
Abstract
Fusaric acid (FA) is a nonhost-selective toxin mainly produced by Fusarium oxysporum, the causal agent of plant wilt diseases. We demonstrate that FA can induce programmed cell death (PCD) in tobacco suspension cells and the FA-induced PCD is modulated by nitric oxide (NO) signalling. Cells undergoing cell death induced by FA treatment exhibited typical characteristics of PCD including cytoplasmic shrinkage, chromatin condensation, DNA fragmentation, membrane plasmolysis, and formation of small cytoplasmic vacuoles. In addition, caspase-3-like activity was activated upon the FA treatment. The process of FA-induced PCD was accompanied by a rapid accumulation of NO in a FA dose-dependent manner. Pre-treatment of cells with NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO) or NO synthase inhibitor N G-monomethyl-arginine monoacetate (L-NMMA) significantly reduced the rate of FA-induced cell death. Furthermore, the caspase-3-like activity and the expression of PAL and Hsr203J genes were alleviated by application of cPTIO or L-NMMA to FA-treated tobacco cells. This indicates that NO is an important factor involved in the FA-induced PCD. Our results also show that pre-treatment of tobacco cells with a caspase-3-specific inhibitor, Ac-DEVD-CHO, can reduce the rate of FA-induced cell death. These results demonstrate that the FA-induced cell death is a PCD and is modulated by NO signalling through caspase-3-like activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Programmed cell death (PCD) is an important physiological process during normal organ development and maintenance of cellular homeostasis in both plants and animals. PCD aims to remove damaged, wrongly produced or misplaced cells in tissues or organs (Danon et al. 2000; Domínguez and Cejudo 2012). It can be induced by a wide range of biotic and abiotic stress stimuli, including various pathogenic toxins (Yao et al. 2001; Duval et al. 2005; Samadi and Behboodi 2006). Numerous studies have demonstrated that PCD is an important plant response against various diseases and is known as a hypersensitive response (Heath 2000; Shirasu and Schulze-Lefert 2000; Greenberg and Yao 2004). The typical hallmarks of plant PCD include cytoplasmic shrinkage, chromatin condensation, DNA fragmentation, and an increase of caspase-like enzyme activity (Danon et al. 2000; Kuriyama and Fukuda 2002; Hughes and Gobe 2007; van Doorn et al. 2011). PCD in plants can be divided into two classes: autolytic PCD and non-autolytic PCD based on the features of cell morphology (van Doorn 2011).
Fusaric acid (FA, 5-butylpicolinic acid) is a well-known nonhost-selective toxin produced by Fusarium species (Bacon et al. 1996), particularly the pathogenic strains of F. oxysporum. FA was shown to have an important role in fungal infection and can cause synergistic effect in plants with other mycotoxins (Davis 1969; Bacon et al. 1996; Dong et al. 2012). The toxic effects of FA on plant cell include alteration of membrane permeability, decrease of mitochondrial activity, inhibition of ATP synthesis, and decrease of production of plant polyphenol oxidase and peroxidase (Marré et al. 1993; Kuźniak 2001; Bouizgarne et al. 2006a, b; Dong et al. 2012). It was documented that low levels of FA could induce PCD, while high levels of FA (>200 μM) promoted necrosis in saffron root-tip cells (Samadi and Behboodi 2006). To date, the molecular mechanism underlying the FA-induced cell death remains unknown.
Nitric oxide (NO), a free radical gas, is known as an important signalling molecule in both plants and animals. It has been shown that NO is involved in plant growth, cell proliferation and maturation, leaf senescence, stomata closure, and regulation of responses to abiotic and biotic stresses (Arasimowicz and Floryszak-Wieczorek 2007; Balestrazzi et al. 2011; Arasimowicz-Jelonek et al. 2012; Zhang et al. 2012). NO can react with other signalling molecules such as superoxide and hydrogen peroxide (H2O2) during plant–pathogen interactions (Delledonne et al. 1998; Wendehenne et al. 2004; Arasimowicz and Floryszak-Wieczorek 2007). NO can also react with a protein cysteine thiol to form an S-nitrosothiol, which emerges as the redox-based post-translational modification (Arasimowicz and Floryszak-Wieczorek 2007; Yu et al. 2012).
The PCD signal transduction pathway requires activation of a cascade of specific cysteine proteases called caspases. Among the known caspases, caspase-3 protease is a crucial executioner of PCD in animals (Kumar 2007). Recently, many studies reported that caspase-3-like activity is involved in PCD and development processes in plants (Wang et al. 2010; Han et al. 2012; Iakimova et al. 2013; Ye et al. 2013). To date, the role of NO signalling in caspase-3-like pathway and its relationship with FA is largely unknown. We provide morphological, biochemical, and molecular evidence to demonstrate that FA can induce PCD in tobacco suspension cells and NO signalling can modulate PCD through caspase-3-like activity.
Materials and methods
Chemicals
FA, cPTIO [2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide], DAPI (4′,6-diamidino-2-phenylindole), and Ac-DEVD-CHO (acetyl-Asp-Glu-Val-Asp-aldehyde) were purchased from Sigma-Aldrich (St. Louis, MO, USA). L-NMMA (N G-monomethyl-arginine monoacetate) and DAF-2 (4,5-diaminofluorescein) were purchased from Enzo Life Sciences (Farmingdale, NY, USA). Evans blue was from Aladdin (Shanghai, China). Reagents for reverse transcription-polymerase chain reaction (RT-PCR) were from TaKaRa (Dalian, China). FA was dissolved in phosphate buffered saline (PBS) to produce a 100-mM stock solution. Ac-DEVD-CHO was dissolved in dimethyl sulfoxide (DMSO) to make a 100-mM stock solution. cPTIO and L-NMMA were all dissolved in sterile water to produce 100 mM stock solutions. All stock solutions were stored at −20 °C.
