
Abstract
NADH-dependent NO scavenging in barley extracts is linked to hemoglobin (Hb) expression and is inhibited by SH-reagents. Barley Hb has a single cysteine residue. To determine whether this cysteine was critical for NO scavenging, barley Hb and a mutated version, in which the single Cys79 was replaced by Ser, were over-expressed in Escherichia coli and purified to near homogeneity. The purified proteins exhibited very low NO-scavenging activity (12–14 nmol min−1 mg−1 protein) in the presence of NADH or NADPH. This activity was insensitive to SH-reagents. Addition of an extract from barley roots to either of the purified proteins resulted in high NADH-dependent NO turnover in a reaction that was sensitive to SH-reagents. A protein was purified from barley roots and identified by mass-spectrometry analysis as a cytosolic monodehydroascorbate reductase. It efficiently supported NADH-dependent NO scavenging in the presence of either native or mutated barley Hb. Ascorbate strongly facilitated the rate of metHb reduction. The K m for Hb was 0.3 μM, for ascorbate 0.6 mM and for NADH 4 μM. The reaction in the presence of monodehydroascorbate reductase was sensitive to SH-reagents with either form of the Hb. We conclude that metHb reduction and NO turnover do not involve direct participation of the Cys79 residue of barley Hb. NO scavenging is facilitated by monodehydroascorbate reductase mediating a coupled reaction involving ferric Hb reduction in the presence of ascorbate and NADH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hemoglobin (Hb) interaction with NO is an important characteristic of the molecule’s physiological function in many species. Some Hbs are apparently specially adapted to the function of NO removal (Poole 2005). Microorganisms and fungi possess flavohemoglobins, containing flavin and Hb domains (Gardner et al. 1998; Kobayashi et al. 2002; Hernandez-Urzua et al. 2003), wherein the Hb domain converts NO to nitrate, while the flavin domain is responsible for rapid reduction of the ferric heme back to its ferrous state. This provides an efficient mechanism for a rapid removal of toxic NO. In species lacking flavohemoglobins, metHb reduction could be accomplished by a separate reductase protein (Moran et al. 2002; Wittenberg and Wittenberg 2003), however, such reductases remain largely unidentified (Gardner 2005). There have been reports suggesting Hb itself may have metHb reductase activity but usually this activity is negligible (Becana and Klucas 1990; Minning et al. 1999) and can represent a slow chemical reduction of Hb by NADH and other reducing agents.
Class-1 plant Hbs are distinguished by their high avidity for oxygen, with the result that they exist as the oxyhemoglobin form under most physiological conditions. They are induced during plant hypoxia (Hill 1998), triggered by ATP decline (Nie and Hill 1997). Expression of the gene results in improved redox and energy status in the hypoxic tissue (Sowa et al. 1998). NO formation and turnover is believed to be involved in the improved plant status (Igamberdiev et al. 2004). NO is formed in relatively large amounts during plant hypoxia (Dordas et al. 2003, 2004). This NO is oxygenated by class 1 oxyhemoglobin, forming nitrate (Igamberdiev and Hill 2004).
The current evidence for the reaction between NO and hemoglobin favors a heme-dioxygenase as opposed to a S-denitrosylase mechanism (Gardner 2005). This was also recently confirmed in kinetic experiments with neuroglobin, where it was demonstrated that the oxyneuroglobin rapidly reacts with NO-forming nitrate while alternative reactions are exceedingly slow (Brunori et al. 2005). Furthermore, the formation of NO–Hb complexes, such as S-nitrosohemoglobin and heme-nitrosylHb formed from deoxyHb, is generally minor under physiological conditions (Xu et al. 2003; Han et al. 2004; Herold and Rock 2005). A recent paper, however, has suggested that the reaction between NO and Hb is facilitated through the formation of S-nitrosohemoglobin (Perazzolli et al. 2004). In this submission, we address two aspects: does the reaction between NO and oxyhemoglobin require an SH group, and; how is methemoglobin, formed in the reaction of NO with oxyhemoglobin, reduced to hemoglobin? Barley Hb has a single cysteine (Cys79). Using site-directed mutagenesis, we have introduced a serine in place of the cysteine and determined the effect of this on NO turnover. To determine how metHb is reduced to Hb, we have isolated an enzyme, monodehydroascorbate reductase, from barley extracts and demonstrate that it, along with ascorbate and NADH, effectively maintain oxygenation of NO in the presence of either purified recombinant or mutated (cysteine79→serine79) barley Hb.
