
Abstract
The elaborate networks and the crosstalk of established signaling molecules like salicylic acid (SA), jasmonic acid (JA), ethylene (ET), abscisic acid (ABA), reactive oxygen species (ROS) and glutathione (GSH) play key role in plant defense response. To obtain further insight into the mechanism through which GSH is involved in this crosstalk to mitigate biotic stress, transgenic Nicotiana tabacum overexpressing Lycopersicon esculentum gamma-glutamylcysteine synthetase (LeECS) gene (NtGB lines) were generated with enhanced level of GSH in comparison with wild-type plants exhibiting resistance to pathogenesis as well. The expression levels of non-expressor of pathogenesis-related genes 1 (NPR1)-dependent genes like pathogenesis-related gene 1 (NtPR1), mitogen-activated protein kinase kinase (NtMAPKK), glutamine synthetase (NtGLS) were significantly enhanced alongwith NtNPR1. However, the expression levels of NPR1-independent genes like NtPR2, NtPR5 and short-chain dehydrogenase/reductase family protein (NtSDRLP) were either insignificant or were downregulated. Additionally, increase in expression of thioredoxin (NtTRXh), S-nitrosoglutathione reductase 1 (NtGSNOR1) and suppression of isochorismate synthase 1 (NtICS1) was noted. Comprehensive analysis of GSH-fed tobacco BY2 cell line in a time-dependent manner reciprocated the in planta results. Better tolerance of NtGB lines against biotrophic Pseudomonas syringae pv. tabaci was noted as compared to necrotrophic Alternaria alternata. Through two-dimensional gel electrophoresis (2-DE) and image analysis, 48 differentially expressed spots were identified and through identification as well as functional categorization, ten proteins were found to be SA-related. Collectively, our results suggest GSH to be a member in cross-communication with other signaling molecules in mitigating biotic stress likely through NPR1-dependent SA-mediated pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A broad range of functions of GSH in plants, including redox control, detoxification of heavy metals and electrophilic xenobiotics, serving as electron donor for biochemical reactions, long-distance transport of reduced sulfur, stress defense gene expression, direct posttranslational modifications of proteins through reversible glutathionylation of thiol residues etc. have been reported so far (Foyer et al. 1997; May et al. 1998; Ball et al. 2004; Pasternak et al. 2008; Ishikawa et al. 2010; Mhamdi et al. 2010). In addition, GSH in relation to tolerance of abiotic stresses like frost, salt, chilling, heavy metal and so on has been studied as well (Kocsy et al. 2000; Ruiz and Blumwald 2002; Gomez et al. 2004; Kumar et al. 2009). Appertaining to the aforementioned functions, the potential role of GSH has been investigated in various defense reactions, demonstrating that GSH participates in plant defense reactions against viral, fungal and bacterial infections. Exogenous application of GSH was found to activate several genes encoding enzymes that participate in the biosynthesis of lignin and phytoalexins in bean cell culture (Wingate et al. 1988). It has long been known that, elicitor stimulation induces chalcone synthase expression, and this effect can be partly mimicked by GSH supplementation (Dron et al. 1988). Treatment of bean and soybean cells with fungal elicitor or GSH causes the rapid insolubilization of hydroxy-proline-rich structural proteins in the cell wall (Bradley et al. 1992). Early study revealed that, the enhanced resistance of melon and tomato roots against Fusarium oxysporum brought about by herbicides coincides with the significant increase of GSH levels (Bolter et al. 1993). In compatible barley–barley powdery mildew interactions the ascorbate-GSH cycle and other antioxidative enzymes (e.g. glutathione S-transferase) are activated and these processes might diminish the damaging effects of oxidative stress. However, in incompatible interactions these antioxidative reactions are not or are only slightly activated (El-Zahaby et al. 1995). A considerable accumulation of GSH and, in particular, oxidized glutathione (GSSG) has been observed in tomato cells carrying Cf-9 or Cf-2 resistance genes after treatment with race-specific elicitors of the fungus Cladosporium fulvum (May et al. 1996). Additionally, previous studies also reported that Arabidopsis pad2-1 mutant with only 22% of wild-type amounts of GSH were susceptible to P. syringae as well as Phytophthora brassicae (Glazebrook and Ausubel 1994; Glazebrook et al. 1997; Parisy et al. 2007). These results cumulatively suggest that the accumulation of GSH may be necessary for disease resistance.
Genetic, physiological and molecular analyses have revealed that the stress-related phytohormones SA, JA and ET play key roles in the complex crosstalk among signaling pathways to mitigate biotic stress (Thomma et al. 1998; Glazebrook 2001; Grant and Jones 2009; Pieterse et al. 2009). The small plant hormone molecule SA plays significant role in plant disease resistance. Active oxygen species has also been found to be involved in SA-mediated systemic acquired resistance (Chen et al. 1993). Considering the interaction of SA with other established signaling molecules, antagonism between SA and JA is a widely accepted fact, synergistic actions of SA and JA/ET have been reported as well (Van Wees et al. 2000; Mur et al. 2006), highlighting the potential significance of SA-JA crosstalk in nature (Leon-Reyes et al. 2010). Further, SA signaling is classically thought to control the resistance to biotrophic pathogens; by contrast, JA and ET-signaling pathways are responsible for resistance to necrotrophic pathogens (Glazebrook 2005; Loake and Grant 2007).
Studies have shown that innate immune responses are associated with changes in cellular redox states sensed by NPR1 (Mou et al. 2003; Tada et al. 2008). After detecting microbial pathogens, plants accumulate SA, subsequently activates NPR1, which play an important regulatory role in plant defense including the expression of PR genes (Cao et al. 1994, 1997; Zhang et al. 1999; Zhou et al. 2000; Pieterse and Van Loon 2004). Increase in SA triggers the reduction of disulphide bonds located on NPR1. This reduction stimulates both the translocation of NPR1 from the cytosol to the nucleus and the physical interaction of NPR1–TGA1 that is necessary for the activation of PR gene transcription (Mou et al. 2003; Després et al. 2003). Previous study also showed that NPR1 plays a crucial role in the cross-communication between SA-JA-dependent defense signaling pathways (Spoel et al. 2003). In addition, the SA-induced NPR1 oligomer-to-monomer reaction regulated by thioredoxin through reduction of its intermolecular disulphide bonds has also been reported (Tada et al. 2008). Further, SA signaling is thought to be mediated by at least two mechanisms, one requiring NPR1 and a second that is independent of NPR1 (Blanco et al. 2009).
In contrast to the overwhelming amount of information with respect to SA, JA, ET, ABA and ROS as well as their crosstalk serving as important regulators of induced disease resistance, the role of GSH cross-communication networks in plant defense is less well understood. The aim of the present study was to obtain an insight into the role of GSH in the crosstalk with other signaling molecules in mitigating biotic stress. Here, the genetic engineering approach has been used to develop and establish transgenic tobacco overexpressing LeECS with enhanced GSH content. Biotic stress tolerance potential of the NtGB lines has also been established. Transcript profiling of selected defense-related genes of the transgenic as well as the wild-type plants and a time-course gene expression analysis of GSH-fed BY2 cell line have been determined. Until now, limited data are available about the stress-elicited changes in plants with enhanced GSH content, at the proteome level. Proteome analysis of the NtGB lines and identification of changes at the protein expression level have been performed. Taken together, present findings suggest that GSH plays an important role in biotic stress tolerance with a possible crosstalk through NPR1-dependent SA-mediated signaling pathway.
Materials and methods
Plant materials and growth conditions
Tobacco (N. tabacum cv. Xanthi) and L. esculentum seeds were sown on MS medium (Murashige and Skoog 1962) containing 3% sucrose and 0.8% agar and allowed to germinate at 22 ± 1°C (16-h of 150 μE m−2 s−1 light and 8-h darkness). Subsequently plants were grown and maintained in MS medium supplemented with 1 mg L−1 IBA and 0.25 mg L−1 kinetin. Tobacco BY2 cells were grown in dark on modified MS medium to which KH2PO4, thiamine and myo-inositiol were added in excess. The medium was supplemented with 0.2 mg L−1 2, 4-D.