Growth and treatment of tobacco suspension cells
All experiments were conducted using tobacco (Nicotiana tabacum cv. NC89) suspension cells grown in a liquid Murashige–Skoog medium, supplemented with 30 g L−1 sucrose, 2 mg L−1 α-naphthalene acetic acid, and 0.2 mg L−1 6-benzyladenine as described (Liu et al. 2010). The suspension cells were sub-cultured once every 7 days by transferring the cultured cells into (1:10, v/v) fresh media. All the experiments were performed using 4-day-old cultures based on the exponential growth phase. The control groups were treated with sterile PBS only. Ac-DEVD-CHO, cPTIO, and L-NMMA, when required, were added them individually into cultured cells at 10 min, 1 h, and 1 h before FA treatment, respectively.
Assay of cell death
Cell death was determined through Evans blue staining as described (Baker and Mock 1994). An aliquot of suspension cells was transferred into a centrifuge tube and pelleted by centrifugation at 10,000×g for 30 s. After three washes with PBS (pH 7.4), the cells were incubated in 1 mL of 0.05 % Evans blue solution for 15 min. After incubation, the cells were washed again with PBS, mounted on microscope slides and examined for cell death under a light microscope (C-35AD-2, Olympus). Cells that accumulated Evans blue dye were considered as dead cells. The experiments were repeated at least three times and each time more than 500 cells were counted per treatment.
DAPI staining
DAPI staining was performed to visualise the changes of nuclear morphology and chromatin condensation after FA treatment. The suspension cells were prepared as described above and treated with 100 μM FA for 24 h. The treated cells were harvested, washed with PBS and then stained with a DAPI (5 μg mL−1) solution for 5 min at room temperature in dark. The stained cells were washed three times with PBS and examined under a fluorescent microscope (Eclipse 90i, Nikon).
DNA extraction and electrophoresis
Cells were harvested and ground in liquid nitrogen. Total genomic DNA was extracted from the cells using the DNeasy Plant Mini Kit as instructed (Qiagen). To visualise the genomic DNA fragments, DNA samples (20 μg DNA per sample) were separated in a 2 % agarose gel at a constant 50 V for 3 h. After electrophoresis, the gel was stained with ethidium bromide followed by visualisation with a gel imaging analysis system (Bio-Best 140E, SIM). DNA ladders (DL5000 DNA marker; TaKaRa, Dalian, China) were used to show the size of DNA.
TUNEL assay
Nuclear DNA fragmentation was analysed using the In Situ Cell Death Detection Kit as instructed (Roche Molecular Chemicals, Switzerland). Briefly, the cell samples were firstly fixed in a 4 % paraformaldehyde solution for 1 h at 15–25 °C followed by three washes in PBS. The cell samples were then resuspended in a solution with 0.1 % Triton X-100 and 0.1 % sodium citrate for 2 min before treatment with RNase (30 μg mL−1; TaKaRa, Dalian, China) and Proteinase K (20 μg mL−1; Sigma-Aldrich, USA) for 30 min. The terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay was done by applying a 50-μL TUNEL kit solution to each sample and the sample was incubated at 37 °C for 1 h in dark. The cells were washed with PBS, mounted on microscope slides and examined under a fluorescent microscope (Eclipse 90i, Nikon).
Electron microscopy
To investigate the ultrastructural changes after FA treatment, the suspension cells were treated with 100 μM FA for 12 h. The cells were harvested by centrifugation at 10,000×g for 30 s and then fixed in a 3.5 % glutaraldehyde solution for 24 h at 4 °C. The cells were then post-fixed in 0.1 M cacodylate buffer containing 1 % osmium tetroxide. The post-fixed cells were dehydrated in a series of ethanol and embedded in Epon812 araldite resin. Ultrathin sections were cut with an LKB ultramicrotome and stained with 2 % uranyl acetate in 50 % methanol for 10 min, followed by lead citrate for 7 min. The sections were examined under a transmission electron microscope (JEM-1200EX, JEOL) at 80 kV.
Assays of nitric oxide (NO) accumulation
Extra- and intra-cellular accumulation of NO was determined using a specific fluorophore DAF-2 and a fluorescent dye 4-amino-5-methylamino-2′,7′-difluorescein diacetate (DAF-FM DA; Beyotime, Haimen, China), respectively. To determine the intracellular NO accumulation, 1 mL suspension cells was mixed with 0.5 μM DAF-FM DA and incubated on a slow shaker for 30 min at 25 °C in dark. The cells were then treated with 100 μM FA for 30 min followed by three washes in PBS. Fluorescence intensity of each sample was determined under a fluorescent microscope (Eclipse 90i, Nikon).
For the extracellular accumulation of NO, the specific fluorophore DAF-2 was used by fluorimetric assay through its binding to NO as described (Nakatsubo et al. 1998). We followed the procedure of Carimi et al. (2005). After cells were treated with FA for 0–90 min, fluorescence measurements were performed using a luminescence spectrometer (LS-55, Perkin Elmer) with an excitation wavelength of 495 nm and an emission wavelength of 515 nm. NO was quantified as fold number compared with control cells (relative fluorescence).