Materials and methods
Mutation of barley recombinant hemoglobin
To accomplish the site-directed mutagenesis of Cys79 (TGC) to Ser79 (AGC) in barley Hb, the QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used according to the manufacturer’s instructions.
PCR amplification of the sequence encoding barley rHb (Duff et al. 1997), cloned into Stratagene’s circular plasmid pUC19, was performed using primers containing the desired base change (forward primer: 5′-CC GTG TCC GTC TTC GTC ATG ACC A GC GAG GCG GC -3′; reverse primer: 5′-GC CGC CTC GC T GGT CAT GAC GAA GAC GGA CAC GG-3′, where the bold letters show the mutated base, and the underlined sequence indicates the triplet coding for the altered amino acid). After digesting the non-mutated template DNA, the new mutated plasmid was transformed into bacterial cells. Colonies were then selected to confirm the mutation by sequencing 3 of 20 positive clones. Sequencing the two chosen full-length mutHb clones confirmed the desired base change; one clone was further used to produce recombinant mutated barley Hb.
Purification of overexpressed recombinant and mutant barley hemoglobin from E. coli
Escherichia coli strain DH-5α (Invitrogen, Canada) was used as a host for both recombinant and nonrecombinant pUC 19 plasmids (Invitrogen, Canada). Preparation of extracts and purification of Hb were conducted as described in (Duff et al. 1997), except that DEAE-Sephacel packing with a 0– 250 mM NaCl gradient was used for ion exchange column and the final step included gel filtration on a prepacked Superose 12 column using 50 mM Tris–HCl, pH 8.5, 150 mM NaCl as column buffer. All procedures were performed at 0–40C, and all chromatographic separations were on a Pharmacia FPLC system. The purified Hb was then either immediately used for analysis or stored at –800C until needed.
Purified Hb fractions had a ratio of absorbance at 412–280 nm in the range 3.0–3.2, which corresponds to a 90–95% degree of purification (Igamberdiev et al. 2004). The native molecular mass of the protein was determined on Superose 12 column using bovine serum albumin, ovalbumin, carbonic anhydrase and cytochome c as standards. SDS-PAGE electrophoresis was performed using a BioRad mini-gel system with acrylamide concentrations of 15%.
Mass spectrometry analysis of tryptic peptides
Coomassie-stained bands were in situ digested with modified trypsin as described (Shevchenko et al. 1996). Prior to digestion, both rHb and mutHb proteins were reduced with 10 mM dithiothreitol (DTT) in 100 mM NH4HCO3 buffer at 56°C for 45 min and then incubated with 55 mM iodoacetamide at 25°C for 30 min. Tryptic peptide mixture was purified on reverse-phase POROS R2 (20–30 μm bead size, PerSeptive Biosystems, Framingham, CA, USA) nanocolumn, eluted onto the MALDI probe with saturated matrix solution (2,5-dihydroxybenzoic acid in 50% (v/v) acetonitrile/5% formic acid) and MS/MS analysis of peptides mixture was performed by MALDI Qq-TOF mass spectrometer (Manitoba/Sciex prototype) (Chernushevich et al. 1999; Loboda et al. 2000). Knexus automation software package (Proteometrics LLC, Canada) with ProFound search engine was used for peptide mass fingerprint analysis of MS spectra. Tandem MS spectra were analyzed by using the software m/z (Proteometrics Ltd., New York, NY, USA) and Sonar MS/MS (Proteometrics, Canada) search engine. The MS/MS spectra with and without the mutation site were verified manually.
Measurement of NO-scavenging activity
NO conversion was measured using an NO electrode (NOMK2, World Precision Instruments, USA). The medium was 50 mM Tris–HCl buffer (pH 7.5) in a 2 ml vial. When FAD was present, an addition of 1 mg ml−1 bovine Cu,Zn-superoxide dismutase (Sigma) was necessary to prevent the formation of peroxynitrite via interaction of NO and superoxide (Gardner et al. 2001). NO was delivered by 1 mM sodium nitroprusside (SNP) (Sigma) added with continuous stirring upon illumination. Alternatively, NO was added either from the solution in the same buffer, which was prepared by bubbling from an NO tank (Matheson Company Inc., USA), or it was delivered by 20 μM sodium 2-(N, N-diethylamino)-diazenolate-2-oxide (DEANO) (Alexis Biochemicals) in darkness to a concentration of ~1 μM. The sample (1–10 μl) was added, followed by the addition of NAD(P)H (0.1 mM). There was no NO decrease with NAD(P)H in the absence of the sample. There was no significant difference in the rates if NO added from the tank, delivered by SNP or DEANO. The inhibitors used were 0.1 mM p-hydroxymercurybenzoate (pHMB) and 1 mM N-ethylmaleimide (NEM). pHMB was added to the solution, and NEM was preincubated with barley Hb for 1 h. The total protein in extracts was measured by the method of Bradford (1976).