Plasmid construction and plant transformation
A cDNA designated as LeECS was isolated from 2-week-old L. esculentum seedlings by RT-PCR. Total RNA was isolated using Trizol reagent (Invitrogen, USA) and first-strand cDNA was synthesized from total RNA using RevertAid H Minus First Strand cDNA Synthesis kit (Fermentas, USA) with oligo(dT)18 primer as standardized by Banerjee and Chattopadhyay (2010). The 1,571-bp full-length DNA sequence of LeECS was amplified using gene-specific primers: forward, 5′-ATCTAGATATGGCCTTGATGTCTCAGGC-3′; and reverse, 5′-AAGAGCTCAATCAGTAGAGAAGCTCCTCAAA-3′ (Sigma, USA). The PCR product was cloned into pGEM-T Easy vector (Promega, USA) and subcloned into the binary vector pBI121 at XbaI and SacI sites. Tobacco leaf discs were infected with Agrobacterium tumefaciens LBA4404 harboring the recombinant plasmid. The regenerated shoots were maintained on MS medium supplemented with 0.1 mg L−1 NAA and 1 mg L−1 BAP alongwith 50 mg L−1 kanamycin and 62.5 mg L−1 cephotaxime. Kanamycin-resistant and PCR positive, transgenic T0 plants were selected, and transferred to greenhouse and maintained up to T2 generation, which were used for further analysis.
Molecular analysis of NtGB lines
PCR was employed to screen the transformants carrying nptII gene. Genomic DNA was extracted from young leaves of wild-type and NtGB lines by cetyltrimethylammonium bromide (CTAB) method. PCR amplification was performed as follows: 94°C for 45 s, 55°C for 45 s and 72°C for 1 min, with an initial denaturation at 94°C for 1 min using the primer pair listed in Supplementary Table S1.
For RT-PCR, total RNA from tobacco leaves was isolated and cDNA synthesized as mentioned earlier. NptII and LeECS transcripts were detected by semiquantitative RT-PCR using specific primers (Supplementary Table S1). PCR amplification of nptII was performed as mentioned earlier and amplification of LeECS was performed as follows: 94°C for 45 s, 63°C for 30 s and 72°C for 1 min, with an initial denaturation at 94°C for 5 min. Equal loading for amplification of each cDNA was determined by actin PCR product.
For Southern blot analysis, a total of 10 μg of genomic DNA was digested with EcoRI and HindIII separately, fractionated on 0.8% agarose gel and then transferred onto Immobilon-NY+ membrane (Millipore, USA). Hybridization was performed at 68°C using a 700-bp fragment of LeECS as probe, labeled with [α-32P] dATP. The blots were washed repeatedly under stringent conditions and exposed to X-ray film. All molecular biology experiments were performed following standard protocols (Sambrook and Russell 2001).
Determination of GSH and GSH:GSSG ratio
GSH was extracted from mature tobacco leaves and quantified (Tsakraklides et al. 2002). High-performance liquid chromatography (HPLC) was conducted using a 515-HPLC pump (Waters, USA) and 2475 fluorescence detector (Waters, USA), at a flow-rate of 1.5 mL min−1 using AccQ.Tag (3.9 × 150 mm) column (Waters, USA) at excitation wavelength of 360 nm and emission wavelength of 450 nm. Briefly, the elution condition was: solvent A, composed of sodium acetate and triethylamine at pH 5.05 at 5% (v/v) dilution and acetonitrile:water (30:70) as solvent B. From 0 to 9 min, A was 94%, from 9 to 16 min a linear gradient of 94 to 91.5% A was applied, from 16 to 22 min a linear gradient of 75% A was applied and from 22 to 30 min A decreased to 0% linearly. GSH (Sigma, USA) was used as standard. Data analyses were performed with Empower 2 software.
GSH:GSSG ratio was measured according to Ishikawa et al. (2010) with NtGB lines and wild-type plants. Total glutathione was measured by glutathione reductase (GR) recycling system coupled with 5, 5′-dithiobis (2-nitrobenzoic acid) (DTNB) by measuring 5′-thio-2-nitrobenzoic acid (TNB) formation at 412 nm. GSSG was measured by pretreating the samples with 2-vinylpyridine to mask GSH and assayed in the presence of GR and DTNB as above. GSH was calculated by subtracting the GSSG content from the total glutathione content and GSH:GSSG ratio was determined as well.
Extraction and quantification of SA
Extraction and quantification of SA was performed as described previously (Freeman et al. 2005). SA was quantified by HPLC with a fluorescence detector, as mentioned earlier using Symmetry C-18 reverse-phase column (5 μm, 4.6 × 250 mm) at excitation wavelength of 254 nm and emission wavelength of 395 nm. The elution condition was methanol gradient (solvent A, water and 1% formate; and solvent B, 100% methanol and 1% formate) of 10 to 40% B (10 min), 40 to 50% B (5 min), 50 to 100% B (2.5 min), 100 to 40% B (2.5 min), 40 to 10% B (1 min) and 10% B (1 min) with a flow-rate of 1 mL min−1 over 22 min.
Treatment of tobacco BY2 cells
Cell suspension culture was established and treated with 100 μM GSH. GSH-induced as well as control cells were harvested after 3, 6 and 12 h following treatment, centrifuged briefly to collect cells for total RNA isolation. Total RNA was isolated using Trizol reagent as mentioned earlier.
Semiquantitative and quantitative real-time RT-PCR analysis
Semiquantitative RT-PCR was carried out using 1 μg total RNA isolated from NtGB lines and wild-type plants and cDNA synthesized as described previously. PCR amplification was performed for different genes at different cycles of 94°C for 30 s, 30 s at varying annealing temperatures for different genes, 72°C for 1 min, with an initial denaturation at 94°C for 1 min. Primer pairs used for semiquantitative RT-PCR are listed in Supplementary Table S1. Actin was used as a loading control. The PCR products were analyzed by 1% agarose gel electrophoresis.
Quantitative RT-PCR was performed using Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, USA) with SYBR Premix Ex Taq (TaKaRa). Primer pairs are listed in Supplementary Table S1. Each PCR reaction contained 1× SYBR Premix Ex Taq, 0.05 nM of each primer, and 1 μL cDNA in a final volume of 20 μL. PCR amplification was performed for 40 cycles at 94°C, 30 s and 60°C, 2:30 min with a preceding initial denaturation of 30 s at 95°C. Relative expression levels were calculated by ∆-∆Ct method, and all quantifications were normalized using actin mRNA as an internal control.
Disease resistance assay of transgenic plants
Pseudomonas syringae pv. tabaci was grown up to an OD of 0.7, centrifuged and the cells were resuspended in 25 mL of 10 mM MgCl2 (Pageau et al. 2006). The cells were injected subepidermally in two leaves of similar position from each of five independent wild-type and NtGB lines. The infection was monitored up to 14 days post inoculation (dpi). Leaf discs were ground in 10 mM MgCl2, subjected to serial dilution and plated on 10 g L−1 yeast extract, 10 g L−1 peptone and 5 g L−1 NaCl containing 15 g L−1 agar (YEP) plates with rifampicin selection. For gene expression analysis, P. syringae pv. tabaci cells at particular ODs were used to infect wild-type and NtGB lines. Disease test with A. alternata (ITCC 6306) was performed according to Zhang et al. 2009a. Briefly, the fungus was grown for 7 days in petridishes containing potato dextrose agar (PDA) at 28°C in the dark. Agar plugs from actively growing culture of the fungus were excised and inverted on different positions of detached leaves and were placed on wet filter paper in petridishes and incubated at 28°C to permit normal disease development under high humidity. After 5 days, disease progression was checked, lesion diameter measured and photographed.