Measurement of caspase-3-like activity
Caspase-3-like activity was determined using the “Caspase-3 Activity Assay Kit” (Beyotime, Haimen, China) as instructed. During the assay, hydrolysis of the peptide substrate [acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA)] by caspase-3 results in a release of p-nitroaniline (pNA) moiety. The absorbance at 405 nm was measured using a microplate reader (Molecular Devices, SpectraMax M2) as instructed. Protein concentration in each sample was determined using the Bradford (1976) assay as described. The experiment was repeated three times.
RNA isolation and RT-PCR analysis
Suspension cells were treated with 100 μM FA for 12 h, in the presence or absence of cPTIO or L-NMMA. Total RNA was extracted from these cells using the Trizol reagent as instructed (Invitrogen). Concentration and quality of each RNA sample were determined using an Ultraspec 2100 spectrophotometer (Amersham Biosciences) and electrophoresis in 1 % agarose gels. For reverse transcription, 1 μg RNA/sample was incubated with M-MLV reverse transcriptase and an oligo (dT) primer as instructed by the manufacturer. The primers used to determine the expression level of PAL gene (GenBank accession no. D17467) were PALF (5′-GTCACACATTACCACAATCAG-3′) and PALR (5′-GTGTTCCATAATAGCAGCAGCCT-3′). The primers used to determine the expression level of Hsr203J gene (GenBank accession no. X77136) were Hsr203JF (5′-TGTACTACACTGTCTACACGC-3′) and Hsr203JR (5′-GATAAAAGCTATGTCCCACTCC-3′). The primers for EF1α (GenBank accession no. AJ223969) were EF1αF (5′-AGACCACCAAGTACTACTGCAC-3′) and EF1αR (5′-CCACCAATCTTGTACACATCC-3′). The tobacco EF1α gene was used as an internal control. The PCR products were separated by electrophoresis on a 1 % agarose gel, stained with ethidium bromide and observed with a gel imaging analysis system (Bio-Best 140E, SIM).
Statistical analysis
Data were presented as the mean ± standard deviations, and the statistical significance was determined by t test with p values <0.05.
Results
Effect of FA on viability of cultured tobacco cells
Evans blue staining was used to determine cell viability after FA treatment. Results in Fig. 1a indicate that the cell death rate increased gradually as the concentration of FA was increased from 10 to 200 μM. For example, cells treated with 100 μM FA showed an approximately 48 % cell death, while the cells treated with 200 μM FA showed more than 70 % cell death. To determine the effect of FA treatment duration on cell viability, the cells were treated with 100 μM FA for up to 24 h. Results of the study show that the cell death rate was increased from about 10 % at 0 h to about 50 % by 24 h (Fig. 1b). Consequently all the following viability experiments were conducted with 100 μM FA and 24 h incubation unless otherwise indicated.

FA treatment induces cell death. a Effect of FA concentration on cell death at 24 h post-FA treatment. b Time course of cell death induced by 100 μM FA. Data represent means of three independent experiments ± standard deviation. All the cells were stained with Evans blue dye (0.05 %) before observation
Cell morphological changes induced by FA treatment
Change of cell morphology is one of the most important features of PCD and is well documented in plants (van Doorn 2011; van Doorn et al. 2011). To investigate the morphological changes during cell death induced by FA, Evans blue staining was examined under a light microscope. Compared with control cells, cytoplasmic shrinkage was obvious in the FA-treated cells (Fig. 2a).

Cytoplasmic shrinkage and chromatin condensation in FA-treated tobacco cells. a Light microscopic images of cells stained with Evans blue dye (0.05 %). b Fluorescence microscopic images of cells stained with DAPI (5 μg mL−1). Cells were examined at 24 h post-treatment with 100 μM FA. Bar 20 μm
To investigate nuclear changes in the FA-treated cells, the cells were stained with DAPI and examined under a fluorescent microscope. Control cells showed round, uniformly stained nuclei with a large central nucleolus. In contrast, cells treated with 100 μM FA for 24 h showed elongated nuclei with irregular staining patterns (Fig. 2b).
Results of electron microscopy further confirmed the ultrastructural changes in the FA-treated tobacco suspension cells. In control cells, the cell organelles remained intact and the heterochromatin was evenly distributed inside the nucleus (Fig. 3a, b). In contrast, cells treated with 100 μM FA for 12 h showed dilated endoplasmic reticulum cisternae (Fig. 3c). Mitochondria in the FA-treated cells also exhibited abnormalities with ruptures of membranes and obscured cristae (Fig. 3d). The plasma membrane in the FA-treated cells was separated from the cell wall and numerous small vacuoles were formed in the cytoplasm of the FA-treated cells when compared with control cells (Fig. 3e). Heterochromatin was aggregated into large masses and located at the periphery of the nucleus (Fig. 3f).

Ultrastructural changes in 100 μM FA-treated tobacco cells. a, b PBS-treated (control) cells showing normal endoplasmic reticulum (ER), nucleus and mitochondria. c Dilation of ER cisternae in a FA-treated cell. d Mitochondria (M) with ruptured membrane and obscured cristae in a FA-treated cell. e Separation of plasma membrane (PM) from cell wall and increase of vacuole (V) number in a FA-treated cell. f Condensed chromatin in a FA-treated cell. The black arrow indicates the dilated ER cisternae, the white arrow points to a ruptured mitochondrial membrane. CW cell wall, Nu nucleolus. Bar a, c 100 nm; bar b, d 200 nm; bar e 1 μm; bar f 500 nm
DNA fragmentation in FA-treated tobacco cells
One of the hallmarks of PCD is nuclear DNA fragmentation. This fragmentation can be detected through electrophoresis of nuclear DNA in agarose gel or by the TUNEL assay. As shown in Fig. 4a, the genomic DNA extracted from the PBS-treated (control) cells appeared intact. The nuclear DNA extracted from the 100 μM FA-treated cells showed, however, fragmented DNA ladders. DNA fragmentation in the FA-treated cells was further confirmed in situ by the TUNEL assay. Compared with control cells, the FA-treated cells contained nuclei with stronger green fluorescence (Fig. 4b). Approximately 31 % of the FA-treated cells contained TUNEL-positive nuclei, whereas <1 % of control cells had TUNEL-positive nuclei (Fig. 4c). These results indicate that FA treatment can cause DNA fragmentation.