Purification of monodehydroascorbate reductase
Barley (Hordeum vulgare L., cv Harrington) was germinated in darkness on filter paper. For the extraction of a protein-mediated reduction of metHb, 40 g of roots of 3-day-old barley seedlings were ground in liquid nitrogen and homogenized in 50 mM Tris–HCl buffer, pH 8.0, containing 1 mM EDTA and 5 mM MgCl2. After filtering through cheesecloth and centrifugation at 10,000 g, the supernatant was fractionated by ammonium sulfate and the pellet between 50 and 70% saturation was redissolved in 10 mM Tris–HCl buffer pH 8.0 and the concentration of ammonium sulfate was adjusted to 30% saturation. The sample was loaded on Phenyl Superose column, eluted by the gradient of ammonium sulfate (30–0% saturation) and NO-scavenging activity was measured in the eluate fractions. No activity was found. Then the fractions were tested for NADH-dependent NO-scavenging activity in the presence of 5 μM recombinant barley Hb. Active fractions were collected, pooled and concentrated on Centricon tubes (Millipore, Nepean, Canada). Fractions were loaded on a DEAE-cellulose column (1×5 cm2) equilibrated with Tris–HCl pH 8.0. A step gradient of NaCl was used to further purify Hb- and NADH-dependent NO-scavenging activity. The major part of activity was eluted between 35 and 50 mM NaCl. Active fractions were collected, concentrated and loaded onto a Blue Sepharose column. Protein was eluted with 2 mM NADH in 10 mM Tris–HCl buffer.
The single band from the Blue Sepharose column fraction with Hb- and NADH-dependent NO-scavenging activity with molecular mass ~40 kDa (Fig. 1c) was in situ digested and sequenced by MALDI MS-MS as described above for Hb. Monodehydroascorbate reductase (MDHAR) activity was determined at 340 nm in 50 mM Tris–HCl buffer pH 8.0 containing 0.1 mM NADH, 2.5 mM ascorbate and the amount of ascorbate oxidase (Sigma) to yield ~3 μM monodehydroascorbate (Hossain et al. 1984). MDHAR activity with the purified recombinant and mutated barley Hb (in metHb form) was determined at 340 nm by the oxidation of NADH in the absence or presence of ascorbate (2 mM) and verified by an increase in absorbance at 576 nm. Extinction coefficients of 17.3 and 9.2 mM−1 cm−1 for the reduced ferrous oxy form and oxidized ferric form of Hb were used to calculate protein concentration (Duff et al. 1997). The K m values were determined in Hanes coordinates (s/v, s) by varying concentrations of Hb (or cytochrome c), NADH, ascorbate at constant concentrations of other substrates (5 μM Hb, 0.1 mM NADH, 2 mM ascorbate).

Electrophoregrams of purified recombinant non-mutated (a) and mutated (b) hemoglobins and monodehydroascorbate reductase (c). The amount of protein loaded to each lane was 1 μg
Results
Purification of recombinant (rHb) and mutated (mutHb) hemoglobins
We have prepared a recombinant, mutated barley Hb in which the unique Cys79 residue is replaced by serine. The purification technique, modified from Duff et al. (1997), permitted preparation of rHb and mutHb with A 412/A 280 ratio of 3.0–3.2, which corresponded to near homogeneity. SDS-PAGE yielded bands with molecular weights of 18.5 kDa (Fig. 1a, b). DNA sequencing revealed the replacement of TGC with AGC, a single nucleotide substitution.
rHb: 270 CACGCCGTGTCCGTCTTCGTCATGACCTGCGAGGCGGCTGC 310
mutHb: CACGCCGTGTCCGTCTTCGTCATGACCAGCGAGGCGGCTGC
To check the quality of the recombinant Hb proteins, the molecular masses of the protein monomers were determined by the nano-electrospray ionization mass spectrometry under denaturing acidic conditions (containing 50% acetonitrile and 2% acetic acid). The average molecular weight of the rHb monomer was determined to be 18,625±2 Da. Translation of the recombinant protein sequence as presented in (Duff et al. 1997) leads to the protein mass of 18,538, excluding the covalently bound heme. The average molecular weight of the mutant Hb monomer was 18,641±3 Da. The measured mass difference of 16 Da between the recombinant and mutant Hb is in agreement with the mass difference expected when cysteine is replaced with serine in the molecule. Analysis of tryptic digests by matrix-assisted laser desorption ionization-time-of-flight proved, indeed, substitution of the cysteine residue in the recombinant Hb to serine in the mutant Hb (data not shown).