Western blotting
Proteins were extracted after homogenizing leaves in 50 mM potassium phosphate buffer, pH 7.8, containing 0.15% (v/v) Triton X-100 at 0°C. Protein samples were quantified by Bradford assay (Bradford 1976), using BSA as standard, resolved in 12% SDS-PAGE gels and transferred onto polyvinylidene difluoride membrane (Millipore, USA), blocked with 5% skimmed milk and gamma-glutamylcysteine synthetase (γ-ECS) protein bands were detected by using a rabbit polyclonal antibody raised against maize γ-ECS (Agrisera, Sweden) as the primary antibody and an anti-rabbit IgG conjugated to horseradish peroxidase (Sigma-Aldrich, USA) as the secondary antibody. Immunoreactive proteins were visualized using the SuperSignal West Pico (Pierce, USA) chemiluminescent reagent.
Protein extraction and 2-DE
Proteins from leaves of three biological replicates at similar positions of wild-type, NtGB9 and NtGB19 plants were extracted. Approximately 2.5 g of tissue samples was ground in liquid nitrogen and suspended in extraction buffer [700 mM Sucrose, 500 mM Tris–HCl, pH 7.5, 50 mM EDTA, 100 mM KCl, 2% (w/v) β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride] and protein extraction was done following the phenol extraction method (Isaacson et al. 2006). The resultant pellet was resuspended in 2-DE sample buffer consisting of 9 M Urea, 4% (w/v) CHAPS, 0.5% (v/v) Triton X-100, 20 mM dithiothreitol (DTT) and 1% (w/v) Bio-Lyte (3/10) ampholyte (BioRad Laboratories, Hercules, CA, USA) as standardized before (Sinha and Chattopadhyay 2010). The total protein concentration was analyzed by Bradford assay. 100 μg of total protein for each sample was used for passive rehydration of 7 cm IPG strip (pH 4–7; BioRad Laboratories, Hercules, CA, USA). Programmed IsoElectric Focusing (IEF) was performed with the Protean IEF cell (BioRad Laboratories, Hercules, CA, USA) using the following conditions: 250 V for 15 min, 4,000 V for 7 h, 500 V for 5 min. The focused strips were equilibrated twice in Equilibration Buffer I and II, respectively (BioRad Laboratories, Hercules, CA, USA), for 15 min each. The equilibrated strips were then subjected to the second dimension, which was carried out in SDS-PAGE in a vertical slab of 12% Acrylamide in BioRad Mini-PROTEAN Tetra Cell. After running for 35 min at 200 V, gels were stained with colloidal Coomasie Brilliant Blue (CBB) G-250 (Neuhoff et al. 1988).
Image and data analysis
The gel images were acquired using Versa-Doc Image system (BioRad Laboratories, Hercules, CA, USA) and image analysis was performed with PD Quest software version 8.0.1 (BioRad Laboratories, Hercules, CA, USA). Detection of spots was performed by matching the gels automatically, followed by manual verification. Protein spots were annotated only if detected in all gels after normalization of the spot densities against the whole gel densities; the percentage volume of each spot was averaged for nine different (three biological replicates of wild-type and NtGB lines) gels and statistical analysis was performed to find out significant protein fold changes between wild-type and NtGB lines.
Protein identification using MALDI TOF-TOF MS/MS
Selected protein spots were excised from 2-DE gels and subjected to in-gel digestion with trypsin following the manufacturer’s instructions (in-gel trypsin digestion kit, Pierce, USA). Digested proteins were further desalted with Zip-Tip μ-C18 (ZipTip, Millipore, USA) and analyzed using a 4800 MALDI TOF-TOF MS/MS analyzer (Applied Biosystems, Foster City, CA, USA). Peptides were evaporated with a ND:YAG laser at 355 nm, using a delayed extraction approach. They were accelerated with 25 kV injection pulse for TOF analysis. Each spectrum was the cumulative average of 1,000 laser shots. The MS/MS spectrum was collected in MS/MS 1 kV positive reflectron mode with fragments generated by post source decay (PSD). The MS/MS mass tolerance was set to ±20 ppm. After processing, ten MS/MS precursor was selected (Minimum signal to noise ratio-50). Before each analysis, the instrument was calibrated with the Applied Biosystems 4700 Proteomics Analyzer Calibration Mixture. Data interpretation was carried out using the GPS Explorer Software (Applied Biosystems), and an automated database search was carried out using the MASCOT program (Matrix Science Ltd., London, UK).
MS/MS data was used to perform protein identification by searching in a non-redundant protein sequence database (NCBI nr—20070216; 4626804 sequences, 1596079197 residues) using a MOWSE algorithm as implemented in the MASCOT search engine version 3.5 (Matrix science: http://www.matrixscience.com). The following parameters were used for database searches: taxonomy, viridiplantae (green plants; 186963 sequences); cleavage specificity, trypsin with one missed cleavages allowed; mass tolerance of 100 ppm for precursor ions and a tolerance of 0.2 da for the fragment ions; allowed modifications, carbamidomethyl (fixed), oxidation of Met (variable), cleavage by trypsin, cuts C-term side of KR unless next residue is P. According to MASCOT probability analysis, only significant hits (P < 0.05) were considered.
To evaluate the functional categories and hierarchies of identified proteins, KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/) was used (Kanehisa and Goto 2000). The theoretical peptide mass and pI of the polypeptides were evaluated at EXPASy (http://www.expasy.org/tools/pi_tool.html) (Bjellqvist et al. 1993; Gasteiger et al. 2005) for final confirmation according to their positions in the 2-DE gel map.
Statistical analysis
All experiments were repeated at least three times, unless otherwise stated, using plants grown independently. Significance of differences between data sets was evaluated by Student’s t test (5% significance, P < 0.05). Calculations were carried out with Microsoft Excel software.
Results
Characteristics of NtGB lines
GSH is synthesized in two steps in which γ-ECS catalyzes the first step and is known to be the rate-limiting enzyme (Hell and Bergmann 1990; May et al. 1998). The gene encoding L. esculentum γ-ECS was cloned into the binary vector pBI121 under the control of CaMV35S promoter (Fig. 1a) and used to transform tobacco (cv. Xanthi) leaves. Kanamycin-resistant and PCR positive transgenic plants constitutively expressing 35S::LeECS were selected. Independent T2 transgenic lines grown in greenhouse conditions were screened by PCR analysis (Fig. 1b). Semiquantitative RT-PCR analysis demonstrated that the levels of nptII and LeECS were increased in multiple T2 lines (Fig. 1c). Southern blot analysis confirmed the stable integration of LeECS in T2 lines (Supplementary Fig. S1a) and the LeECS protein was detected in significantly higher level in leaves of NtGB plants compared with that of wild-type plants which showed lesser reactivity (Fig. 1d). The growth and appearance of NtGB were almost the same as those of wild-type plants throughout the growth period under normal growth conditions (data not shown). These findings suggest that enhanced GSH content through the overexpression of LeECS is not directly involved with the phenotypic alterations (Noctor et al. 1998; Zhu et al. 1999; Gullner et al. 2001; Xiang et al. 2001; Gomez et al. 2004; Liedschulte et al. 2010). Furthermore, previous study showed that transgenic plants having 500- to 1,000-fold increased SA and SAG accumulation did not show any phenotypic variation but constitutively expressed PR genes (Verberne et al. 2000) which can be corroborated by our study.

Analysis of NtGB lines. a Construct design of 35S::LeECS that was inserted into the tobacco genome to constitutively express LeECS in NtGB lines. LeECS was cloned into the binary vector pBI121 at XbaI and SacI sites after removing the gus gene, under CaMV35S promoter and nos terminator. b PCR screening of nptII in T2 independently transformed NtGB plant lines grown in greenhouse conditions. Leaves from 6-week-old plants were used for analysis. c Semiquantitative RT-PCR analyses of wild-type and NtGB lines harboring LeECS. Expression of nptII and LeECS mRNA in different NtGB lines is shown. Actin was used as a loading control. d Western blot analysis showing the LeECS protein levels in wild-type and NtGB lines. γ-ECS protein bands were detected by western blotting using specific antibody
To determine the total glutathione content in NtGB9 and NtGB19 alongwith wild type, HPLC analysis was performed which revealed that the GSH content was 2.6- and 2.07-fold higher in NtGB9 and NtGB19, respectively (Supplementary Fig. S1b). These results clearly indicated that as a result of overexpression of LeECS in tobacco, GSH accumulation was enhanced in agreement with the findings reported previously (Noctor et al. 1998; Gomez et al. 2004). The GSH:GSSG ratio was 13.2 and 11.04 in NtGB9 and NtGB19, respectively, as compared with 8.8 in wild-type plant (Supplementary Fig. S1c). The leaves of NtGB lines were observed to accumulate more SA in comparison with that of wild-type (Supplementary Fig. S1d).