DNA fragmentation visualised by DNA laddering and TUNEL assay. a DNA laddering was determined in 2 % agarose gel through electrophoresis. Genomic DNA was extracted from FA-treated tobacco cells at 24 h post-treatment. M, DL5000 DNA marker. b TUNEL assay in FA-treated tobacco cells at 24 h post-FA treatment. Bar 20 μm. c Percentage of TUNEL-positive cells and dead cells determined by TUNEL assay or Evans blue staining. The cells analysed were all treated with 100 μM FA for 24 h before microscopic examination
Quantitative and qualitative analyses of NO and its effect on the FA-induced cell death
It is well documented that NO produced during various stress responses participates in signalling events leading to cell death (Delledonne et al. 1998; Arasimowicz and Floryszak-Wieczorek 2007). To investigate the role of NO in FA-induced PCD in tobacco cells, we analysed NO production in FA-treated cells using NO-specific fluorescent probes. Tobacco cells were harvested at 30 min post-FA treatment and probed with a NO-specific probe (DAF-FM DA). Under the florescent microscope, stronger green fluorescence was observed in the FA-treated cells when compared with control cells. This indicates that FA treatment induced the production of NO in cells (Fig. 5a). Extracellular NO release was subsequently measured using the specific fluorophore DAF-2. It was evident that the production of extracellular NO was increased significantly when the concentrations of FA increased, when compared with control cells. The production of extracellular NO was rapid and sustained over at least 90 min. This observation indicates that NO production is FA dosage dependent (Fig. 5b).

Accumulation of NO and effect of inhibitors on cell death. a Accumulation of intercellular NO was detected using a NO-specific fluorescent probe, DAF-FM DA. Cells were treated with 100 μM FA for 30 min and then photographed under a fluorescence microscope. Bar 20 μm. b Dose-and time-response curves of extracellular NO production were detected using a NO-specific fluorophore, DAF-2. Fluorescence was shown as relative fluorescence units (RFU). c Effects of cPTIO (NO scavenger) and L-NMMA (NO synthase inhibitor) on tobacco cell death at 24 h after FA treatment. Cells were treated with cPTIO or L-NMMA at 1 h prior to the FA treatment. The cells were treated with 100 μM FA alone, 100 μM FA + 1 mM cPTIO, 100 μM FA + 1 mM L-NMMA, 1 mM cPTIO alone or 1 mM L-NMMA alone. Data are shown as means of three independent experiments ± standard deviation
The effects of NO scavenger cPTIO and NO synthase inhibitor L-NMMA on cell death were also investigated with Evans blue staining. Pre-treatment of cells with cPTIO and L-NMMA reduced the FA-induced cell death by approximately 73 and 59 %, respectively. Cells treated with either cPTIO or L-NMMA alone did not affect the cell death rate (Fig. 5c). These results suggest that the FA-induced cell death is modulated by the release of NO.
Effect of caspase-3-like protease on cell death
To better understand the role of caspase-3-like protease on cell death, cells were treated with FA followed by analysis of activation of caspase-3-like activity using a fluorescent substrate. The activity of caspase-3-like protease was increased gradually and reached its maximum after 12 h treatment with 100 μM FA. After 12 h, the activity of caspase-3-like protease decreased quickly and maintained steady till 48 h after the treatment. At 12 h post-FA treatment, the activity of caspase-3-like protease was almost fourfold higher than that in control cells (Fig. 6a). To determine whether NO can regulate the activity of caspase-3-like protease in tobacco cells, we treated the cells with cPTIO or L-NMMA prior to the FA treatment. Results shown in Fig. 6b indicate that the presence of cPTIO or L-NMMA can significantly decrease the FA-induced caspase-3-like protease activity. Cells treated with cPTIO or L-NMMA alone showed similar caspase-3-like protease activities to control cells.

Activation and inhibition of caspase-3-like activity in tobacco cells. a Time course of caspase-3-like activity in 100 μM FA-treated tobacco cells. b Effect of inhibitors on caspase-3-like activity at 12 h post-FA treatment. Cells were treated with cPTIO (NO scavenger) or L-NMMA (NO synthase inhibitor) at 1 h prior to the FA treatment. The cells were treated with 100 μM FA alone, 100 μM FA + 1 mM cPTIO, 100 μM FA + 1 mM L-NMMA, 1 mM cPTIO alone or 1 mM L-NMMA alone. Different letters above the bars indicate a significant difference by t test (P < 0.05)
To further confirm the role of caspase-3-like protease in FA-induced PCD, we analysed cells treated with a specific caspase-3 inhibitor, Ac-DEVD-CHO. As shown in Fig. 7, treatment of cells with Ac-DEVD-CHO prior to the FA treatment showed a reduced cell death rate by approximately 56 %. In contrast, cells treated with Ac-DEVD-CHO alone showed a cell death rate similar to that observed for control cells. These results indicate that caspase-3-like protease is indeed involved in the FA-induced PCD and the activity of caspase-3-like protease is regulated by NO signalling.

