
Abstract
Amidase 1 (AMI1) from Arabidopsis thaliana converts indole-3-acetamide (IAM), into indole-3-acetic acid (IAA). AMI1 is part of a small isogene family comprising seven members in A. thaliana encoding proteins which share a conserved glycine- and serine-rich amidase-signature. One member of this family has been characterized as an N-acylethanolamine-cleaving fatty acid amidohydrolase (FAAH) and two other members are part of the preprotein translocon of the outer envelope of chloroplasts (Toc complex) or mitochondria (Tom complex) and presumably lack enzymatic activity. Among the hitherto characterized proteins of this family, AMI1 is the only member with indole-3-acetamide hydrolase activity, and IAM is the preferred substrate while N-acylethanolamines and oleamide are not hydrolyzed significantly, thus suggesting a role of AMI1 in auxin biosynthesis. Whereas the enzymatic function of AMI1 has been determined in vitro, the subcellular localization of the enzyme remained unclear. By using different GFP-fusion constructs and an A. thaliana transient expression system, we show a cytoplasmic localization of AMI1. In addition, RT-PCR and anti-amidase antisera were used to examine tissue specific expression of AMI1 at the transcriptional and translational level, respectively. AMI1-expression is strongest in places of highest IAA content in the plant. Thus, it is concluded that AMI1 may be involved in de novo IAA synthesis in A. thaliana.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Although indole-3-acetic acid (IAA) is one of the most prominent plant growth promoting substances with well-characterized functions in the regulation of nearly every step in plant development, knowledge about the molecular mechanisms of IAA action and its synthesis remain fragmentary. Several biosynthetic routes of de novo biosynthesis of IAA in plants have been proposed, but to the present day no complete in vivo pathway has been proven beyond doubt or established on the genetic level (Woodward and Bartel 2005).
Several lines of evidence led to the suggestion that one possible IAA-biosynthetic pathway may operate from l-tryptophan (l-Trp) via indole-3-acetamide (IAM) to IAA (Pollmann et al. 2002). Earlier reports suggested the occurrence of IAM in higher plants like rice (Kawaguchi et al. 1991), Poncirus trifoliata (Kawaguchi et al. 1993), or Prunus jamasakura (Saotome et al. 1993), but used nonsterile plant material and thus did not exclude plant-associated microorganisms as the source of the compound. However, recently, the occurrence of IAM as an endogenous compound has been proven in leaves of sterile-grown Arabidopsis thaliana plants (Pollmann et al. 2002). Proof for IAM as a substrate of enzymatic conversion in this tissue came from the cloning of a specific IAM-hydrolase (AMI1) from the same species and the demonstration of AMI1 expression in leaves using RT-PCR (Pollmann et al. 2003). Thus, a pathway leading from l-Trp via IAM to IAA, hitherto known only from bacteria such as Agrobacterium tumefaciens (Weiler and Schröder 1987), must be considered in higher plants. Nevertheless, the subcellular localization and tissue specific expression pattern of AMI1 remained unclear.
In this study we report that AMI1 is part of a small enzyme family in Arabidopsis, members of which are characterized by an amidase-signature motif localized within a stretch of approximately 130 conserved amino acids. This amidase-signature has earlier been described for enzymes from mammals (Patricelli and Cravatt 2000) and bacteria (Shin et al. 2002). The A. thaliana amidase-signature family comprises seven different members each encoded by a different gene from which only two proteins, AMI1 (Pollmann et al. 2003) and a fatty acid amide hydrolase (FAAH; Shrestha et al. 2003), were characterized for their enzymatic activity. The third isoform (At5g09420) seems to be localized in the outer mitochondrial membrane in complex with a protein translocase (Chew et al. 2004) and a fourth one (At3g17970) has been found associated with proteins of the chloroplast Toc-complex after protein cross-linking (Sohrt and Soll 2000). No information is available so far about the function and expression of the other three polypeptides.
In order to elucidate the intracellular location(s) of AMI1, AMI1:GFP-fusion proteins were transiently expressed from their chimeric genes, and the localization of the fusion proteins was detected by confocal laser scanning microscopical imaging of GFP-fluorescence. In addition, we analyzed the tissue specific expression of AMI1 by RT-PCR as well as by Western-blot analysis. Moreover, additional experiments comparing the enzymatic activities of recombinant AMI1 and FAAH (At5g64440) provide insight into the completely different substrate preferences of these two polypeptides and thus the high specificity of the two enzymes.
Materials and methods
Plant material
If not stated otherwise, all experiments were carried out using A. thaliana (L.) Heynh. Col-0 (originally from Nottingham Arabidopsis Stock Centre, NASC, stock N1092). All plant organs were harvested from plants grown in a greenhouse on a mixture of soil and sand (2:1) for 4–5 weeks in short days (8-h photoperiod). Thereafter, plants were transferred to long day conditions (16-h photoperiod). The greenhouse was maintained under constant climatic conditions, 22–24°C during daytime and 18–20°C over night. The photosynthetically active radiation (PAR) was no less than 150 μmol photons m−2 s−1 (supplementary light, if required, from sodium-vapor lamps).
