2006) Plant fertility defects induced by the enhanced expression of microRNA167. Cell Res 16:457–465
Sato Y, Wuli B, Sederoff R, Whetten R (2001) Molecular cloning and expression of eight laccase cDNAs in loblolly pine (Pinus taeda). J Plant Res 114:147–155
Shen Y, Zhang Z, Lin H, Liu H, Chen J, Peng H, Cao M, Rong T, Pan G (2011) Cytoplasmic male sterility-regulated novel microRNAs from maize. Funct Integr Genomics 11:179–191
Sunkar R, Kapoor A, Zhu JK (2006) Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 18:2051–2065
Thomine S, Wang R, Ward JM, Crawford NM, Schroeder JI (2000) Cadmium and iron transport by members of a plant metal transporter family in Arabidopsis with homology to Nramp genes. Proc Natl Acad Sci USA 97:4991–4996
Tsuji H, Aya K, Ueguchi-Tanaka M, Shimada Y, Nakazono M, Watanabe R, Nishizawa NK, Gomi K, Shimada A, Kitano H, Ashikari M, Matsuoka M (2006) GAMYB controls different sets of genes and is differentially regulated by microRNA in aleurone cells and anthers. Plant J 47:427–444
Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:12
Wei LQ, Yan LF, Wang T (2011) Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol 12:R53
Wei M, Wei H, Wu M, Song M, Zhang J, Yu J, Fan S, Yu S (2013) Comparative expression profiling of miRNA during anther development in genetic male sterile and wild type cotton. BMC Plant Biol 13:66
Wu MF, Tian Q, Reed JW (2006) Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 133:4211–4218
Wu L, Zhang Q, Zhou H, Ni F, Wu X, Qi Y (2009) Rice microRNA effector complexes and targets. Plant Cell 21:3421–3435
Xing S, Salinas M, Hohmann S, Berndtgen R, Huijser P (2010) miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 22:3935–3950
Yamaguchi A, Wu MF, Yang L, Wu G, Poethig RS, Wagner D (2009) The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev Cell 17:268–278
Yan J, Tian H, Wang S, Shao J, Zheng Y, Zhang H, Guo L, Ding Y (2014) Pollen developmental defects in ZD-CMS rice line explored by cytological, molecular and proteomic approaches. J Proteomics 108C:110–123
Yang J, Liu X, Xu B, Zhao N, Yang X, Zhang M (2013) Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genomics 14:9
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297
Yi P, Wang L, Sun Q, Zhu Y (2002) Discovery of mitochondrial chimeric-gene associated with cytoplasmic male sterility of HL-rice. Chin Sci Bull 47:744–747
Yi R, Zhu Z, Hu J, Qian Q, Dai J, Ding Y (2013) Identification and expression analysis of microRNAs at the grain filling stage in rice (Oryza sativa L.) via deep sequencing. PLoS ONE 8:e57863
Zhang YC, Yu Y, Wang CY, Li ZY, Liu Q, Xu J, Liao JY, Wang XJ, Qu LH, Chen F, Xin P, Yan C, Chu J, Li HQ, Chen YQ (2013) Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat Biotechnol 31:848–852
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
This study is supported by “973” Program of China (2013CB126900) and National Nature Science Foundation of China (NSFC 30971740).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Size distribution of sRNAs in the libraries of the sterile line MxA and its maintainer line MxB in this study. Supplementary material 1 (TIFF 1931 kb)
Fig. S2
Distributions of the small RNA tags processed from the miRNA precursors. Supplementary material 2 (PDF 155 kb)
Fig. 3
Characteristics of the novel miRNAs. (a) Distribution of the novel small RNA length. (b) Distribution of the first nucleotidesBelow is the link to the electronic supplementary material. Supplementary material 3 (TIFF 428 kb)
Supplemental materials
Table S1 Primers used in this study
Table S2 The filtering for small RNA clean reads in the libraries of MxA and MxB
Table S3 Summary of the known miRNAs identified in the libraries MxA and MxB
Table S4 Summary of the novel miRNAs predicted in the libraries of MxA and MxB
Table S5 Summary of miRNA editing type, position, snp count and editing rate in the sterile line MxA and its maintainer line MxB
Table S6 Comparison of the expression levels for the known miRNAs between the sterile line MxA and its maintainer line MxB
Table S7 Targets predicted for the known miRNAs
Table S8 Targets predicted for the novel miRNAs
Table S9 Predicted targets of the differentially expressed miRNAs
Rights and permissions
About this article

Cite this article
Yan, J., Zhang, H., Zheng, Y. et al. Comparative expression profiling of miRNAs between the cytoplasmic male sterile line MeixiangA and its maintainer line MeixiangB during rice anther development. Planta 241, 109–123 (2015). https://doi.org/10.1007/s00425-014-2167-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-014-2167-2