NO-scavenging activity of recombinant and mutated Hb
Escherichia coli extracts with overexpressed rHb and mutHb possessed NO-scavenging activities in the presence of NADH or NADPH. The activity in E. coli extracts was dependent on NADH without addition of external FAD for exhibiting maximum rates (not shown). At later steps of purification FAD was required for maximum rates. The activity was strongly inhibited by the SH reagents pHMB (Fig. 2a) and NEM (not shown). After the size exclusion step, the specific activity decreased threefold to fourfold. The specific activity was not improved by the addition of FAD and the reaction was not inhibited by pHMB (Fig. 2b). In the absence of NADH, purified Hbs in the oxy ferrous form reacted stoichiometrically with NO. The addition of NADH permitted a low, sustained rate of NO scavenging (Fig. 2c). The rate of metHb reduction in the presence of NADH (6–8 nmol min−1 mg−1 protein) stoichiometrically corresponded to a half rate of NO decline (12–14 nmol min−1 mg−1 protein).

NO scavenging by rHb recorded on NO electrode. a Scavenging by the partially purified rHb fraction (after Phenyl- Superose column). Addition of NADH (0.1 mM), FAD (5 μM) and pHMB (0.1 mM) is shown by arrows. b Scavenging by the purified rHb fraction (after Superose 12 column). Addition of oxyHb (0.3 μM), NADH (0.1 mM), FAD (5 μM) and pHMB (0.1 mM) is shown by arrows. Mutated Hb patterns were identical. c NO scavenging by rHb before and after the addition of purified monodehydroascorbate reductase (MDHAR). Points of addition of oxyHb (0.1 μM), NADH (0.1 mM) or MDHAR (0.15 U ml−1) are shown by arrows
Identification of a reductase associated with barley Hb
When the barley root extract was fractionated with ammonium sulfate, the major portion of NADH-dependent NO-scavenging activity was found between 50 and 70% ammonium sulfate saturation. After Phenyl Superose column chromatography of this fraction, the activity disappeared. Addition of external barley Hb restored activity to certain fractions (Fig. 3). These fractions were further purified using DEAE-cellulose and Blue Sepharose columns. The NO-scavenging activity upon the barley Hb addition was monitored. A single protein band from the SDS gel (Fig. 1c) was analyzed by MALDI Qq-TOF single and tandem mass spectrometry and identified as monodehydroascorbate reductase. Two sequenced peptides had 100% identity of the rice cytosolic MDHAR (Table 1) and very close similarity to monodehydroascorbate reductases from Brassica, pea, Arabidopsis and other plant species (not shown). Six more peptides matched to the same protein by peptide mass fingerprinting analysis. Oxidation of NADH by the purified enzyme was slow in the presence of MetHb and NADH alone (Table 2). Addition of ascorbate resulted in close to a 20-fold increase in the reaction rate. This rate was independent of whether the MetHb was the native or mutated form. Replacement of Hb with cytochrome C gave roughly the same rate of NADH oxidation. The reaction was inhibited by SH-reagents. Purified MDHAR exhibited a threefold higher rate with NADH than with NADPH, using either MDHA or metHb. NO scavenging was highest in the presence of MDHAR, NADH and Hb (Fig. 2c) and sensitive to pHMB (Fig. 3). We could not assess the effect of ascorbate on NO turnover in the presence of the three other components as it interferes with the amperometric analysis of NO. There was no detectable NAD(P)H-dependent NO-scavenging activity of purified MDHAR in the absence of Hb or other hemeproteins (not shown).

NO scavenging by barley root extracts after ammonium sulfate fractionation followed by Phenyl Superose column chromatography. Recovery of NADH-dependent NO-scavenging activity was observed after the addition of non-mutated (a) or mutated Hbs (b) (50 nM). The rate of reaction with the same amount of Hb (non-mutated and mutated) without the extract was low (corresponded to the rates in the figures after addition pHMB)
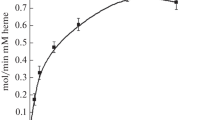
Measuring NADH oxidation, the K m values in the presence of ascorbate were the same for non-mutated and mutated metHb (0.3±0.1 μM) while for cytochrome c, the K m was higher (3±1 μM). The K m for ascorbate was determined as 0.6±0.3 mM (Fig. 4) and the K m for NADH was 4±1 μM. Ascorbate interferes with NO amperometric measurements, precluding the possibility of measuring NO disappearance with all reagents present.