Generation of plant expression vectors for fluorescence analysis
All molecular techniques were performed using standard protocols according to Sambrook et al. (1989) or Ausubel et al. (2000). To generate vectors for fluorescence analysis in transiently transformed plant cells, the expression cassettes containing the genes of the two fluorescent proteins, EGFP and DsRed were excised with HindIII/EcoRI from the vectors pFF19-GFP and pFF19-RFP (Wachter et al. 2005), respectively, and integrated into the HindIII/EcoRI site of the vector pQE-30Xa (Qiagen), which was first modified by XbaI/NheI restriction followed by religation to eliminate an additional XbaI site, resulting in pSP-EGFP and pSP-DsRed. To create a control construct (pSP-CRY2(Δ1-500)-DsRed) for proteins with nuclear localization, a fragment of cryptochrome 2 was amplified which encodes the C-terminal amino acids 501–612. These are sufficient for nuclear import as shown by Kleiner et al. (1999). In the course of PCR, restriction sites for SacI and KpnI were added to the cDNA fragment, which were afterwards used for fusing the CRY2 fragment with the DsRed gene in pSP-DsRed. Besides the EGFP expression vector (pSP-EGFP), a second construct (pEGG) with an additional GUS gene in front of the EGFP coding sequence was produced. To achieve this, the GUS-gene was amplified by PCR, thus adding SalI and PstI restriction sites that allowed cloning into the multiple cloning site of pSP-EGFP using the enzyme combination SalI and PstI. Finally, the cDNA region encoding amidase 1 (AMI1) was amplified by PCR from a plasmid-template containing the corresponding cDNA using gene-specific primers (5′-TATGCTAGCACTAGTATGGCGACCAATAATGATTTTGGAG, 5′-TATCTCGAGAATAAATGCAGCAAGGGAATCC) with additional SpeI and XhoI sites. This fragment was cloned into the XbaI and SalI restriction sites of pSP-EGFP and pEGG, making use of the compatible ends of SpeI/XbaI and XhoI/SalI, respectively, resulting in pSP-AMI1-EGFP and pEGG-AMIf. The sequences of all nucleotide fragments obtained by PCR as well as the translational fusions of the cDNA fragments were verified by commercial DNA sequencing (GATC, Konstanz, Germany).
Plant transformation and microscopic techniques for fluorescence analysis
Four- to six-week-old A. thaliana seedlings grown under sterile conditions on solidified 1/2 MS-medium supplemented with 2% (w/v) sucrose in Petri dishes (Murashige and Skoog 1962) were bombarded with 1-μm gold particles at 6.5 bar pressure. The particles were emitted from a particle inflow gun as described by Finer et al. (1992). The conditions for the bombardment of plant cells were set to −0.8 bar chamber vacuum, 7 cm distance of particle outlet to the dispersive grid and 12 cm distance to the plantlets (Bruce et al. 1989). After bombardment the plants were kept under constant conditions (8-h light at 24°C, 16-h darkness at 20°C, PAR 105 μmol photons m−2 s−1 from standard white fluorescent tubes) for 1 day. Single leaves of the transformed seedlings were then analyzed with the confocal laser scanning system ZEISS LSM 510 Meta. Excitation light of 488 nm produced by a krypton/argon laser allowed the simultaneous detection of GFP-mediated and chlorophyll-mediated fluorescence using a meta detector system with emission filters set to 500–530 nm for GFP and 650–798 nm for the chlorophyll-autofluorescence, respectively. An additional excitation of the samples at 543 nm (helium/neon laser) permitted a concurrent analysis of DsRed-derived fluorescence by recording the light passing an emission filter in the range of 575–595 nm.
Analysis of mRNA expression by RT-PCR
Total RNA was prepared from 100 mg of plant tissue. The samples were homogenized in 1 ml of Trizol reagent (Invitrogen) in a prechilled mortar. For RT-PCR analysis, total RNA was extracted and converted to cDNA by using M-MLV reverse transcriptase (Promega) with oligo-d(T)15 primers (Promega) according to the manufacturer’s instructions. Thereafter, genomic DNA residuals were eliminated by a DNase (Promega) treatment of 30 min following the manufacturer’s advices. Two cDNA fragments of 518 bp (AMI1) (At1g08980) and 736 bp (FAAH) (At5g64440), respectively, were amplified from 80 ng of total RNA by 30 PCR cycles using intron spanning specific primer sets for AMI1 (5′-ATGGCGACCAATAATGATTTTGG, 5′-CCAACTGTGTCGAAGCTCTG) and FAAH (5′-ACCGGAGCATGATGTTGTC, 5′-ACCCTGTCATATCTGTCCGAC), allowing to distinguish between genomic DNA and cDNA. As controls, cDNA fragments with sizes of 696 bp (ACT2), 682 bp (EF-1α), and 721 bp (eIF4-A1) of constitutively expressed genes representing, respectively, actin 2 (An et al. 1996), elongation factor 1-α (At5g60390; Reddy et al. 2002) and eukaryotic translation initiation factor 4A-1 (At3g13920; Metz et al. 1992) were simultaneously amplified from 40 ng total RNA by 30 PCR cycles with the following, likewise intron spanning oligonucleotide pairs: ACT2 (5′-GTCGCCATCCAAGCTGTTCT, 5′-CTGCCTCATCATACTCGGCC), EF-1α (5′-ATGGGTAAAGAGAAGTTTCAC, 5′-CTTGGGCTCGTTGATCTGG), and eIF4-A1 (5′-ATGGCAGGATCTGCACCAG, 5′-GATTCTCACTGGCTTGCTC). To ensure a linear range of PCR amplification the amount of initial mRNA used was varied, 80 ng for low abundant AMI1 and FAAH fragments or 40 ng for the control fragments, respectively.
Cloning and expression of [His]6-tagged amidases
Recombinant fatty acid amide hydrolase (FAAH) was expressed in Escherichia coli TOP10 harboring the expression vector pTrcHis2 (Invitrogen) containing the full-length FAAH-cDNA, kindly provided by Dr. Rhidaya Shrestha and Dr. Kent D. Chapman, as described by Shrestha et al. (2003). Hexahistidine-tagged amidase 1 (AMI1) was expressed in E. coli M15 harboring the vector pQE-30 (Qiagen) with a BamHI/SalI-integrated AMI1-cDNA according to the manufacturer’s protocol with the following exceptions: Bacterial cultures were incubated at 37°C until they reached an OD600 of 0.85. Thereafter, they were transferred to 30°C and protein expression was induced by adding 200 μM IPTG. Hereafter the cultures were incubated for another 16 h at 30°C before the proteins were harvested.