Transcript profiling in enhanced GSH condition
Gene expression profile of NtGB lines
To investigate the role of GSH in the signaling pathway crosstalk with SA, JA and ET in mitigating biotic stress, the expression profiles of selected genes involved with various defense-signaling pathways were taken into account. Experiments were also designed to understand the specific involvement of GSH with SA, and even if it is so, whether it follows NPR1-dependent or -independent pathway.
In this study, the expression level of NtNPR1 was higher in NtGB lines as compared with the wild-type plants. The NPR1-dependent homologues in tobacco were also found to be enhanced as shown in Fig. 2. PR1, a marker for SA-mediated plant defense (Blanco et al. 2009); GLS, which catalyzes the first reaction in the main path of ammonia assimilation in higher plants and known to be induced upon infection with P. syringae (Pérez-García et al. 1995); MAPKK, a mitogen-activated protein kinase kinase that interacts with SA-induced protein kinase in tobacco, which may play a role in SA signaling through NPR1 (Liu et al. 2000); TRXh, which further catalyzes an SA-induced NPR1 activation by oligomer-to-monomer conversion in Arabidopsis (Tada et al. 2008); and GSNOR1, that catalyzes the catabolism of GSNO which further facilitates oligomerization of NPR1 in Arabidopsis (Tada et al. 2008) were considerably enhanced at transcript level. However, the transcript level of NtICS1 was decreased in NtGB lines due to the suppression of its expression by nuclear NPR1 through a feedback loop, thus inhibiting the further elevation in the SA content in accordance with a recent report with Arabidopsis (Zhang et al. 2009b). Interestingly, the expressions of NPR1-independent genes like NtPR2, NtPR5 and NtSDRLP were unchanged or downregulated (Fig. 3a). These findings suggest the role of GSH in mitigating biotic stress, which is likely to be regulated through NPR1-dependent SA-mediated pathway. However, further pharmacological experimental evidences are required to confirm this.

Expression profile of NPR1-dependent SA-mediated pathway genes. Quantitative RT-PCR analysis was carried out to determine the expression levels of NPR1-dependent SA-mediated pathway. Actin was used as an internal control. Data are the mean ± SD for three individual experiments (n = 3) using plants grown independently

Effect of LeECS overexpression on transcript levels of NPR1-independent SA-mediated pathway alongwith JA and ET pathway genes. a Quantitative RT-PCR analyses of NPR1-independent SA-mediated pathway genes. b Expression levels of NtAOS (enzyme of the first step in the biosynthesis of JA), NtPR4 (ET marker) and NtACCOx (ET biosynthetic gene). Actin was used as an internal control. Data are the mean ± SD for three individual experiments (n = 3) using plants grown independently
To assess for any involvement of GSH with other signaling pathways viz. JA and ET, the expression pattern of allene oxide synthase (AOS) homolog in tobacco, an enzyme of JA biosynthetic pathway (Laudert and Weiler 1998) as well as NtPR4, the marker for ET signaling (Lawton et al. 1994) and ACC oxidase (ACCOx) that oxidizes ACC to ET was also monitored. The expression of NtAOS remained unchanged in NtGB lines, whereas, the expression level of NtPR4 and NtACCOx, in NtGB lines was fairly increased as compared with the wild-type plants (Fig. 3b).
Gene expression profile of GSH-fed tobacco BY2 cells
To gain further understanding of the mechanisms of how GSH is responsible for the expression of the aforementioned set of genes, an experiment was designed on exogenously GSH-fed tobacco BY2 cells at transcript level in a time-dependent manner and compared with in planta results. To get exact comparative profile, semiquantitative RT-PCR analysis was performed which reciprocated the gene expression level with NtGB lines (Fig. 4). Interestingly, in GSH-fed tobacco BY2 cells the expression of NtNPR1, NtPR1, NtGLS, NtMAPKK, NtTRXh, NtGSNOR1 and NtPR4 were upregulated, whereas NtPR5 and NtSDRLP were unchanged. NtICS1, NtPR2 and NtACCOx were not detected at all. However, the expression level of NtAOS was decreased with time (Fig. 4).

Changes in the transcript levels of NPR1-dependent and -independent SA-mediated pathway, alongwith JA and ET pathway genes by the overexpression of LeECS and in GSH-fed tobacco BY2 cells. Semiquantitative RT-PCR analyses were carried out to determine the expression levels of a NPR1-dependent b NPR1-independent SA-mediated pathway. c Expression levels of NtAOS, NtPR4 and NtACCOx. Actin was used as an internal control
Resistance patterns of NtGB lines to biotrophic P. syringae pv. tabaci and necrotrophic A. alternata
Whole plants of NtGB9 and NtGB19, exhibiting enhanced GSH contents, were infected with the biotrophic bacterial pathogen P. syringae pv. tabaci. On the other hand, necrotrophic fungus A. alternata was used to infect detached leaves of wild-type and NtGB lines. Enhanced resistance to P. syringae pv. tabaci was noted with NtGB lines in comparison with wild-type (Fig. 5a). In contrast, the disease progression of A. alternata-infected NtGB9 and NtGB19 was close to that of the wild type (Fig. 5b). These disease test assays support that SA-mediated defense response comes into play more prominently in NtGB lines.

Disease resistance analysis of NtGB lines against the biotrophic and necrotrophic pathogens, P. syringae pv. tabaci and A. alternata. a Reduction of P. syringae pv. tabaci infection in whole plants of NtGB lines as compared with wild-type. Bacterial suspension was injected subepidermally and disease scored after 14 days. Bacteria were extracted from leaf discs, diluted and spread on YEP medium for colony count. b A. alternata actively growing culture was applied on different positions of detached leaves of NtGB and wild-type plants. Disease progression and lesion size was noted at 5 dpi. Data are the mean ± SD for five individual experiments (n = 5) using plants grown independently
A time-course study of gene expression was performed using NtPR1, NtPR2, NtPR4 and NtPR5 after infecting wild-type and NtGB lines with P. syringae pv. tabaci. Gene expression analysis of plants infected with P. syringae pv. tabaci grown up to an OD of 0.05 was monitored up to 48 h (Supplementary Fig. S2).
Proteomic analysis under enhanced GSH condition
Altered proteome profile in NtGB lines
To gain an insight into the nature of protein involved during GSH-mediated biotic stress tolerance, proteins isolated from leaves of NtGB9, NtGB19 and that of wild-type plant were separated using 2-DE. Figure 6 represents gel images for both wild-type and NtGB samples in a pI range of 4–7. The results showed dramatic difference in global expression pattern of proteins in wild-type and NtGB lines. 48 spots were detected by statistical evaluation (Student’s t test, at 5% significance, P < 0.05) of relative spot densities. These spots were further categorized as induced, i.e., uniquely expressed, upregulated (above twofold) and downregulated (below twofold).

Comparison of proteome patterns of NtGB lines and wild-type. Representative 2-DE gels of proteins extracted from young leaves of three biological replicates at similar positions of wild-type, NtGB9 and NtGB19 6-week-old plants
Identification of differentially expressed proteins and functional classification
To identify the differentially expressed proteins, we used MALDI TOF-TOF MS/MS approach. The identified proteins for the corresponding 48 spots were sorted out into various categories. Protein functions were assigned according to KEGG and the functional categorization revealed that 38% of the differentially expressed proteins identified belong to the stress and defense category (Table 1; Figs. 7, 8). However, the classification of the protein is only tentative, since the biological function of many proteins identified has not yet been established experimentally.