Effect of caspase-3 inhibitor (Ac-DEVD-CHO) on FA-induced cell death. Cells were treated with 100 μM Ac-DEVD-CHO at 10 min prior to the 100 μM FA treatment. The number of dead cells was counted at 24 h post-FA treatment
Effects of cPTIO and L-NMMA on gene expression
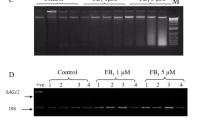
To evaluate the molecular mechanism underlying the effect of NO on FA-induced cell death, we assayed the expression of PAL and Hsr203J genes, in the presence of cPTIO or L-NMMA, through semi-quantitative RT-PCR. PAL gene encodes phenylalanine ammonia lyase, a key enzyme functioning in the phenylpropanoid biosynthetic pathway. Hsr203J gene was proposed as a PCD marker gene in plants (Pontier et al. 1998). As shown in Fig. 8, the expression of PAL and Hsr203J was significantly increased after FA treatment when compared with control cells. Furthermore, treatment of cells with cPTIO or L-NMMA prior to the FA treatment clearly reduced the expression of both genes. As controls, cells treated with cPTIO or L-NMMA alone did not affect the expression of these two genes. This finding supports the above conclusion that the NO signalling pathway is involved in the expression of defence- and PCD-related genes in tobacco suspension cells.

Effects of cPTIO and L-NMMA on the expression of PAL and Hsr203J genes in FA-treated tobacco cells. Cells were treated with cPTIO (NO scavenger) or L-NMMA (NO synthase inhibitor) at 1 h prior to the 100 μM FA treatment. Total RNA was isolated from tobacco cells at 12 h post-FA treatment and the RNA transcripts were measured by RT-PCR. The expression level of EF1α was used as a positive internal control
Discussion
In the present study, we investigated morphological, biochemical, and molecular changes in tobacco suspension cells treated with FA. Our results indicate that the changes observed in the FA-treated cells resemble the PCD hallmarks reported previously for plant cells. In a previous work published by Samadi and Behboodi (2006), FA treatment of saffron root-tip cells was shown to cause chromatin condensation, DNA fragmentation, cytochrome c release, and H2O2 accumulation. Taken together, our study results and the results published by Samadi and Behboodi (2006) strongly indicate that the changes in cells treated with FA are typical hallmarks of PCD. In this paper, we also presented molecular evidence of PCD by demonstrating the activation of caspase-3-like activity modulated through NO signalling in FA-treated tobacco cells.
FA induced cytoplasmic shrinkage, chromatin condensation, and DNA fragmentation, which are morphological characteristics of plant PCD. The FA-induced PCD in tobacco cells is shown to be dosage-dependent and is similar to that observed in the saffron root-tip cells. For example, after 24 h treatment with 100 μM FA, approximately 31 % of the tobacco cells showed TUNEL-positive nuclei and 17 % cells showed necrosis (Fig. 3c). However, after 24 h treatment with 200 μM FA, the numbers of TUNEL-positive cells decreased to 23 % while the necrotic cell number increased up to 46 % (Fig. S1), a trend similar to that reported previously by Samadi and Behboodi (2006). On the other hand, the FA-induced PCD was further evidenced by DNA laddering in the FA-treated cells.
Ultrastructural analysis of FA-treated cells showed significant changes in cell morphology. For example, by 12 h post-FA treatment, the plasma membrane in FA-treated cells was separated from cell walls known as plasmolysis (Fig. 3e). Similar changes were also observed in Thellungiella halophila or tobacco cells under the salt or aluminium stresses (Panda et al. 2008; Wang et al. 2010). In addition, mitochondria in the FA-treated tobacco cells showed ruptured membrane and obscured cristae (Fig. 3d). Mitochondria are known to regulate PCD in plants and the integrity of mitochondrial membrane is crucial for maintaining this function (Lam et al. 2001; Li and Xing 2010, 2011; Godbole et al. 2012). Under an electron microscope, the FA-treated tobacco cells showed heterochromatin aggregation, similar to that observed in tobacco cells treated with H2O2 and in oat cells treated with victorin (Houot et al. 2001; Yao et al. 2001; Reape and McCabe 2010). In the FA-treated tobacco cells, numerous vacuoles similar to that reported in the tobacco cells under the aluminium stress (Panda et al. 2008) were also observed. This increase of vacuole number and later clearance of cytoplasm lead ultimately to cell death and the process is termed as autolytic PCD (van Doorn 2011). Based on the observed cell morphological changes, we propose that the FA-induced PCD is autolytic.
NO is an important signalling molecule produced during PCD. Many reports have demonstrated that NO plays a critical role in biotic and abiotic stress-induced PCD in plants (Arasimowicz and Floryszak-Wieczorek 2007; De Michele et al. 2009; Ma et al. 2010; Arasimowicz-Jelonek et al. 2012; Ye et al. 2013). In this work, we determined that the increase of intra- and extra-cellular NO levels correlated with the increase of FA concentration (Fig. 5a, b). To further verify the role of NO in PCD, we applied L-NMMA and cPTIO to FA-treated cells followed by observation of cell death. L-NMMA is a NO synthase inhibitor that proved to be effective in plants (Foissner et al. 2000; Garcês et al. 2001; Carimi et al. 2005; De Michele et al. 2009). cPTIO (NO scavenger) was also used to investigate the role of NO in plant PCD and growth of pollen tubes (Laxalt et al. 2007; De Michele et al. 2009; Wang et al. 2009). Our results show that presence of cPTIO or L-NMMA alone did not cause PCD in tobacco suspension cells. In the FA-treated cells, however, the presence of cPTIO or L-NMMA indeed reduced cell death rate to significant levels (Fig. 5c). Interestingly in the presence of both FA and cPTIO or FA and L-NMMA, the cell death rate was much higher than that observed in control cells. This finding indicates that NO is indeed involved in the FA-induced cell death. The contribution of other signalling molecules, particularly H2O2 to cell death, has to be studied. H2O2 production was previously observed in FA-treated saffron cells (Samadi and Behboodi 2006); therefore, we propose that H2O2 and NO may synergistically be involved in the activation of FA-induced PCD.