Preparation of plant protein extracts
To prepare tissue specific plant protein extracts for subsequent assays, organs were harvested from 4 to 8-week-old plants and homogenized in 1 ml g−1 prechilled extraction buffer (50 mM HEPES pH 7.5, 200 mM sucrose, 3 mM DTT, 3 mM EDTA). The extract was filtered through miracloth and centrifuged at 12,000g for 45 min. The resulting supernatants were desalted using PD-10 columns (Amersham Biosciences) equilibrated in 50 mM potassium phosphate buffer, pH 7.5, and consecutively concentrated by ultrafiltration with the help of Centricon YM-30 units. Afterwards the samples were centrifuged at 100,000g for 45 min, separating the soluble proteins (supernatant) from the microsomal fraction (pellet). After determination of the protein concentration, 35 μg of protein from the soluble fraction of each tissue was precipitated with three volumes of MeOH, and the proteins were separated by exponential-gradient PAGE.
Enzymatic assay
The enzymatic activity of the recombinant amidohydrolases was determined by monitoring the release of ammonia during the reaction as previously described (Vorwerk et al. 2001). In brief, the different substrates (5 mM) were incubated with 5 μg of purified protein in 50 mM potassium phosphate buffer at pH 7.5, at 30°C in a total volume of 0.3 ml. For background control, heat-denatured enzyme (20 min, 100°C) was used. After 3–4 h of incubation aliquots of 0.1 ml were taken, and the reaction was stopped by adding 0.1 ml each of 0.33 M sodium phenolate, 0.02 M sodium hypochlorite, and 0.01% (w/v) sodium pentacyanonitrosyl ferrate(III) (sodium nitroprusside). After incubating for 2 min at 100°C (water bath), each sample was diluted with 0.6 ml of water, and the absorbance was read at 640 nm. Each experiment was calibrated utilizing NH4Cl solutions of known concentrations.
In case of analyzing the 16:0 and 18:2 N-acylethanolamine conversion, the amount of enzymatically produced free fatty acids was examined. For this reason, 5 μg affinity-purified protein was incubated with 1 mM substrate over a time period of 1 h at 30°C in 50 mM potassium phosphate buffer (pH 7.5), in a total reaction volume of 0.1 ml. Thereafter, the reaction was stopped by acidifying with 5 μl of 3 M HCl. Produced free fatty acids were twice extracted with 0.1 ml of chloroform, and the combined solution was then brought to dryness. The precipitate was re-dissolved in 20 μl methanol, treated with 0.1 ml ethereal diazomethane and subsequently the solvent was removed under a gentle stream of nitrogen. The methylated samples were then taken up in 20 μl chloroform and transferred to GC vials. The GC–MS analysis was performed according to Stelmach et al. (2001). Typical retention times (methyl esters) were as follows: hexadecanoic acid (palmitic acid), 14.17 min (m/z = 217 [M + H]+), (9Z,12Z)-octadecadienoic acid (linoleic acid), 15.46 min (m/z = 245 + 263 [M + H]+). The amount of free fatty acids was calculated by comparing results to external standards (free fatty acids) of known concentrations. As a negative control, a tryptic digest of the sample protein was used.
General biochemical procedures
Protein concentrations were determined according to Bradford (1976) with bovine serum albumin as protein standard. Plant protein extracts were separated by 10–20% (w/v) SDS-polyacrylamide-gradient gels as described by Scharf and Nover (1982). To visualize the separated proteins in the gels, they were stained with Coomassie brilliant blue. For immunoblotting, the polypeptides were transferred to nitrocellulose (Towbin et al. 1979) after their electrophoretical separation. The immunological detection of AMI1 was carried out according to Pollmann et al. (2003) with an anti-AMI1-serum used at a final dilution of 1:25,000. The difference in anti-serum dilution (previously 1:1,000) is due to the development of the serum over the boosting-time. The serum for these experiments was taken after the 15th boost of the rabbit, instead of serum harvested after the 1st boost in the publication mentioned above.
Results
Amidase signature proteins in Arabidopsis thaliana
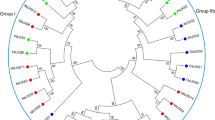
Members of the amidase-signature family (Mayaux et al. 1990) are widespread in nature, ranging from mammals (Cravatt et al. 1996; Giang and Cravatt 1997) to bacteria, e.g. of the species Rhodococcus (Kobayashi et al. 1993) or Bradyrhizobium (Kim and Kang 1994). Even though the proteins in this family exhibit a variety of biological functions, the core structure of the enzymes is highly conserved throughout the family (Shin et al. 2002). When examining the proteome of the model plant A. thaliana (Superfamily 1.69; http://www.supfam.org/SUPERFAMILY/index.html; Madera et al. 2004) one can find seven putative members of this enzyme superfamily, all comprising the characteristic eponymous glycine- and serine-rich amidase-signature motif (Fig. 1a). The construction of a phylogenetic tree revealed the clustering of the Arabidopsis amidase-signature proteins into two sub-groups and additionally two single proteins. The first group encompasses FAAH and a so far uncharacterized protein, At3g25660. The second group contains AMI1 along with the proteins Toc64-III and mtOM64. The two remaining enzymes do not cluster in another group, but rather branch directly from the origin (Fig. 1b).