Determination of K m values of monodehydroascorbate reductase with non-mutated and mutated Hb, cytochrome c and ascorbate. ΔA change in absorbance at 340 nm
Discussion
In addition to the traditional functions of Hbs as carriers and stores of oxygen, it is becoming increasingly apparent that they have a major role in controlling cellular levels of reactive oxygen species such as nitric oxide and hydroperoxides (reviewed in Gardner 2005). Such a role requires that the resulting oxidized Hb (ferric Hb or metHb) should be effectively and rapidly reduced to sustain the reaction. NAD(P)H and other reductants, such as glutathione and ascorbate, without the apparent intervention of a reductase protein, can reduce ferric heme of metHb at an exceedingly slow rate (Becana and Klucas 1990; Minning et al. 1999). This contrasts with a value of several thousands nanomole per minute per milligram protein for the bacterial flavoHb-carrying NO dioxygenase activity (Mills et al. 2001), a protein that appears to have been engineered by nature for NO scavenging. In flavoHbs, the flavin domain facilitates this reaction many fold (Kaur et al. 2002). If the flavin domain is absent, rapid reduction of ferric Hb must be achieved by different mechanisms. The coordination of the heme domain of flavoHbs in oxygen ligation is pentacoordinate. No examples of a hexacoordinate flavoHb have been found and no organism appears to possess both hexacoordinate Hb and flavoHb genes (Kundu et al. 2003). Hexacoordinate Hbs are more easily reduced by dithionite than pentacoordinate Hbs because the reduction is not limited by the rate constant for electron transfer but only by the binding of the reductant (Weiland et al. 2004).
Hemoglobins have been long known to bind NO and the conversion of NO to nitrate has been accepted as a mechanism for NO detoxification (Gardner 2005). Among the Hbs, hexacoordinated Hbs have extremely high affinity to O2 and are, therefore, capable of NO scavenging with high efficiency even under conditions that are fully anaerobic for mitochondria (Igamberdiev and Hill 2004). Depending on the reaction mechanism, NO conversion activity can be classified as an NO dioxygenase (Gardner et al. 1998), or a denitrosylase (Hausladen et al. 2001). The role for Cys in maintaining heme coordination and NO conversion has not been shown (Hargrove et al. 2000). In the denitrosylase mechanism it may serve in intramolecular transfer of NO between heme and cysteine, which is rather unlikely (Hausladen et al. 2001). The results for human Hb exclude any possibility of intramolecular NO transfer between heme and cysteine (β93) (Hrinczenko et al. 2000). The NO oxygenase reaction is a fast and predominant reaction of oxyHb with NO (Hrinczenko et al. 2000; Brunori et al. 2005), and the NO–β93Cys reaction is minor under normal physiological conditions (Hrinczenko et al. 2000). In human Hb, S-nitrosohemoglobin formation is important for NO delivery but not for NO conversion (Gladwin et al. 2000). In neuroglobin, mutation of both cysteines yielded identical results to the non-mutated form with respect to NO scavenging (Brunori et al. 2005).
The ferric Hb- and ascorbate-dependent NADH oxidation (Table 2) and the corresponding NO scavenging in the presence of MDHAR (Fig. 3) are comparable with the rates of NO scavenging shown for bacterial flavohemoglobins (Mills et al. 2001). The mutation of the only cysteine (Cys79) in barley Hb has no direct effect on its NO scavenging activity. This argues against a role for Cys79 in facilitating NO conversion to nitrate. While flavohemoglobins are capable of sustaining a rapid NADH-supported oxygenation of NO, hemoglobins lacking the flavin domain demonstrate a very slow NADH-supported reaction that can be construed to be the result of slow chemical reduction of ferric Hb (Becana and Klucas 1990). The slow NADH-supported NO-scavenging activity is not affected by the replacement of the cysteine residue of barley Hb. The activity is insensitive to SH reagents and does not require FAD. It's specific activity is considerably lower than that observed in root homogenates (Igamberdiev et al. 2004) suggesting that, for physiologically relevant rates, this reaction alone cannot totally account for the observed rates of NO turnover.
Monodehydroascorbate reductase (EC 1.6.5.4) has a sequence similar to dihydrolipoamide dehydrogenase (Sano and Asada 1994) and can function in combination with alternative electron acceptors, including ferricyanide and cytochromes (Bérczi and Møller 1998). In peroxisomes, membrane-bound MDHAR was suggested to be the main protein (PMP32) oxidizing internal NADH (del Río et al. 2002). An isoform of the MDHAR faces the internal side of the plasma membrane and may also be involved in oxidation of NADH (Bérczi and Møller 1998). MDHAR contains FAD- and NADH-binding domains (Hossain and Asada 1985) and has some similarity to the flavoprotein part of bacterial flavohemoglobins, to ferredoxin and thioredoxin reductases (Sano and Asada 1994).