Representative proteome profile of NtGB9 and wild type. a Close up of the area with spots of SA-related proteins that were induced in NtGB lines. b Close-up view of the SA-related protein spots that were upregulated in NtGB line in comparison with wild-type plants

Functional classification of a induced, b upregulated and c downregulated protein categories identified in NtGB lines
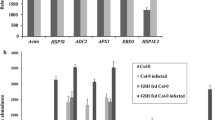
The expression levels of six SA-related genes were confirmed by quantitative RT-PCR. For example, NtPR10, calmodulin-binding protein (NtCBP), arginine decarboxylase (NtADC) and carbonic anhydrase (NtCA) were found to be upregulated. These genes were therefore found to be possibly regulated at the transcription level as compared with others like heat-shock protein (NtHsp70), the expression of which was nearly unchanged and alcohol dehydrogenase (NtADH), which was downregulated (Fig. 9).

Effect of LeECS overexpression on transcript levels of genes related to SA that were upregulated at the protein level in 2-DE in NtGB lines as a result of overexpression of LeECS. The experimental conditions were the same as in Fig. 4. Quantitative RT-PCR analyses were performed to determine the expression level of NtPR10, NtCBP, NtHsp70, NtADH, NtADC and NtCA. Actin was used as an internal control. Data are the mean ± SD for three individual experiments (n = 3) using plants grown independently
Discussion
Plant’s resistance to pathogen attack is the consequence of a complex regulatory network that connects the individual signaling pathways, enabling each to assist or antagonize the others (Grant and Jones 2009; Pieterse et al. 2009). The role of GSH in plant defense has long been known (Dron et al. 1988; Wingate et al. 1988) in addition to substantial numbers of recent reports. A growing body of evidence also supports the notion that interplay between GSH and various established signaling components leads to the establishment of disease resistance. Current studies have also implicated that GSH is one of the important players in biotic stress management through its interaction with various established messengers. These prompted us to develop NtGB lines exhibiting enhanced GSH content, stable integration and higher expression of LeECS and more resistance to the biotroph P. syringae pv. tabaci in comparison with the necrotroph A. alternata, without any phenotypic variation to wild-type plants.
According to previous studies, higher level of GSH due to the overexpression of γ-ECS either of bacterial or of plant origin did not show any distinguishable difference in visual appearance in comparison with the wild-type plants as noted in poplar, mustard, Arabidopsis or tobacco (Noctor et al. 1998; Gullner et al. 2001; Zhu et al. 1999; Xiang et al. 2001; Gomez et al. 2004; Liedschulte et al. 2010), which can be corroborated by our results. However, some reports represented variation in phenotype as a result of overexpression of γ-ECS in tobacco and poplar, respectively (Creissen et al. 1999; Herschbach et al. 2000).
A recent report has shown that GSH status regulates SA and other pathways involved in biotic stress responses at several levels in Arabidopsis, including SA-accumulation and JA-linked gene expression (Mhamdi et al. 2010). Ball et al. (2004) reported that 32 stress-responsive genes were altered due to changed GSH metabolism in Arabidopsis rax1-1 and cad2-1, mutants of γ-ECS. Additionally, previous studies also reported that Arabidopsis pad2-1 mutant with only 22% of wild-type amounts of GSH were susceptible to P. syringae as well as Phytophthora brassicae (Glazebrook and Ausubel 1994; Glazebrook et al. 1997; Parisy et al. 2007). May et al. (1996) demonstrated a considerable accumulation of GSH and GSSG in tomato cells after treatment with race-specific elicitors of the fungus Cladosporium fulvum. Enhanced resistance to Fusarium oxysporum due to increase in GSH level has been revealed in melon and tomato roots (Bolter et al. 1993). In another study, constitutive overexpression of SA was shown to induce GSH-mediated nickel tolerance in Thlaspi hyperaccumulators (Freeman et al. 2005). In this investigation, disease resistance tests revealed that NtGB lines showed considerable resistance to P. syringae pv. tabaci; however, in case of A. alternata, disease progression was observed to be almost similar to wild type. Therefore, it can be deduced that resistance of NtGB lines was effective against biotrophic pathogens, which involve SA (Loake and Grant 2007).
With a view to obtain further insight into the specific interactions of GSH with other established messengers, the expression profile of selected marker/pathway genes of the SA, JA and ET was investigated in transgenic T2 generation of NtGB lines. This was further validated in 100 μM GSH-fed tobacco BY2 cells as well. Previous finding showed that the intensity of NtNPR1 transcript was approximately twofold greater in the INA/SA induced samples (Cao et al. 1997). In corroboration with this, our study revealed significant upregulation of NtNPR1 in NtGB lines as compared with wild type. Furthermore, onset of NtNPR1 transcript accumulation was noted at 3 h, which was enhanced potentially up to 12 h in a time-dependent manner in GSH-fed BY2 cells. Interestingly, our results also demonstrated the enhanced expression of NtPR1, the marker gene of NPR1-dependent SA-mediated pathway, alongwith some other genes of the same category like NtGLS, NtMAPKK, NtTRXh, NtGSNOR1 and NtICS1. However, SDRLP reported to be a NPR1-independent SA-induced gene in Arabidopsis (Blanco et al. 2009) showed similar level of expression to that of wild type in our study. It has been observed that during attack with PsmES4326 the PR1 expression levels in Arabidopsis npr1-2 and npr1-3 mutants were greatly reduced but that of PR2 and PR5 were not significantly affected (Glazebrook et al. 1996), which can be correlated well with our study. Similarly in another study, the expression of NAD(P)-induced PR2 and PR5 in Arabidopsis npr1-1 mutant was not affected (Zhang and Mou 2009). Similarly, the transcript levels of NPR1-independent PR2 and PR5 in both overexpressor and disruptant of ADP-ribose/NADH pyrophosphohydrolase were noted to be mostly similar to control plants (Ishikawa et al. 2010). However, in Arabidopsis npr1-1 as well as in sai1 mutant, which is allelic to npr1, neither PR1 nor PR2 and PR5 were expressed as a result of SA treatment (Cao et al. 1994; Shah et al. 1997).
Additionally, to catch a more dynamic picture of how GSH interacts with other signaling molecules, the gene expression profiles of NtPR1, NtPR2, NtPR4 and NtPR5 were determined in P. syringae pv. tabaci-infected NtGB lines with respect to time, which eventually exhibited variable expression pattern. The increased expression profiles of NtPR1 and NtPR4 with time after pathogen infection can be corroborated with previous studies in Arabidopsis and Chinese cabbage, respectively (Wildermuth et al. 2001; Du et al. 2009; Ryang et al. 2002). However, in infected condition the change in the expression level of NtPR2 and NtPR5 was not that impressive. Previous reports have variable views on the coordinated regulation between PR1, PR2 and PR5. Some studies showed that they are coordinately regulated (Uknes et al. 1992; Görlach et al. 1996; Cao et al. 1994; Delaney et al. 1995; Shah et al. 1997; Bowling et al. 1997; Clarke et al. 1998). In contrast, some other reports hinted an SA-independent PR2/PR5 expression after inoculation with P. syringae (Glazebrook et al. 1996; Rogers and Ausubel 1997; Zhou et al. 1998; Nawrath and Métraux 1999). Further, a recent study in Arabidopsis demonstrated that PR2 and PR5 did not show any induction after P. syringae infection while PR1 was strongly induced in enhanced SA condition (Beak et al. 2010). Thus, to clarify further the mode and path of action of PR2 and PR5, which still is a mystery, needs further investigation.
ET is known to enhance SA/NPR1-dependent defense responses and potentiates SA/NPR1-dependent PR1 transcription and also GSH content is known to be related in some way or the other to ET (de Vos et al. 2006; Yoshida et al. 2009). In our investigation, the expression levels of NtPR4, as well as NtACCOx, were also noted to be upregulated in NtGB lines. However, there was no change in expression of NtAOS.