Sequence alignments of the seven members of the amidase-signature family from Arabidopsis thaliana (a). The analyzed sequences are AMI1 (At1g08980), At5g07360 (amidase family protein), mtOM64 (At5g09420), Toc64-III (At3g17970), At4g34880 (amidase family protein), At3g25660 (putative glutamyl-tRNA(Gln) amidotransferase), FAAH (At5g64440). Amino acid positions are indicated. Identities and similarities among the polypeptides are indicated by black or gray boxes, respectively. The assumed catalytic triad of the protein family members is marked by asterisks, the glycine- and serine-rich amidase-signature motif is underlined. b A phylogenetic tree of the amidase-signature family members from A. thaliana is shown. The above-mentioned enzymes were aligned using the optimal alignment method of the DNAMAN software (Lynnon BioSoft, Vaudreuil, Canada) with default parameters. The branch length indicates the relative observed divergency among the polypeptides
Subcellular localization of AMI1
Preliminary studies suggested a cytosolic localization for AMI1, since the immunological signal was exclusively found in the soluble fraction of protein preparations (Pollmann et al. 2003). Nevertheless, these data are not sufficient to prove a cytoplasmic localization nor do they exclude AMI1 from organelles, which could have been disrupted during the preparation procedure, thus releasing their soluble proteins. Some recent publications (Vojta et al. 2004; Chew et al. 2004) assumed a possible plastidial or mitochondrial localization of AMI1, associating AMI1 with two proteins, Toc64-III and mtOM64, mainly based on their primary amino acid sequence homology, emphasized by the clustering of the three proteins into one branch of a phylogenetic tree (Fig. 1b). However, these assumptions were solely based on in silico studies and thus ambiguous. Here we demonstrate unequivocally that AMI1 is located in the cytoplasm of plant cells transiently expressing AMI1:GFP fusion constructs.
To study AMI1-localization, clones in which EGFP was fused to the 3′-end of the AMI1-cDNA were initially constructed and EGFP-fluorescence was analyzed with a confocal laser scanning microscope after introduction of the plasmid construct by particle bombardment into 4 to 6-week-old Arabidopsis seedlings. Coexpression of either EGFP alone or DsRed fused to the nuclear localization site (NLS) of CRY2 (cryptochrome 2) served as controls, the latter for nuclear targeting (Kleiner et al. 1999). Whereas the GFP protein alone showed an expected cytoplasmic as well as a nuclear localization (Fig. 2a) and DsRed fused to the NLS of CRY2 was targeted to the nucleus (Fig. 2b), AMI1:GFP fusion-proteins were found in the cytoplasm and intriguingly to some extent in the nucleus (Fig. 2c). Considering a calculated molecular weight of the fusion protein of approximately 72 kDa, an unspecific localization of AMI1 in the nucleus, in consequence of a diffusion process through nuclear pores, seemed unlikely but not impossible as earlier reports, e.g. by Saslowsky et al. (2005) showed identical findings. Addressing the question of a specific targeting of AMI1 to the nucleus, a second EGFP-construct was generated which contained a GUS-gene inserted between the AMI1- and the GFP-coding sequences in order to increase the apparent size of the fusion protein. The GFP-fluorescence in cells receiving this second EGFP-construct and, at the same time, a nuclear-localization positive control (CRY2(Δ1-500):DsRed) showed an exclusive localization of EGFP-fluorescence in the cytoplasm and DsRed-fluorescence in the nucleus (Fig. 2d). The AMI1-GUS-GFP fusion protein, exhibiting a calculated molecular weight of ca. 142 kDa, apparently is no longer able to enter the nucleus. Rather, EGFP-fluorescence was found restricted to the cytoplasm and the cytoplasmic pocket harboring the nucleus, thus eliminating the possibility of specific nuclear import of the fusion protein and, by inference, of AMI1 itself. Furthermore, no particular affinity of the AMI1-GFP fusion polypeptide to either chloroplasts or mitochondria was observed (data not shown). Therefore, a participation of AMI1 in the chloroplastidic or the mitochondrial protein import machineries can be ruled out. This conclusion is further underlined by the lack of a tetratricopeptide repeat motif in AMI1, which seems to be a characteristic domain mediating protein–protein interaction in the two translocon-associated members of the amidase-signature protein family, Toc64-III (Sohrt and Soll 2000) and mtOM64 (Chew et al. 2004).

Localization of AMI1-EGFP fusion proteins in epidermis cells of A. thaliana. The upper part (I) schematically illustrates the chimeric constructs. The lower part (II, a–d) gives typical results of the confocal laser scanning microscopy, using differential emission light-filter sets. The first row on the left (EGFP-fluorescence) shows the GFP channel (500–530 nm), followed by the DsRed channel (DsRed-fluorescence), showing the fluorescence of the DsRed-fluorophore between 575 and 595 nm. The chlorophyll-autofluorescence is shown in a separate channel (650–798 nm, third row from left). On the right side, an overlay of all channels is depicted. a Single transformation with the empty EGFP-vector, cytoplasmic control. b Single transformation with the prepared CRY2-DsRed vector, nuclear control according to Kleiner et al. (1999). c Double transformation of an A. thaliana epidermis cell with the AMI1:EGFP construct and the nuclear control CRY2:DsRed. d A double transformation of AMI1:GUS:EGFP and the chimeric CRY2:DsRed construct highlights the exclusive cytoplasmic localization of AMI1
Analysis of tissue specific AMI1 expression by RT-PCR
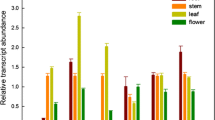
In the initial work proposing AMI1 as an enzyme converting IAM to IAA, thus being of particular interest for plant auxin biosynthesis, we documented that AMI1 cDNA was obtained by RT-PCR from leaf tissues. The mRNA-level of AMI1 in this tissue seemed fairly constant in plants between 3 and 8 weeks after germination (Pollmann et al. 2003). To obtain more detailed information and to compare the expression pattern of AMI1 and the related isogene FAAH (Shrestha et al. 2003), total RNA was extracted from different tissues of A. thaliana. As the expression level of AMI1 was expected to be too low to be detected by Northern-blot analysis RT-PCR was used to analyze the specific expression patterns of the two genes in different organs. AMI1-expression was found in all tissues analyzed, albeit at different levels. Highest transcript levels were found in flowers and young sink leaves (Fig. 3). As depicted in Fig. 4, a similar profile for the AMI1 expression is found using the MPSS database server (Brenner et al. 2000). This expression pattern is compatible with a role of AMI1 in auxin de novo biosynthesis which is supposed to take place in tissues with highest meristematic activity. Intriguingly, AMI1 expression is highest in areas of high IAA content in the plant, as visualized in the IAA map created by Müller et al. (2002) for A. thaliana.