Caspases are conserved cysteine proteases and play central roles in the progression of PCD in animal cells (del Pozo and Lam 1998; Earnshaw et al. 1999; Kumar 2007). Several studies have indicated that the progression of plant PCD may also require activation of caspase-like activities (De Jong et al. 2000; Lam and del Pozo 2000; Mlejnek and Procházka 2002; Danon et al. 2004; Vacca et al. 2006; Reape and McCabe 2010). To determine if caspase-3-like activity also has a role in the FA-induced PCD, we applied Ac-DEVD-CHO, a specific caspase-3 inhibitor, to the FA-treated cells. Results of this study indicate that a caspase-3-like protease was activated in the FA-treated cells (Fig. 6a). In addition, the activation of caspase-3-like protease was also significantly reduced after the addition of cPTIO or L-NMMA to FA-treated cells (Fig. 6b). This result is consistent with a previous report in Arabidopsis thaliana cells induced by cadmium (Ye et al. 2013). This finding demonstrates that NO plays a key role in regulating the activity of caspase-3-like protease. Moreover, application of Ac-DEVD-CHO to FA-treated cells significantly reduced cell death rate (Fig. 7), further indicating that the caspase-3-like protease participated in the FA-induced PCD. However, the cell death induced by FA in the presence of Ac-DEVD-CHO did not reduce to the control level, which is similar as that of the tomato cells treated with camptothecin, fumonisin B1 or CdSO4 (Iakimova et al. 2013), suggesting that caspase-3-independent mechanisms may be involved in the FA-induced PCD.
It is well documented that PCD occurs not only during plant growth, but also development and differentiation; it is also involved in plant defence responses. In previous studies, low concentrations of FA were used as an elicitor to induce plant defence responses. Application of low concentrations of FA to cells resulted in production of H2O2, increase of cytosolic calcium, and synthesis of phytoalexin (Kuźniak 2001; Bouizgarne et al. 2006a, b). Considering PCD can alter gene expression in plants, we opted to investigate the effect of NO on the expression of PAL and Hsr203J genes during the FA-induced PCD. PAL gene was used previously as a molecular marker for the activation of plant defence responses, and Hsr203J gene was used as a marker of PCD (Pontier et al. 1998). As previously reported by Arasimowicz and Floryszak-Wieczorek (2007), the expression of PAL gene was increased after the treatment of tobacco and soybean suspension cells with NO donors. In the present study, the expression of tobacco PAL and Hsr203J genes were both increased after the cells were treated with FA. Furthermore, the expression of these two genes was significantly inhibited by cPTIO or L-NMMA in FA-treated cells (Fig. 8). Based on these results, we propose that NO is involved in regulating gene expression during FA-induced PCD.
Taken together, we conclude that FA treatment can induce production of hallmarks of PCD including chromatin condensation, DNA fragmentation, appearance of small vacuoles, and activation of caspase-3-like protease. We also present evidence showing that the FA-induced PCD is modulated by the NO signalling pathway and caspase-3-like protease activity. Our results shed more lights on the mechanisms underlying the FA-induced PCD. We also indicate that additional factor(s) may participate in this PCD and require further studies.