NADH oxidation of MDHAR, associated with hemeproteins, was markedly increased in the presence of ascorbate (Table 2), suggesting that it could be an intermediate in the reaction, generating MDHA radicals as a result of the reduction of oxidized hemeprotein (Hossain and Asada 1985). It is interesting to note that there was only a slight NADH oxidation with MDHAR in the presence of ascorbate and metmyoglobin, a pentacoordinate Hb. The results are consistent with MDHAR operating to couple ascorbate reduction of metHb to the removal of MDHA, a strong oxidant. The reverse reaction of MDHA acting as a strong oxidant in the course of ascorbate-driven reduction of cytochrome b 561 has been shown (Takigami et al. 2003).
There are several other points that suggest MDHAR may have a function regenerating Hb from metHb in barley. The MDHAR gene is induced after 2 h of hypoxia (Klok et al. 2002) as is barley Hb (Taylor et al. 1994). The enzyme is a monomer and contains 1 mol of FAD per mole of enzyme. It has higher affinity to NADH as compared to NADPH and has optimum pH near 8.0 (Table 2 and Hossain and Asada 1985). It is strongly inhibited by pHMB and preserves its activity in the presence of FAD due to the suppression of electron transport from NADH to the bound FAD by SH-reagents (Hossain and Asada 1985). All these properties correspond to characteristics described for NAD(P)H-dependent NO-scavenging activity in root extracts (Igamberdiev et al. 2004).
While we have presented evidence supporting the role of barley MDHAR in NO scavenging, other reductases may also contribute to sustaining the Hb/NO cycle. The limitations to involvement would be the redox potential of the reductant relative to methemoglobin and the availability of the reductase protein. Thus, glutathione/glutathione reductase or thioredoxin/thioredoxin reductase are also possibilities.
To summarize, the molecular mechanism of Hb action in NO turnover does not directly involve the participation of the surface Cys79. The metHb produced as a result of NO turnover is reduced via a coupled reaction in which ascorbate is oxidized and regenerated via an NADH-dependent cytosolic monodehydroascorbate reductase. Both Hb and MDHAR constitute a system metabolizing NO to nitrate, referred to as an Hb/NO cycle, in conjunction with enzyme(s) forming NO from nitrate (Fig. 5). This cycle serves to maintain the redox and energy status of plant cells under hypoxic conditions (Igamberdiev and Hill 2004).

The Hb/NO cycle. NR Nitrate reductase, E NO sources including nitrate reductase and/or nitrite: NO reductase. NO is oxidized to nitrate by oxyhemoglobin [Hb(Fe2+)O2], which turns to metHb [Hb(Fe3+)]. MetHb is reduced in the ascorbate (ASC)-mediated monodehydroascorbate reductase (MDHAR) reaction. High affinity of Hb(Fe2+) to O2 results in its immediate oxygenation even at very low (nanomolar) O2 concentration. MDHA monodehydroascorbate (ascorbate free radical)
Abbreviations
- DEANO:
-
2-(N, N-diethylamino)-diazenolate-2-oxide
- DTT:
-
Dithiothreitol
- metHb:
-
Methemoglobin (ferric hemoglobin)
- rHb:
-
Recombinant non-mutated barley Hb
- mutHb:
-
Recombinant barley Hb with replacement of Cys79 by Ser
- MDHA:
-
Monodehydroascorbate (ascorbate free radical)
- MDHAR:
-
Monodehydroascorbate reductase
- NEM:
-
N-ethylmaleimide
- pHMB:
-
p-Hydroxymercuribenzoate
- SNP:
-
Sodium nitroprusside
- TCEP:
-
Tris(2-carboxyethyl)phosphine
References
Becana M, Klucas RV (1990) Enzymatic and nonenzymatic mechanisms for ferric leghemoglobin reduction in legume root nodules. Proc Natl Acad Sci USA 87:7295–7299
Bérczi A, Møller IM (1998) NADH-monodehydroascorbate oxidoreductase is one of the redox enzymes in spinach leaf plasma membranes. Plant Physiol 116:1029–1036
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brunori M, Giuffré A, Nienhaus K, Nienhaus GU, Scandurra FM, Vallone B (2005) Neuroglobin, nitric oxide, and oxygen: functional pathways and conformational changes. Proc Natl Acad Sci USA 102:8483–8488