AMI1 and FAAH mRNA expression in different Arabidopsis organs. RNAs from separate tissues were extracted and converted to cDNAs by reverse transcription. Fragments of 518 bp (AMI1) and 736 bp (FAAH), respectively, were amplified from 80 ng total RNA by 30 cycles of PCR using gene specific primers. As control, fragments of actin 2 (696 bp), elongation factor 1α (682 bp) and eukaryotic translation initiation factor 4A-1 (721 bp) were amplified simultaneously from 40 ng RNA by 30 PCR cycles

AMI1 mRNA abundance in dependency of different tissues. AMI1 expression profile as deposited in the MPSS database (http://www.mpss.udel.edu/at/java.html). The following tissues are shown: inflorescence (INF), mixed stage, immature buds, classic MPSS; root (ROS), 21 days, untreated; young leaves (LES), 21 days, untreated; silique (SIS), 24–48 h postfertilization, signature MPSS; germinating seeds (GSE). The axis of ordinates shows the sum of abundance for the signature classes 1, 2, 5, and 7
FAAH expression was highest in flowers, lower in roots, and still lower in sink leaves, while unlike AMI1, FAAH-mRNA was not detected in source leaves, stems and siliques, emphasizing differential functions for the two gene products.
Examination of AMI1 content in different plant tissues
Analysis of transcript levels alone does not allow to draw conclusions about the levels of the corresponding polypeptides, as for example protein turnover in different plant tissues or organs can be variable. Therefore, protein extracts from different plant tissues were prepared and their AMI1 content was analyzed by immunodetection, utilizing a further developed, previously described anti-AMI1-serum (Pollmann et al. 2003).
The levels of AMI1 protein in the different organs indeed differ somewhat from the transcript levels described above. Whereas high AMI1 levels in sink leaves reflected the transcriptional situation, the immunological detection revealed very low amounts of AMI1 in all other tissues analyzed (Fig. 5b). The major difference between transcript and protein levels was found in flowers where AMI1-transcript levels are comparable to those in sink leaves. In root tissue, RT-PCR experiments showed low levels of AMI1-mRNA, while in Western blots AMI1-polypeptide was not detectable. Interestingly, in siliques—in contrast to all other organs—AMI1-immunoreactivity is represented as a faint double band (Fig. 5b). Thus, leaving open the possibility of a post-translational modification of the amidase polypeptide in this specific tissue.

Western-blot analysis of organ specific AMI1 distribution in 4 to 8-week-old A. thaliana plantlets. Soluble protein fractions were prepared from the different tissues as described in the experimental procedures. Aliquots of 35 μg of each of the corresponding protein samples were separated by SDS-PAGE using exponential gradient gel systems (10–20%). a Gel stained with Coomassie Brilliant Blue; b cutout of the corresponding Western-blot section, focusing onto the deduced molecular mass of AMI1 (∼45 kDa). The anti-AMI1-serum, used as the primary antibody in this experiment, was utilized at a dilution of 1:25,000
Comparison of AMI1 and FAAH substrate preferences
In the plant kingdom, only two members of the amidase-signature family of hydrolytic enzymes are assigned for their specific enzymatic function so far. The first identified enzyme was AMI1 (Pollmann et al. 2003), the second a fatty acid amide hydrolase (FAAH), capable of converting N-acylethanolamine (NAE) to ethanolamine and the free acid (Shrestha et al. 2003). Moreover, it had previously been demonstrated that orthologous FAAHs also cleave fatty acid derivatives like oleamide (cis-9,10-octadecenoamide), releasing fatty acids and ammonia (Patricelli and Cravatt 2000).
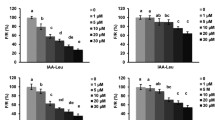
In the first study on AMI1 (Pollmann et al. 2003), neither fatty acid amides nor N-acylethanolamines were tested as potential substrates, and, as a consequence, a function of AMI1 as a FAAH cannot be excluded. Thus, the substrate specificities of the two recombinant proteins were compared in this study. Whereas the A. thaliana FAAH showed a very narrow substrate spectrum, more or less refusing all offered substrates except N-acylethanolamines and oleamide (Fig. 6), AMI1 shows to be specific for IAM, but the enzyme shows high activity also with 1-naphthaleneacetamide (NAM), a compound which is not occurring in plants. However, the cleavage product, 1-naphthaleneacetic acid (NAA) is a strong synthetic auxin in plants. Interestingly, auxin activity has been reported for NAM (Upadhyaya et al. 2000). Our results show that the auxin activity may not reside in NAM itself, even more so that NAM does not follow Thimann’s 0.55-nm rule for auxin activity, but that it may result from NAA liberated from NAM via IAM hydrolases.

Relative substrate conversions by recombinant AMI1 and FAAH polypeptides. The enzymatical reactions were carried out at pH 7.5 and 30°C under standard conditions (5 mM substrate concentration, reaction time 4 h) and the amount of released NH3 was analyzed thereafter (a). In case of N-acylethanolamines as substrates (b), the produced amount of free fatty acids was examined by GC–MS. The substrate concentration was reduced to 1 mM and the reaction time was shortened to 1 h. The data shown are means ± SD derived from n = 3 independent experiments
Discussion
Several pathways of IAA biosynthesis have been proposed in the past, but to date, none of these pathways has been experimentally proven to be completely operating in vivo. Nevertheless, it is generally accepted that IAA can, at least partially, be synthesized from l-tryptophan (l-Trp), which is, as a product of the shikimate pathway, synthesized in the chloroplasts. l-Trp is then released from the plastids and further converted to IAA by so far undefined enzymatic reaction steps in the cytoplasm, e.g. with the help of a proposed IAA-synthase multienzyme complex (Müller and Weiler 2000).