Abbreviations
- Ac-DEVD-CHO:
-
Acetyl-Asp-Glu-Val-Asp-aldehyde
- cPTIO:
-
2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide
- DAF-2:
-
4,5-Diaminofluorescein
- DAF-FM DA:
-
4-Amino-5-methylamino-2′,7′-difluorescein diacetate
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- FA:
-
Fusaric acid
- L-NMMA:
-
N G-monomethyl-arginine monoacetate
- NO:
-
Nitric oxide
- PBS:
-
Phosphate buffered saline
- PCD:
-
Programmed cell death
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling
References
Arasimowicz M, Floryszak-Wieczorek J (2007) Nitric oxide as a bioactive signalling molecule in plant stress responses. Plant Sci 172:876–887
Arasimowicz-Jelonek M, Floryszak-Wieczorek J, Deckert J, Rucińska-Sobkowiak R, Gzyl J, Pawlak-Sprada S, Abramowski D, Jelonek T, Gwóźdź EA (2012) Nitric oxide implication in cadmium-induced programmed cell death in roots and signaling response of yellow lupine plants. Plant Physiol Biochem 58:124–134
Bacon CW, Porter JK, Norred WP, Leslie JF (1996) Production of fusaric acid by Fusarium species. Appl Environ Microbiol 62:4039–4043
Baker CJ, Mock NM (1994) An improved method for monitoring cell death in cell suspension and leaf disc assay using Evans blue. Plant Cell, Tissue Organ Cult 39:7–12
Balestrazzi A, Agoni V, Tava A, Avato P, Biazzi E, Raimondi E, Macovei A, Carbonera D (2011) Cell death induction and nitric oxide biosynthesis in white poplar (Populus alba) suspension cultures exposed to alfalfa saponins. Physiol Plant 141:227–238
Bouizgarne B, El-Maarouf-Bouteau H, Frankart C, Reboutier D, Madiona K, Pennarun AM, Monestiez M, Trouverie J, Amiar Z, Briand J, Brault M, Rona JP, Ouhdouch Y, El Hadrami I, Bouteau F (2006a) Early physiological responses of Arabidopsis thaliana cells to fusaric acid: toxic and signaling effects. New Phytol 169:209–218
Bouizgarne B, El-Maarouf-Bouteau H, Madiona K, Biligui B, Monestiez M, Pennarun AM, Amiar Z, Rona JP, Ouhdouch Y, El Hadrami I, Bouteau F (2006b) A putative role for fusaric acid in biocontrol of the parasitic angiosperm Orobanche ramosa. Mol Plant-Microbe Interact 19:550–556
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Carimi F, Zottini M, Costa A, Cattelan I, De Michele R, Terzi M, Lo Schiavo F (2005) NO signalling in cytokinin-induced programmed cell death. Plant, Cell Environ 28:1171–1178
Danon A, Delorme V, Mailhac N, Gallois P (2000) Plant programmed cell death: a common way to die. Plant Physiol Biochem 38:647–655
Danon A, Rotari VI, Gordon A, Mailhac N, Gallois P (2004) Ultraviolet-C overexposure induces programmed cell death in Arabidopsis, which is mediated by caspase-like activities and which can be suppressed by caspase inhibitors, p35 and defender against apoptotic death. J Biol Chem 279:779–787
Davis D (1969) Fusaric acid in selective pathogenicity. Phytopathology 59:1391–1395
De Jong AJ, Hoeberichts FA, Yakimova ET, Maximova E, Woltering EJ (2000) Chemical-induced apoptotic cell death in tomato cells: involvement of caspase-like proteases. Planta 211:656–662
De Michele R, Vurro E, Rigo C, Costa A, Elviri L, Di Valentin M, Careri M, Zottini M, Sanità di Toppi L, Lo Schiavo F (2009) Nitric oxide is involved in cadmium-induced programmed cell death in Arabidopsis suspension cultures. Plant Physiol 150:217–228
del Pozo O, Lam E (1998) Caspases and programmed cell death in the hypersensitive response of plants to pathogens. Curr Biol 8:1129–1132
Delledonne M, Xia YJ, Dixon RA, Lamb C (1998) Nitric oxide functions as a signal in plant disease resistance. Nature 394:585–588
Domínguez F, Cejudo FJ (2012) A comparison between nuclear dismantling during plant and animal programmed cell death. Plant Sci 197:114–121
Dong X, Ling N, Wang M, Shen QR, Guo SW (2012) Fusaric acid is a crucial factor in the disturbance of leaf water imbalance in Fusarium-infected banana plants. Plant Physiol Biochem 60:171–179
Duval I, Brochu V, Simard M, Beaulieu C, Beaudoin N (2005) Thaxtomin A induces programmed cell death in Arabidopsis thaliana suspension-cultured cells. Planta 222:820–831
Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspase: structure, activation, substrate, and functions during apoptosis. Annu Rev Biochem 68:383–424
Foissner I, Wendehenne D, Langebartels C, Durner J (2000) In vivo imaging of an elicitor-induced nitric oxide burst in tobacco. Plant J 23:817–824
Garcês H, Durzan D, Pedroso MC (2001) Mechanical stress elicits nitric oxide formation and DNA fragmentation in Arabidopsis thaliana. Ann Bot 87:567–574
Godbole A, Dubey AK, Reddy PS, Udayakumar M, Mathew MK (2012) Mitochondrial VDAC and hexokinase together modulate plant programmed cell death. Protoplasma. doi:10.1007/s00709-012-0470-y
Greenberg JT, Yao N (2004) The role and regulation of programmed cell death in plant-pathogen interactions. Cell Microbiol 6:201–211
Han JJ, Lin W, Oda Y, Cui KM, Fukuda H, He XQ (2012) The proteasome is responsible for caspase-3-like activity during xylem development. Plant J 72:129–141
Heath MC (2000) Hypersensitive response-related death. Plant Mol Biol 44:321–334
Houot V, Etienne P, Petitot AS, Barbier S, Blein JP, Suty L (2001) Hydrogen peroxide induces programmed cell death features in cultured tobacco BY-2 cells, in a dose-dependent manner. J Exp Bot 52:1721–1730
Hughes J, Gobe G (2007) Identification and quantification of apoptosis in the kidney using morphology, biochemical and molecular markers. Nephrology 12:452–458
Iakimova ET, Michaeli R, Woltering EJ (2013) Involvement of phospholipase D-related signal transduction in chemical-induced programmed cell death in tomato cell cultures. Protoplasma. doi:10.1007/s00709-013-0497-8