It is worthwhile to mention that about 38% of induced and upregulated proteins belonged to the category of stress and defense as revealed from functional classification of the differentially expressed proteins. Many of the proteins identified here were reported previously to be induced/accumulated through SA-mediated pathway. Out of these, calcineurin, identified in our study, is a calcium sensor protein and is involved in stress and calcium signaling in plants. Transcript levels of calcineurin B-like proteins have been found to be upregulated in response to various stresses including SA in Pisum sativum (Tuteja and Mahajan 2007). Most PRs and related proteins are induced through the action of the signaling compounds SA, JA or ET (van Loon et al. 2006). Here, in NtGB lines, the pathogenesis-related protein, PR10, was identified in induced category. PR10 is known to be induced by acibenzolar-S-methyl (ASM), a functional analog of SA (Ziadi et al. 2001). Calmodulin regulates SA-mediated plant immunity (Du et al. 2009). The protein identified here was calmodulin-binding protein, which enhances the ability of calmodulin to bind Ca2+ and activate other enzymes (Zielinski 1998). Other than these, KR4 having conserved motifs of R gene products such as NBS and LRR domains was induced by exogenous SA (Wang et al. 2004) and was also identified in our study. In addition, SA enhances heat-induced Hsp/Hsc70 accumulation in plants (Snyman and Cronjé 2008), which can be correlated with the present study. In addition, SA-induced accumulation of ADH transcripts in Solanum tuberosum (Matton et al. 1990) and significant increase in the expression of ADC gene as a result of SA treatment (Jang et al. 2009) were reported previously which was identified at protein level in this study. CA is the most abundant soluble zinc-containing protein in the chloroplast of plant. It has been reported earlier that tobacco chloroplast CA acts as a SA-binding protein like SABP3, which was isolated from stroma of chloroplast (Slaymaker et al. 2002) and also identified in our study. Legume lectin protein was reported as NPR1-dependent SA-binding protein (Blanco et al. 2009), which can be correlated with our data.
In conclusion, the present study demonstrates that biotic stress tolerance against biotrophic P. syringae pv. tabaci is more effective than necrotophic A. alternata in NtGB lines exhibiting enhanced GSH content. Comprehensive analysis of transcriptomic and proteomic data derived from our study suggests that GSH plays an important role to mitigate biotic stress in induced disease resistance, which probably occurs through NPR1-dependent SA-mediated pathway. This knowledge provides further insight into this expanding area on the involvement of GSH in the cross-communication with other established signaling molecules in defense response.
Abbreviations
- ET:
-
Ethylene
- GSH:
-
Glutathione
- JA:
-
Jasmonic acid
- NPR1:
-
Non-expressor of pathogenesis-related genes 1
- PR:
-
Pathogenesis-related protein gene
- ROS:
-
Reactive oxygen species
- SA:
-
Salicylic acid
References
Ball L, Accotto GP, Bechtold U, Creissen G, Funck D, Jimenez A, Kular B, Leyland N, Mejia-Carranza J, Reynolds H, Karpins S, Mullineaux PM (2004) Evidence for a direct link between glutathione biosynthesis and stress defense gene expression in Arabidopsis. Plant Cell 16:2448–2462
Banerjee A, Chattopadhyay S (2010) Effect of over-expression of Linum usitatissimum PINORESINOL LARICIRESINOL REDUCTASE (LuPLR) gene in transgenic Phyllanthus amarus. Plant Cell Tissue Organ Cult 103:315–323
Beak D, Pathange P, Chung JS, Jiang J, Gao L, Oikawa A, Hirai MY, Saito K, Pare PW, Shi H (2010) A stress-inducible sulphotransferase sulphonates salicylic acid and confers pathogen resistance in Arabidopsis. Plant Cell Environ 33:1383–1392
Bjellqvist B, Hughes GJ, Pasquali C, Paquet N, Ravier F, Sanchez JC, Frutiger S, Hochstrasser D (1993) The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 14:1023–1031
Blanco F, Salinas P, Cecchini NM, Jordana X, Van Hummelen P, Alvarez ME, Holuigue L (2009) Early genomic responses to salicylic acid in Arabidopsis. Plant Mol Biol 70:79–102
Bolter C, Brammall RA, Cohen R, Lazarovits G (1993) Glutathione alterations in melon and tomato roots following treatment with chemicals which induce disease resistance to Fusarium wilt. Physiol Mol Plant Pathol 42:321–336
Bowling SA, Clarke JD, Liu Y, Klessig DF, Dong X (1997) The cpr5 mutant of Arabidopsis expresses both NPR1-dependent and NPR1-independent resistance. Plant Cell 9:1573–1584
Bradford M (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Bradley DJ, Kjellbom P, Lamb CJ (1992) Elicitor- and wound-induced oxidative cross-linking of a proline-rich plant cell wall protein: a novel, rapid defense response. Cell 70:21–30
Cao H, Bowling SA, Gordon AS, Dong X (1994) Characterization of an Arabidopsis mutant that is nonresponsive to inducers of systemic acquired resistance. Plant Cell 6:1583–1592
Cao H, Glazebrook J, Clarke JD, Volko S, Dong X (1997) The Arabidopsis NPR1 gene that controls systemic acquired resistance encodes a novel protein containing ankyrin repeats. Cell 88:57–63
Chen Z, Silva H, Klessig DF (1993) Active oxygen species in the induction of plant systemic acquired resistance by salicylic acid. Science 262:1883–1886
Clarke JD, Liu Y, Klessig DF, Dong X (1998) Uncoupling PR gene expression from NPR1 and bacterial resistance: characterization of the dominant Arabidopsis cpr6-1 mutant. Plant Cell 10:557–569
Creissen G, Firmin J, Fryer M, Kular B, Leyland N, Reynolds H, Pastori G, Wellburn F, Baker N, Wellburn A, Mullineaux P (1999) Elevated glutathione biosynthetic capacity in the chloroplasts of transgenic tobacco plants paradoxically causes increased oxidative stress. Plant Cell 11:1277–1291
De Vos M, Van Zaanen W, Koornneef A, Korzelius JP, Dicke M, Van Loon LC, Pieterse CMJ (2006) Herbivore-induced resistance against microbial pathogens in Arabidopsis. Plant Physiol 142:352–363
Delaney T, Friedrich L, Ryals J (1995) Arabidopsis signal transduction mutant defective in chemically and biologically induced disease resistance. Proc Natl Acad Sci USA 92:6602–6606
Després C, Chubak C, Rochon A, Clark R, Bethune T, Desveaux D, Fobert PR (2003) The Arabidopsis NPR1 disease resistance protein is a novel cofactor that confers redox regulation of DNA binding activity to the basic domain/leucine zipper transcription factor TGA1. Plant Cell 15:2181–2191
Dron M, Clouse SD, Dixon RA, Lawton MA, Lamb CJ (1988) Glutathione and fungal elicitor regulation of a plant defense gene promoter in electroporated protoplasts. Proc Natl Acad Sci USA 85:6738–6742
Du L, Ali GS, Simons KA, Hou J, Yang T, Reddy ASN, Poovaiah BW (2009) Ca2+/calmodulin regulates salicylic-acid-mediated plant immunity. Nature 457:1154–1158
El-Zahaby HM, Gullner G, Kiraly Z (1995) Effects of powdery mildew infection of barley on the ascorbate–glutathione cycle and other antioxidants in different host–pathogen interactions. Phytopathol 85:1225–1230
Foyer CH, Lopez-Delgado H, Dat JF, Schott IM (1997) Hydrogen peroxide- and glutathione-associated mechanisms of acclamatory stress tolerance and signalling. Physiol Plant 100:241–254
Freeman JL, Garcia D, Kim D, Hopf A, Salt DE (2005) Constitutively elevated salicylic acid signals glutathione-mediated nickel tolerance in Thlaspi nickel hyperaccumulators. Plant Physiol 137:1082–1091