Chernushevich IV, Ens W, Standing KG (1999) Orthogonal-injection TOFMS for analyzing biomolecules. Anal Chem 71:452A–461A
Dordas C, Hasinoff BB, Igamberdiev AU, Manac’h N, Rivoal J, Hill RD (2003) Expression of a stress-induced hemoglobin affects NO levels produced by alfalfa under hypoxic stress. Plant J 35:763–770
Dordas C, Hasinoff BB, Rivoal J, Hill RD (2004) Class-1 hemoglobins, nitrate and NO levels in anoxic maize cell-suspension cultures. Planta 219:66–72
Duff SMG, Wittenberg JB, Hill RD (1997) Expression, purification, and properties of recombinant barley (Hordeum sp.) hemoglobin. Optical spectra and reactions with gaseous ligands. J Biol Chem 272:16746–16752
Gardner PR (2005) Nitric oxide dioxygenase function and mechanism of flavohemoglobin, hemoglobin and their associated reductases. J Inorg Biochem 99:247–266
Gardner PR, Gardner AM, Martin LA, Salzman AL (1998) Nitric oxide dioxygenase: an enzymic function for flavohemoglobin. Proc Natl Acad Sci USA 95:10378–10383
Gardner PR, Martin LA, Hall D, Gardner AM (2001) Dioxygen-dependent metabolism of nitric oxide in mammalian cells. Free Radical Biol Med 31:191–204
Gladwin MT, Ognibene FP, Pannell LK, Nichols JS, Pease-Fye ME, Shelhamer JH, Schechter AN (2000) Relative role of heme nitrosylation and β-cysteine 93 nitrosation in the transport and metabolism of nitric oxide by hemoglobin in the human circulation. Proc Natl Acad Sci USA 97:9943–9948
Han TH, Fukuto JM, Liao JC (2004) Reductive nitrosylation and S-nitrosation of hemoglobin in inhomogeneous nitric oxide solutions. Nitric Oxide 10:74–82
Hargrove MS, Brucker EA, Stec B, Sarath G, Arredondo-Peter R, Klucas RV, Olson JS, Phillips GN Jr (2000) Crystal structure of a nonsymbiotic plant hemoglobin. Structure 8:1005–1014
Hausladen A, Gow A, Stamler JS (2001) Flavohemoglobin denitrosylase catalyzes the reaction of a nitroxyl equivalent with molecular oxygen. Proc Natl Acad Sci USA 98:10108–10112
Hernandez-Urzua E, Mills CE, White GP, Contreras-Zentella ML, Escamilla E, Vasudevan SG, Membrillo-Hernandez J, Poole RK (2003) Flavohemoglobin Hmp, but not its individual domains, confers protection from respiratory inhibition by nitric oxide in Escherichia coli. J Biol Chem 278:34975–34982
Herold S, Rock G (2005) Mechanistic studies of the oxygen-mediated oxidation of nitrosylhemoglobin. Biochemistry 44:6223–6231
Hill RD (1998) What are hemoglobins doing in plants? Can J Bot 76:707–712
Hossain MA, Asada K (1985) Monodehydroascorbate reductase from cucumber is a flavin adenine dinucleotide enzyme. J Biol Chem 260:12920–12926
Hossain MA, Nakano Y, Asada K (1984) Monodehydroascorbate reductase in spinach chloroplasts and its participation in regeneration of ascorbate for scavenging hydrogen peroxide. Plant Cell Physiol 25:385–395
Hrinczenko BW, Schechter AN, Wojtkowski TL, Pannell LK, Cashon RE, Alayash AI (2000) Nitric oxide-mediated heme oxidation and selective beta-globin nitrosation of hemoglobin from normal and sickle erythrocytes. Biochem Biophys Res Commun 275:962–967
Igamberdiev AU, Hill RD (2004) Nitrate, NO and haemoglobin in plant adaptation to hypoxia: an alternative to classic fermentation pathways. J Exp Bot 55:2473–2482
Igamberdiev AU, Seregélyes C, Manac’h N, Hill RD (2004) NADH-dependent metabolism of nitric oxide in alfalfa root cultures expressing barley hemoglobin. Planta 219:95–102
Kaur R, Pathania R, Sharma V, Mande SC, Dikshit KL (2002) Chimeric Vitreoscilla hemoglobin (VHb) carrying a flavoreductase domain relieves nitrosative stress in Escherichia coli: new insight into the functional role of VHb. Appl Environ Microbiol 68:152–160
Klok EJ, Wilson IW, Wilson D, Chapman SC, Ewing RM, Somerville SC, Peacock WJ, Dolferus R, Dennis ES (2002) Expression profile analysis of the low-oxygen response in Arabidopsis root cultures. Plant Cell 14:2481–2494