Kumar S (2007) Caspase function in programmed cell death. Cell Death Differ 14:32–43
Kuriyama H, Fukuda H (2002) Developmental programmed cell death in plants. Curr Opin Plant Biol 5:568–573
Kuźniak E (2001) Effects of fusaric acid on reactive oxygen species and antioxidants in tomato cell cultures. J Phytopathol 149:575–582
Lam E, del Pozo O (2000) Caspase-like protease involvement in the control of plant cell death. Plant Mol Biol 44:417–428
Lam E, Kato N, Lawton M (2001) Programmed cell death, mitochondria and the plant hypersensitive response. Nature 411:848–853
Laxalt AM, Raho N, ten Have A, Lamattina L (2007) Nitric oxide is critical for inducing phosphatidic acid accumulation in xylanase-elicited tomato cells. J Biol Chem 282:21160–21168
Li Z, Xing D (2010) Mitochondrial pathway leading to programmed cell death induced by aluminium phytotoxicity in Arabidopsis. Plant Signal Behav 5:1660–1662
Li Z, Xing D (2011) Mechanistic study of mitochondria-dependent programmed cell death induced by aluminim phytotoxicity using fluorescence techniques. J Exp Bot 62:331–343
Liu F, Wei FF, Wang L, Liu H, Zhu XP, Liang YC (2010) Riboflavin activates defense responses in tobacco and induces resistance against Phytophthora parasitica and Ralstonia solanacearum. Physiol Mol Plant Pathol 74:330–336
Ma WW, Xu WZ, Xu H, Chen YS, He ZY, Ma M (2010) Nitric oxide modulates cadmium influx during cadmium-induced programmed cell death in tobacco BY-2 cells. Planta 232:325–335
Marré MT, Vergani P, Albergoni FG (1993) Relationship between fusaric acid uptake and its binding to cell structure in leaves of Egeria densa and its toxic effects on membrane permeability and respiration. Physiol Mol Plant Pathol 42:141–157
Mlejnek P, Procházka S (2002) Activation of caspase-like proteases and induction of apoptosis by isopentenyladenosine in tobacco BY-2 cells. Planta 215:158–166
Nakatsubo N, Kojima H, Kikuchi K, Nagoshi H, Hirata Y, Maeda D, Imai Y, Irimura T, Nagano T (1998) Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: diaminofluoresceins. FEBS Lett 427:263–266
Panda SK, Yamamoto Y, Kondo H, Matsumoto H (2008) Mitochondrial alterations related to programmed cell death in tobacco cells under aluminium stress. C R Biol 331:597–610
Pontier D, Tronchet M, Rogowsky P, Lam E, Roby D (1998) Activation of hsr203, a plant gene expressed during incompatible plant-pathogen interactions, is correlated with programmed cell death. Mol Plant Microbe Interact 11:544–554
Reape TJ, McCabe PF (2010) Apoptosis-like regulation of programmed cell death in plants. Apoptosis 15:249–256
Samadi L, Behboodi BS (2006) Fusaric acid induces apoptosis in saffron root-tip cells: roles of caspase-like activity, cytochrome c, and H2O2. Planta 225:223–234
Shirasu K, Schulze-Lefert P (2000) Regulators of cell death in disease resistance. Plant Mol Biol 44:371–385
Vacca RA, Valenti D, Bobba A, Merafina RS, Passarella S, Marra E (2006) Cytochrome c is released in a reactive oxygen species-dependent manner and is degraded via caspase-like proteases in tobacco Bright-Yellow 2 cells en route to heat shock-induced cell death. Plant Physiol 141:208–219
van Doorn WG (2011) Classes of programmed cell death in plants, compared to those in animals. J Exp Bot 62:4749–4761
van Doorn WG, Beers EP, Dangl JL, Franklin-Tong VE, Gallois P, Hara-Nishimura I, Jones AM, Kawai-Yamada M, Lam E, Mundy J, Mur LAJ, Petersen M, Smertenko A, Taliansky M, Van Breusegem F, Wolpert T, Woltering E, Zhivotovsky B, Bozhkov PV (2011) Morphological classification of plant cell deaths. Cell Death Diff 18:1241–1246
Wang YH, Chen T, Zhang CY, Hao HQ, Liu P, Zheng MZ, Baluška F, Šamaj J, Lin JX (2009) Nitric oxide modulates the influx of extracellular Ca2+ and actin filament organization during cell wall construction in Pinus bungeana pollen tubes. New Phytol 182:851–862
Wang J, Li XR, Liu YB, Zhao X (2010) Salt stress induces programmed cell death in Thellungiella halophila suspension-cultured cells. J Plant Physiol 167:1145–1151
Wendehenne D, Durner J, Klessig DF (2004) Nitric oxide: a new play in plant signaling and defence responses. Curr Opin Plant Biol 7:449–455
Yao N, Tada Y, Park P, Nakayashiki H, Tosa Y, Mayama S (2001) Novel evidence for apoptosis cell response and differential signal in chromatin condensation and DNA cleavage in victorin-treated oats. Plant J 28:13–26
Ye Y, Li Z, Xing D (2013) Nitric oxide promotes MPK6-mediated caspase-3-like activation in cadmium-induced Arabidopsis thaliana programmed cell death. Plant, Cell Environ 36:1–15
Yu M, Yun BW, Spoel SH, Loake GJ (2012) A sleigh ride through the SNO: regulation of plant immune function by protein S-nitrosylation. Curr Opin Plant Biol 15:1–7
Zhang B, Zheng LP, Wang JW (2012) Nitric oxide elicitation for secondary metabolite production in cultured plant cells. Appl Microbiol Biotechnol 93:455–466
Acknowledgments
This work was supported by the National Natural Science Foundation (31171806, 30871620), Shandong Provincial Natural Science Foundation (ZR2012CM032), and Anhui Province Tobacco Company Project (20100551002, 20100551005) in China.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Jiao and B. Zhou contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Jiao, J., Zhou, B., Zhu, X. et al. Fusaric acid induction of programmed cell death modulated through nitric oxide signalling in tobacco suspension cells. Planta 238, 727–737 (2013). https://doi.org/10.1007/s00425-013-1928-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-013-1928-7