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the expasy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, pp 571–607
Glazebrook J (2001) Genes controlling expression of defense responses in Arabidopsis: 2001 status. Curr Opin Plant Biol 4:301–308
Glazebrook J (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 43:205–227
Glazebrook J, Ausubel FM (1994) Isolation of phytoalexin-deficient mutants of Arabidopsis thaliana and characterization of their interactions with bacterial pathogens. Proc Natl Acad Sci USA 91:8955–8959
Glazebrook J, Rogers EE, Ausubel FM (1996) Isolation of Arabidopsis mutants with enhanced disease susceptibility by direct screening. Genetics 143:973–982
Glazebrook J, Zook M, Merrit F, Kagan I, Rogers EE, Crute IR, Holub EB, Hammerschmidt R, Ausubel FM (1997) Phytoalexin-deficient mutants of Arabidopsis reveal that PAD4 encodes a regulatory factor and that four PAD genes contribute to downy mildew resistance. Genetics 146:381–392
Gomez LD, Noctor G, Knight MR, Foyer CH (2004) Regulation of calcium signaling and gene expression by glutathione. J Exp Bot 55:1851–1859
Görlach J, Volrath S, Knauf-Beiter G, Hengy G, Beckhove U, Kogel KH, Oostendorp M, Staub T, Ward E, Kessmann H, Ryals J (1996) Benzothiadiazole, a novel class of inducers of systemic acquired resistance, activates gene expression and disease resistance in wheat. Plant Cell 8:629–643
Grant M, Jones J (2009) Hormone (dis)harmony moulds plant health and disease. Science 324:750–752
Gullner G, Komives T, Rennenberg H (2001) Enhanced tolerance of transgenic poplar plants overexpressing γ-glutamylcysteine synthetase towards chloroacetanilide herbicides. J Exp Bot 52:971–979
Hell R, Bergmann L (1990) γ-Glutamylcysteine synthetase in higher plants: catalytic properties and subcellular localization. Planta 180:603–612
Herschbach C, van der Zalm E, Schneider A, Jouanin L, De Kok LJ, Rennenberg H (2000) Regulation of sulfur nutrition in wild-type and transgenic poplar over-expressing γ-glutamylcysteine synthetase in the cytosol as affected by atmospheric H2S. Plant Physiol 124:461–473
Isaacson T, Damasceno CMB, Saravanan RS, He Y, Catalá C, Saladié M, Rose JKC (2006) Sample extraction techniques for enhanced proteomic analysis of plant tissues. Nat Protoc 1:769–774
Ishikawa K, Yoshimura K, Harada K, Fukusaki E, Ogawa T, Tamoi M, Shigeoka S (2010) AtNUDX6, an ADP-ribose/NADH pyrophosphohydrolase in Arabidopsis, positively regulates NPR1-dependent salicylic acid signalling. Plant Physiol 152:2000–2012
Jang EK, Min KH, Kim SH, Nam SH, Zhang S, Kim YC, Cho BH, Yang KY (2009) Mitogen-activated protein kinase cascade in the signaling for polyamine biosynthesis in tobacco. Plant Cell Physiol 50:658–664
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kocsy G, Szalai G, Vagujfalvi A, Stehli L, Orosz G, Galiba G (2000) Genetic study of glutathione accumulation during cold hardening in wheat. Planta 210:295–301
Kumar A, Chakraborty A, Ghanta S, Chattopadhyay S (2009) Agrobacterium-mediated genetic transformation of mint with E. coli glutathione synthetase gene. Plant Cell Tissue Organ Cult 96:117–126
Laudert D, Weiler EW (1998) Allene oxide synthase: a major control point in Arabidopsis thaliana octadecanoid signalling. Plant J 15:675–684
Lawton KA, Potter SL, Uknes S, Ryals J (1994) Acquired resistance signal transduction in Arabidopsis is ethylene independent. Plant Cell 6:581–590
Leon-Reyes A, Van der Does D, De Lange ES, Delker C, Wasternack C, Van Wees SCM, Ritsema T, Pieterse CMJ (2010) Salicylate-mediated suppression of jasmonate-responsive gene expression in Arabidopsis is targeted downstream of the jasmonate biosynthesis pathway. Planta 232:1423–1432
Liedschulte V, Wachter A, Zhigang A, Rausch T (2010) Exploiting plants for glutathione (GSH) production: uncoupling GSH synthesis from cellular controls results in unprecedented GSH accumulation. Plant Biotechnol J 8:1–14
Liu Y, Zhang S, Klessig DF (2000) Molecular cloning and characterization of a tobacco MAP kinase kinase that interacts with SIPK. Mol Plant Microbe Interact 13:118–124
Loake G, Grant M (2007) Salicylic acid in plant defence—the players and protagonists. Curr Opin Plant Biol 10:466–472
Matton DP, Constabel P, Brisson N (1990) Alcohol dehydrogenase gene expression in potato following elicitor and stress treatment. Plant Mol Biol 14:775–783
May MJ, Hammond-Kosack KE, Jones JDG (1996) Involvement of reactive oxygen species, glutathione metabolism, and lipid peroxidation in the Cf-gene-dependent defense response of tomato cotyledons induced by race-specific elicitors of Cladosporium fulvum. Plant Physiol 110:1367–1379
May MJ, Vernoux T, Leaver C, Van Montagu M, Inzé D (1998) Glutathione homeostasis in plants: implications for environmental sensing and plant development. J Exp Bot 49:649–667
Mhamdi A, Hager J, Chaouch S, Queval G, Han Y, Taconnat L, Saindrenan P, Gouia H, Issakidis-Bourguet E, Renou JP, Noctor G (2010) Arabidopsis GLUTATHIONE REDUCTASE 1 plays a crucial role in leaf responses to intracellular H2O2 and in ensuring appropriate gene expression through both salicylic acid and jasmonic acid signaling pathways. Plant Physiol 153:1144–1160
Mou Z, Fan W, Dong X (2003) Inducers of plant systemic acquired resistance regulate NPR1 function through redox changes. Cell 27:935–944
Mur LAJ, Kenton P, Atzorn R, Miersch O, Wasternack C (2006) The outcomes of concentration-specific interactions between salicylate and jasmonate signaling include synergy, antagonism, and oxidative stress leading to cell death. Plant Physiol 140:249–262
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Nawrath C, Métraux JP (1999) Salicylic acid induction–deficient mutants of Arabidopsis express PR-2 and PR-5 and accumulate high levels of camalexin after pathogen inoculation. Plant Cell 11:1393–1404
Neuhoff V, Arold N, Taube D (1988) Improved staining of proteins in polyacrylamide gels including isoelectric-focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9:255–262
Noctor G, Arisi ACM, Jouanin L, Foyer CH (1998) Manipulation of glutathione and amino acid biosynthesis in the chloroplast. Plant Physiol 118:471–482
Pageau K, Reisdorf-Cren M, Morot-Gaudry JF, Masclaux-Daubresse C (2006) The two senescence-related markers GS1 (cytosolic glutamine synthetase) and GDH (glutamate dehydrogenase), involved in nitrogen mobilisation are differentially regulated during pathogen attack, by stress hormones and reactive oxygen species in Nicotiana tabacum L. leaves. J Exp Bot 57:547–557
Parisy V, Poinssot B, Owsianowski L, Buchala A, Glazebrook J, Mauch F (2007) Identification of PAD2 as a γ-glutamylcysteine synthetase highlights the importance of glutathione in disease resistance of Arabidopsis. Plant J 49:159–172
Pasternak M, Lim B, Wirtz M, Hell R, Cobbett CS, Meyer AJ (2008) Restricting glutathione biosynthesis to the cytosol is sufficient for normal plant development. Plant J 53:999–1012
Pérez-García A, Cánovas FM, Gallardo F, Hirel B, de Vicente A (1995) Differential expression of glutamine synthetase isoforms in tomato detached leaflets infected with Pseudomonas syringae pv. tomato. Mol Plant Microbe Interact 8:96–103
Pieterse CMJ, Van Loon LC (2004) NPR1: the spider in the web of induced resistance signaling pathways. Curr Opin Plant Biol 7:456–464