Kobayashi G, Nakamura T, Ohmachi H, Matsuoka A, Ochiai T, Shikama K (2002) Yeast flavohemoglobin from Candida norvegensis. Its structural, spectral, and stability properties. J Biol Chem 277:42540–42548
Kundu S, Trent JT III, Hargrove MS (2003) Plants, humans and hemoglobins. Trends Plant Sci 8:387–393
Loboda AV, Krutchinsky AN, Bromirski M, Ens W, Standing KG (2000) A tandem quadrupole/time-of-flight mass spectrometer with a matrix-assisted laser desorption/ionization source: design and performance. Rapid Commun Mass Spectrom 14:1047–1057
Mills CE, Sedelnikova S, Soballe B, Hughes MN, Poole RK (2001) Escherichia coli flavohaemoglobin (Hmp) with equistoichiometric FAD and haem contents has a low affinity for dioxygen in the absence or presence of nitric oxide. Biochem J 353:207–213
Minning DM, Gow AJ, Bonaventura J, Braun R, Dewhirst M, Goldberg DE, Stamler JS (1999) Ascaris haemoglobin is a nitric oxide-activated 'deoxygenase'. Nature 401:497–502
Moran JF, Sun Z, Sarath G, Arredondo-Peter R, James EK, Becana M, Klucas RV (2002) Molecular cloning, functional characterization, and subcellular localization of soybean nodule dihydrolipoamide reductase. Plant Physiol 128:300–313
Nie XZ, Hill RD (1997) Mitochondrial respiration and hemoglobin gene expression in barley aleurone tissue. Plant Physiol 114:835–840
Perazzolli M, Dominici P, Romero-Puertas MC, Zago E, Zeier J, Sonoda M, Lamb C, Delledonne M (2004) Arabidopsis nonsymbiotic hemoglobin AHb1 modulates nitric oxide bioactivity. Plant Cell 16:2785–2794
Poole RK (2005) Nitric oxide and nitrosative stress tolerance in bacteria. Biochem Soc Trans 33:176–180
del Río LA, Corpas FJ, Sandalio LM, Palma JM, Gómez M, Barroso JB (2002) Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. J Exp Bot 53:1255–1272
Sano S, Asada K (1994) cDNA cloning of monodehydroascorbate radical reductase from cucumber—a high degree of homology in terms of amino acid sequence between this enzyme and bacterial flavoenzymes. Plant Cell Physiol 35:425–437
Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen A, Shevchenko A, Boucherie H, Mann M (1996) Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci USA 93:14440–14445
Sowa AW, Duff SMG, Guy PA, Hill RD (1998) Altering hemoglobin levels changes energy status in maize cells under hypoxia. Proc Natl Acad Sci USA 95:10317–10321
Takigami T, Takeuchi F, Nakagawa M, Hase T, Tsubaki M (2003) Stopped-flow analyses on the reaction of ascorbate with cytochrome b 561 purified from bovine chromaffin vesicle membranes. Biochemistry 42:8110–8118
Taylor ER, Nie XZ, MacGregor AW, Hill RD (1994) A cereal haemoglobin gene is expressed in seed and root tissues under anaerobic conditions. Plant Mol Biol 24:853–862
Xu XL, Cho M, Spencer NY, Patel N, Huang Z, Shields H, King SB, Gladwin MT, Hogg N, Kim-Shapiro DB (2003) Measurements of nitric oxide on the heme iron and beta-93 thiol of human hemoglobin during cycles of oxygenation and deoxygenation. Proc Natl Acad Sci USA 100:11303–11308
Weiland TR, Kundu S, Trent JT, Hoy JA, Hargrove MS (2004) Bis-histidyl hexacoordination in hemoglobins facilitates heme reduction kinetics. J Am Chem Soc 126:11930–11935
Wittenberg JB, Wittenberg BA (2003) Myoglobin function reassessed. J Exp Biol 206:2011–2020
Acknowledgements
The skilful technical assistance of Doug Durnin is gratefully acknowledged. We thank Professor Werner Ens for mass spectrometry support. This work was supported by the Natural Sciences and Engineering Research Council of Canada (OGP 4689).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Igamberdiev, A.U., Bykova, N.V. & Hill, R.D. Nitric oxide scavenging by barley hemoglobin is facilitated by a monodehydroascorbate reductase-mediated ascorbate reduction of methemoglobin. Planta 223, 1033–1040 (2006). https://doi.org/10.1007/s00425-005-0146-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-005-0146-3