One of the discussed mechanisms of IAA-synthesis is the IAM pathway, which is known to be used by several plant pathogen bacteria. New interest was cast onto this pathway when its intermediate, IAM, was shown to be an endogenous compound in A. thaliana (Pollmann et al. 2002), and a specific IAM-amidohydrolase (AMI1) was later on identified from the same species (Pollmann et al. 2003). AMI1 was previously described to be found in the cytosolic protein fraction, not ruling out a subcellular localization within organelles, e.g. in chloroplasts or mitochondria. Recent work carried out by Vojta et al. (2004), based on in silico analysis, provided the assumption that AMI1, or Toc64-I following their nomenclature, is possibly part of the preprotein translocon of the outer envelope of chloroplasts (Toc complex). This presumption would imply a plastidic localization of AMI1. Although the affinity of the polypeptide to the Toc complex might be low, a higher local concentration of an interacting protein around the plastids has to be taken for granted in this case, resembling the previously described localization of Toc64-III (Lee et al. 2004).
In order to examine the subcellular localization of AMI1 we cloned and analyzed a couple of chimeric EGFP and DsRed constructs, either translationally fused to AMI1, AMI1:GUS or the 3′-end of the cryptochrome 2 gene, encoding the NLS within the last 112 C-terminal amino acid residues of CRY2, the latter used as a control for proteins imported into the nucleus. Utilizing confocal laser scanning microscopy a cytoplasmic localization of AMI1 could be shown (Fig. 2). Immunofluorescence microscopy also showed that AMI1 was localized in the cytoplasm (data not shown), thus confirming the GFP-fusion experiments. In addition, the AMI1 signal was exclusively found in the soluble protein fraction in a slight alkaline milieu (pH 8–8.5) known to deprotonize AMI1 (Pollmann et al. 2003). All these results strongly indicate that AMI1 is located in the cytoplasm where IAA synthesis is thought to take place. Intriguingly, the ca. 72 kDa AMI1:EGFP protein co-localized to some extent with the CRY2:DsRed fusion construct in the nucleus, notwithstanding that nuclear pores are supposed to exclude proteins with a size of more than approximately 40–60 kDa, unless they carry a NLS. However, the localization of a nearly double sized AMI1:GUS:EGFP construct of ca 142 kDa was completely restricted to the cytoplasm. Thus, one can conclude that AMI1 is not specifically imported into nuclei. The observed, presumably unspecific, diffusion of AMI1:EGFP into the nucleus, which has been reported for other proteins as well (Saslowsky et al. 2005) may result from a highly compacted tertiary structure of the fusion protein, its diameter being below the size exclusion limit of the nuclear pores, since a general nucleoplasmic localization of GFP has been described previously (Haseloff et al. 1997). The results of the present study rule out a plastidic localization of AMI1. Furthermore, our findings imply a co-localization of AMI1 with NIT1 in the cytoplasm, previously described by Cutler and Somerville (2005), thus with an enzyme capable of producing IAM as a side product (Pollmann et al. 2002). In accordance with this observation, AMI1 shows some structural differences when compared to the two organelle-associated members of the amidase-signature family, Toc64-III and mtOM64, which are known to be involved in preprotein translocation into chloroplasts or mitochondria, respectively. In contrast to Toc64-III and mtOM64, AMI1 does not contain tetratricopeptide repeat (TPR) motifs in its C-terminal domain. Toc64-III and mtOM64 do not contain the essential residues of the supposed amidase catalytic triad (Lys-cisSer-Ser; Shin et al. 2003; Bracey et al. 2002) as pin-pointed in Fig. 1a (for AMI1: K36, S113, and S137). Interestingly, Toc64-III and mtOM64 orthologous proteins, containing TPR-domains on their C-terminus, can already be found in the ancient moss Physcomitrella patens (Hofmann and Theg 2004). These proteins may be links to an ancestral protein class, from which the amidase-signature family members in plants, especially those lacking a TPR-motif on their C-terminus, might have evolved during evolution.
Besides AMI1, a second member of the A. thaliana amidase-signature family was assigned to its enzymatic function just recently. Like the known fatty acid amide hydrolases from mammals (Cravatt et al. 1996; Giang and Cravatt 1997), an orthologous protein from Arabidopsis (FAAH), capable of converting N-acylethanolamine to ethanolamine and the corresponding free fatty acid has been identified and characterized (Shrestha et al. 2003). In our initial experiments, AMI1 was not tested for this kind of reaction, yet a function as a fatty acid amide hydrolase could not be excluded per se. When comparing the substrate preferences of the two heterologously expressed (His)6-tagged proteins, the different enzymatic functions of AMI1 and FAAH became obvious. As can be seen in Fig. 6, FAAH accepts only oleamide (Fig. 6a) and the tested N-acylethanolamines (Fig. 6b) from a range of substrates, and does not hydrolyze IAM detectably. In contrast, AMI1 utilizes IAM as the preferred substrate and shows only a weak reactivity towards oleamide (Fig. 6a), NAE-16:0, and NAE-18:2 (Fig. 6b), the two major N-acylethanolamine derivatives of plants (Shrestha et al. 2002). AMI1 shows a slightly broader substrate range and also converts NAM at a rate 60% that of IAM. NAM is a synthetic structural homologue of IAM and the reaction product, NAA, like IAA is a strong auxin. In agreement with this observation, a similar behavior has previously been reported for the IaaH gene product of A. tumefaciens which shows enzymatic activity not only against IAM, but also against NAM and phenylacetamide to nearly the same extend (Kemper et al. 1985). The AMI1-catalyzed conversion of all other tested substrates was under 15% of the IAM value, thus, pin-pointing AMI1 to be a specific IAM hydrolase with distinct enzymatic features, differing from those of FAAH. Considering the NAM-converting activity of IAM hydrolases as a common feature of this group of proteins, it is likely that the auxin activity of exogenously added NAM, e.g. in rice (Upadhyaya et al. 2000) is not a direct one, but rather due to NAA, which is enzymatically formed from NAM by the action of IAM hydrolases. An appropriate IAM hydrolyzing activity has been reported to occur in rice (Arai et al. 2004).