Pieterse CMJ, Leon-Reyes A, Van der Ent S, Van Wees SCM (2009) Networking by small-molecule hormones in plant immunity. Nat Chem Biol 5:308–316
Rogers EE, Ausubel FM (1997) Arabidopsis enhanced disease susceptibility mutants exhibit enhanced susceptibility to several bacterial pathogens and alterations in PR-1 gene expression. Plant Cell 9:305–316
Ruiz JM, Blumwald E (2002) Salinity-induced glutathione synthesis in Brassica napus. Planta 214:965–969
Ryang SH, Chung SY, Lee SH, Cha JS, Kim HY, Cho TJ (2002) Isolation of pathogen-induced Chinese cabbage genes by subtractive hybridization employing selective adaptor ligation. Biochem Biophys Res Commun 299:352–359
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Shah J, Tsui F, Klessig DF (1997) Characterization of a salicylic acid–insensitive mutant (sai1) of Arabidopsis thaliana, identified in a selective screen utilizing the SA-inducible expression of the tms2 gene. Mol Plant Microbe Interact 10:69–78
Sinha R, Chattopadhyay S (2010) Changes in the leaf proteome profile of Mentha arvensis in response to Alternaria alternata infection. J Prot. doi:10.1016/j.jprot.2010.11.009
Slaymaker DH, Navarre DA, Clark D, del Pozo O, Martin GB, Klessig DF (2002) The tobacco salicylic acid-binding protein 3 (SABP3) is the chloroplast carbonic anhydrase, which exhibits antioxidant activity and plays a role in the hypersensitive defense response. Proc Natl Acad Sci USA 99:11640–11645
Snyman M, Cronjé MJ (2008) Modulation of heat shock factors accompanies salicylic acid-mediated potentiation of Hsp70 in tomato seedlings. J Exp Bot 59:2125–2132
Spoel SH, Koornneef A, Claessens SMC, Korzelius JP, Van Pelt JA, Mueller MJ, Buchala AJ, Me′traux J-P, Brown R, Kazan K, Van Loon LC, Dong X, Pieterse CMJ (2003) NPR1 modulates crosstalk between salicylate- and jasmonate-dependent defense pathways through a novel function in the cytosol. Plant Cell 15:760–770
Tada T, Spoel SH, Pajerowska-Mukhtar K, Mou Z, Song J, Wang C, Zuo J, Dong X (2008) Plant immunity requires conformational changes of NPR1 via S-nitrosylation and thioredoxins. Science 321:952–956
Thomma B, Eggermont K, Penninckx I, Mauch-Mani B, Vogelsang R, Cammue B, Broekaert W (1998) Separate jasmonate-dependent and salicylate-dependent defense-response pathways in Arabidopsis are essential for resistance to distinct microbial pathogens. Proc Natl Acad Sci USA 95:15107–15111
Tsakraklides G, Martin M, Chalam R, Tarczynski MC, Schmidt A, Leustek T (2002) Sulfate reduction is increased in transgenic Arabidopsis thaliana expressing 5′-adenylylsulfate reductase from Pseudomonas aeruginosa. Plant J 32:879–889
Tuteja N, Mahajan S (2007) Further characterization of calcineurin B-like protein and its interacting partner CBL-interacting protein kinase from Pisum sativum. Plant Signal Behav 2:358–361
Uknes S, Mauch-Mani B, Moyer M, Potter S, Williams S, Dincher S, Chandler D, Slusarenko A, Ward E, Ryals J (1992) Acquired resistance in Arabidopsis. Plant Cell 4:645–656
Van Loon LC, Rep M, Pieterse CMJ (2006) Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol 44:135–162
Van Wees SCM, De Swart EAM, Van Pelt JA, Van Loon LC, Pieterse CMJ (2000) Enhancement of induced disease resistance by simultaneous activation of salicylate- and jasmonate-dependent defense pathways in Arabidopsis thaliana. Proc Natl Acad Sci USA 97:8711–8716
Verberne MC, Verpoorte R, Bol JF, Mercado-Blanco J, Linthorst HJM (2000) Overproduction of salicylic acid in plants by bacterial transgenes enhances pathogen resistance. Nat Biotechnol 18:779–783
Wang B, Wang Y, Wang Q, Luo G, Zhang Z, He C, He SJ, Zhang J, Gai J, Chen S (2004) Characterization of an NBS-LRR resistance gene homologue from soybean. J Plant Physiol 161:815–822
Wildermuth MC, Dewdney J, Wu G, Ausubel FM (2001) Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 414:562–571
Wingate VMP, Lawton MA, Lamb CJ (1988) Glutathione causes a massive and selective induction of plant defense genes. Plant Physiol 87:206–210
Xiang C, Werner BL, Christensen EM, Oliver DJ (2001) The biological functions of glutathione revisited in Arabidopsis transgenic plants with altered glutathione levels. Plant Physiol 126:564–574
Yoshida S, Tamaoki M, Ioki M, Ogawa D, Sato Y, Aono M, Kubo A, Saji S, Saji H, Satoh S, Nakajima N (2009) Ethylene and salicylic acid control glutathione biosynthesis in ozone-exposed Arabidopsis thaliana. Physiol Plant 136:284–298
Zhang X, Mou Z (2009) Extracellular pyridine nucleotides induce PR gene expression and disease resistance in Arabidopsis. Plant J 57:302–312
Zhang YL, Fan WH, Kinkema M, Li X, Dong X (1999) Interaction of NPR1 with basic leucine zipper protein transcription factors that bind sequences required for salicylic acid induction of the PR-1 gene. Proc Natl Acad Sci USA 96:6523–6528
Zhang G, Chen M, Li L, Xu Z, Chen X, Guo J, Ma Y (2009a) Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. J Exp Bot 60:3781–3796
Zhang X, Chen S, Mou Z (2009b) Nuclear localization of NPR1 is required for regulation of salicylate tolerance, isochorismate synthase 1 expression and salicylate accumulation in Arabidopsis. J Plant Physiol 167:144–148
Zhou N, Tootle TL, Tsui F, Klessig DF, Glazebrook J (1998) PAD4 functions upstream from salicylic acid to control defense responses in Arabidopsis. Plant Cell 10:1021–1030
Zhou JM, Trifa Y, Silva H, Pontier D, Lam E, Shah J, Klessig DF (2000) NPR1 diffrerentially interacts with members of the TGA/OBF family of transcription factors that bind an element of the PR-1 gene required for induction by salicylic acid. Mol Plant Microbe Interact 13:191–202
Zhu YL, Pilon-Smits EAH, Tarun AS, Weber SU, Jouanin L, Terry N (1999) Cadmium tolerance and accumulation in Indian mustard is enhanced by overexpressing γ-glutamylcysteine synthetase. Plant Physiol 121:1169–1177
Ziadi S, Poupard P, Brisset M, Paulin JP, Simoneau P (2001) Characterization in apple leaves of two subclasses of PR-10 transcripts inducible by acibenzolar-S-methyl, a functional analogue of salicylic acid. Physiol Mol Plant Pathol 59:33–43
Zielinski RE (1998) Calmodulin and calmodulin-binding proteins in plants. Annu Rev Plant Physiol Plant Mol Biol 49:697–725
Acknowledgments
We gratefully acknowledge Prof. Jack M. Widholm, Department of Crop Science, University of Illinois, USA for providing us seeds of Nicotiana tabacum cv. Xanthi, Dr. Geoffrey Duby, Université Catholique de Louvain, Belgium for providing tobacco BY2 cells and Prof. Siddhartha Roy for sharing his expertise regarding the application of 2-DE and its analysis. We acknowledge the help and support provided by Dr. Maitreyee Banerjee and the staffs of Greenhouse, West Bengal State Council of Science and Technology, Salt Lake, Kolkata, India, throughout the study period. This work was supported by the Department of Science and Technology (DST), New Delhi, India, and partly by the Council of Scientific and Industrial Research (CSIR), New Delhi, India. Research activities by Srijani Ghanta, Dipto Bhattacharyya, Ragini Sinha and Anindita Banerjee have been supported by fellowships from CSIR, New Delhi, India.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ghanta, S., Bhattacharyya, D., Sinha, R. et al. Nicotiana tabacum overexpressing γ-ECS exhibits biotic stress tolerance likely through NPR1-dependent salicylic acid-mediated pathway. Planta 233, 895–910 (2011). https://doi.org/10.1007/s00425-011-1349-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-011-1349-4