In order to further analyze potential differences between the two amide hydrolases, AMI1 and FAAH, the tissue specific transcription profiles of their genes were compared. A completely different distribution of the mature mRNA of the two amidases has been recognized. Whereas FAAH is predominantly expressed in flowers and just weakly in roots and sink leaves, AMI1-mRNA can be detected in all analyzed tissues showing highest levels in flowers and sink leaves (Fig. 3). The AMI1-expression profile matches tissues with high IAA levels as previously mapped by Müller et al. (2002). In A. thaliana leaves, both, AMI1-mRNA and IAA levels are highest in young, expanding sink leaves and levels decline sharply with leaf development into source leaves. When checking the AMI1-transcriptional data at the translational level, we observed that, in contrast to the data obtained by RT-PCR, an AMI1 immunoreactive signal was neither detected in roots nor was the AMI1 content in flowers as high as expected. This might be due to differentially regulated protein turnover rates in the different plant tissues. Nonetheless, AMI1 content in sink leaves was high and declined in adult source leaves. Western-blot analysis revealed a faint double band in case of silique tissue. Further studies are needed to enlighten putative post-translational modifications of AMI1 in this specific tissue, or a putative cross-reactivity of the antibody.
With the hypothesis of AMI1 being involved in plant auxin synthesis, substantiated by the expression of AMI1 in tissues of highest meristematic activity and the subcellular localization in the cytoplasm corresponding with the assumed compartment of IAA biosynthesis, it will be challenging to unravel the physiological relevance of AMI1 by analyzing ami1 knock-out mutants and transgenic lines ectopically overexpressing AMI1. The detailed dissection of the regulation of AMI1 expression, making use of stable transformed AMI1-promoter::uidA lines, will be a matter of particular interest in the future.
Abbreviations
- DsRed:
-
Red fluorescent protein from Discosoma sp.
- GFP:
-
Green fluorescent protein
- IAA:
-
Indole-3-acetic acid
- IAM:
-
Indole-3-acetamide
- NAA:
-
1-Naphthaleneacetic acid
- NAE:
-
N-acylethanolamine
- NAM:
-
1-Naphthaleneacetamide
References
An YQ, McDowell JM, Huang S, McKinney EC, Chambliss S, Meagher RB (1996) Strong, constitutive expression of the Arabidopsis ACT2/ACT8 actin subclass in vegetative tissues. Plant J 10:107–121
Arai Y, Kawaguchi M, Syono K, Ikuta A (2004) Partial purification of an enzyme hydrolyzing indole-3-acetamide from rice cells. J Plant Res 117:191–198
Ausubel FA, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (2000) Current protocols in molecular biology. Wiley, New York
Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF (2002) Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 298:1793–1796
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, Roth R, George D, Eletr S, Albrecht G, Vermaas E, Williams SR, Moon K, Burcham T, Pallas M, DuBridge RB, Kirchner J, Fearon K, Mao J, Corcoran K (2000) Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol 18:630–634
Bruce WB, Christensen AH, Klein T, Fromm M, Quail PH (1989) Photoregulation of a phytochrome gene promotor from oat transferred into rice by particle bombardment. Proc Natl Acad Sci USA 86:9692–9696
Chew O, Lister R, Qbadou S, Heazlewood JL, Soll J, Schleiff E, Millar AH, Whelan J (2004) A plant outer mitochondrial membrane protein with high amino acid sequence identity to a chloroplast protein import receptor. FEBS Lett 557:109–114
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87
Cutler SR, Somerville CR (2005) Imaging plant cell death: GFP-Nit1 aggregation marks an early step of wound and herbicide induced cell death. BMC Plant Biol 5:4
Finer JJ, Vain P, Jones MW, McMullen MD (1992) Development of the particle inflow gun for DNA delivery to plant cells. Plant Cell Rep 11:323–328
Giang DK, Cravatt BF (1997) Molecular characterization of human and mouse fatty acid amide hydrolases. Proc Natl Acad Sci USA 94:2238–2242
Haseloff J, Siemering KR, Prasher DC, Hodge S (1997) Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc Natl Acad Sci USA 94:2122–2127
Hofmann NR, Theg SM (2004) Physcomitrella patens as a model for the study of chloroplast protein transport: conserved machineries between vascular and non-vascular plants. Plant Mol Biol 53:621–632
Kawaguchi M, Kobayashi M, Sakurai A, Syono K (1991) The presence of an enzyme that converts indole-3-acetamide into IAA in wild and cultivated rice. Plant Cell Physiol 32:143–149
Kawaguchi M, Fujioka S, Sakurai A, Yamaki YT, Syono K (1993) Presence of a pathway for the biosynthesis of auxin via indole-3-acetamide in trifoliata orange. Plant Cell Physiol 34:121–128
Kemper E, Waffenschmidt S, Weiler EW, Rausch T, Schröder J (1985) T-DNA coded auxin formation in crown gall cells. Planta 163:257–262
Kim YS, Kang SW (1994) Novel malonamidases in Bradyrhizobium japonicum. Purification, characterization, and immunological comparison. J Biol Chem 269:8014–8021
Kleiner O, Kircher S, Harter K, Batschauer A (1999) Nuclear localization of the Arabidopsis blue light receptor cryptochrome 2. Plant J 19:289–296
Kobayashi M, Komeda H, Nagasawa T, Nishiyama M, Horinouchi S, Beppu T, Yamada H, Shimizu S (1993) Amidase coupled with low-molecular-mass nitrile hydratase from Rhodococcus rhodochrous J1. Sequencing and expression of the gene and purification and characterization of the gene product. Eur J Biochem 217:327–336
Lee YJ, Sohn EJ, Lee KH, Lee DW, Hwang I (2004) The transmembrane domain of AtToc64 and its C-terminal lysine-rich flanking region are targeting signals to the chloroplast outer envelope membrane. Mol Cells 17:281–291
Madera M, Vogel C, Kummerfeld SK, Chothia C, Gough J (2004) The SUPERFAMILY database in 2004: additions and improvements. Nucleic Acids Res 32:D235–D239
Mayaux J-F, Cerbelaud E, Soubrier F, Faucher D, Pétré D (1990) Purification, cloning and primary structure of an enantiomer-selective amidase from Brevibacterium sp. strain R312: evidence for genetic coupling with nitrile hydrolase. J Bacteriol 172:6764–6773
Metz AM, Timmer RT, Browning KS (1992) Sequences for two cDNAs encoding Arabidopsis thaliana eukaryotic protein synthesis initiation factor 4A. Gene 120:313–314
Müller A, Weiler EW (2000) IAA-synthase, an enzyme complex from Arabidopsis thaliana catalyzing the formation of indole-3-acetic acid from (S)-tryptophan. Biol Chem 381:679–686
Müller A, Düchting P, Weiler EW (2002) A multiplex GC–MS/MS technique for the sensitive and quantitative single-run analysis of acidic phytohormones and related compounds, and its application to Arabidopsis thaliana. Planta 216:44–56
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Patricelli MP, Cravatt BF (2000) Clarifying the catalytic roles of conserved residues in the amidase signature family. J Biol Chem 275:19177–19184
Pollmann S, Müller A, Piotrowski M, Weiler EW (2002) Occurrence and formation of indole-3-acetamide in Arabidopsis thaliana. Planta 216:155–161
Pollmann S, Neu D, Weiler EW (2003) Molecular cloning and characterization of an amidase from Arabidopsis thaliana capable of converting indole-3-acetamide into the plant growth hormone, indole-3-acetic acid. Phytochemistry 62:293–300
Reddy VS, Ali GS, Reddy AS (2002) Genes encoding calmodulin-binding proteins in the Arabidopsis genome. J Biol Chem 277:9840–9852
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Saotome M, Shirahata K, Nishimura R, Yakaba M, Kawaguchi M, Syono K, Kitsuwa T, Ishii Y, Nakamura T (1993) The identification of indole-3-acetic acid and indole-3-acetamide in the hypocotyls of Japanese cherry. Plant Cell Physiol 34:157–159
Saslowsky DE, Warek U, Winkel BSJ (2005) Nuclear localization of flavonoid enzymes in Arabidopsis. J Biol Chem 280:23735–23740
Scharf KD, Nover L (1982) Heat-shock-induced alterations of ribosomal protein phosphorylation in plant cell cultures. Cell Sep 30:427–437
Shin S, Lee TH, Ha NC, Koo HM, Kim SY, Lee HS, Kim YS, Oh BH (2002) Structure of malonamidase E2 reveals a novel Ser-cisSer-Lys catalytic triad in a new serine hydrolase fold that is prevalent in nature. EMBO J 21:2509–2516
Shin S, Yun YS, Koo YS, Kim YS, Choi KY, Oh BH (2003) Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad. J Biol Chem 278:24937–24943
Shrestha R, Noordermeer MA, Van der Stelt M, Veldink GA, Chapman KD (2002) N-acylethanolamines are metabolized by lipoxygenase and amidohydrolase in competing pathways during cottonseed imbibition. Plant Physiol 130:391–401
Shrestha R, Dixon RA, Chapman KD (2003) Molecular identification of a functional homologue of the mammalian fatty acid amide hydrolase in Arabidopsis thaliana. J Biol Chem 278:34990–34997
Sohrt K, Soll J (2000) Toc64, a new component of the protein translocon of chloroplasts. J Cell Biol 148:1213–1221
Stelmach BA, Müller A, Hennig P, Gebhardt S, Schubert-Zsilavecz M, Weiler EW (2001) A novel class of oxylipins, sn1-O-(12-oxophytodienoyl)-sn2-O-(hexadecatrienoyl)-monogalactosyl diglyceride, from Arabidopsis thaliana. J Biol Chem 276:12832–12838
Towbin H, Staehlin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354
Upadhyaya NM, Zhou XR, Wu L, Ramm K, Dennis ES (2000) The tms gene as a negative selection marker in rice. Plant Mol Biol Rep 18:227–233
Vojta A, Alavi M, Becker T, Hormann F, Kuchler M, Soll J, Thomson R, Schleiff E (2004) The protein translocon of the plastid envelopes. J Biol Chem 279:21401–21405
Vorwerk S, Biernacki S, Hillebrand H, Janzik I, Müller A, Weiler EW, Piotrowski M (2001) Enzymatic characterization of the recombinant Arabidopsis thaliana nitrilase subfamily encoded by the NIT2/NIT1/NIT3-gene cluster. Planta 212:508–516
Wachter A, Wolf S, Steininger H, Bogs J, Rausch T (2005) Differential targeting of GSH1 and GSH2 is achieved by multiple transcription initiation: implications for the compartmentation of glutathione biosynthesis in the Brassicaceae. Plant J 41:15–30
Weiler EW, Schröder J (1987) Hormone genes and the crown gall disease. Trends Biochem Sci 12:271–275
Woodward AW, Bartel B (2005) Auxin: regulation, action and interaction. Ann Bot 95:707–735
Acknowledgements
We thank Dr. Rhidaya Shrestha and Dr. Kent D. Chapman (Department of Biological Sciences, Division of Biochemistry and Molecular Biology, University of North Texas, Denton, Texas, USA) for providing the (His)6-tagged FAAH clone. Furthermore, the authors thank Dr. Andreas Wachter (Institute of Plant Sciences, Ruprecht-Karls-University, Heidelberg, Germany) for kindly providing the vectors pFF19-GFP and pFF19-RFP. This work was supported by the Deutsche Forschungsgemeinschaft, Bonn, Germany (Schwerpunktprogramm ‘Molecular Mechanisms of Phytohormone Action’, and subsequently Sonderforschungsbereich 480 ‘Molekulare Biologie komplexer Leistungen von botanischen Systemen’).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pollmann, S., Neu, D., Lehmann, T. et al. Subcellular localization and tissue specific expression of amidase 1 from Arabidopsis thaliana . Planta 224, 1241–1253 (2006). https://doi.org/10.1007/s00425-006-0304-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-006-0304-2