
Abstract
Soybean [Glycine max (L.) Merr.] contains two proteins called vegetative storage proteins (VSPs) that function as temporary storage reserves, but are also closely related to plant acid phosphatases of the haloacid dehalogenase (HAD) superfamily. This study examined the biochemical basis for the relatively low catalytic activity previously reported for these VSPs. The specific activity of purified recombinant VSPα on GMP was about 40-fold lower than for a related soybean root nodule acid phosphatase (APase), which had a specific activity of 845 U mg−1 protein. Conversion of Ser106 to Asp increased VSPα activity about 20-fold. This Asp residue is present in nodule APase and is a highly conserved nucleophile in the HAD superfamily. Related VSPs from cultivated soybean and from three wild perennial soybeans, as well as a pod storage protein (PSP) from Phaseolus vulgaris L. all lack the catalytic Asp, suggesting they too are catalytically inefficient. Phylogenetic analysis showed the VSPs and PSP are more closely related to each other than to 21 other VSP-like proteins from several plant species, all of which have the nucleophilic Asp. This study suggests that loss of catalytic activity may be a requirement for the VSPs and PSP to function as storage proteins in legumes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A wide variety of plant species abundantly accumulate proteins that function as storage reserves. The best studied of these are the seed storage proteins synthesized during seed development and then degraded during germination (Derbyshire et al. 1976). Also well documented are numerous vegetative storage proteins (VSPs) that accumulate in other plant tissues. These proteins are often synthesized in sink tissues, such as developing leaves, and then may be degraded within the same growing season to contribute to the needs of developing seeds or other plant sinks (Wittenbach 1983; for review, see Staswick 1994). Other VSPs accumulate in overwintering tissues, such as tree bark (O’Kennedy and Titus 1979), tubers and non-tuberous roots (for review, see Bewely 2002), and can supply amino acids to reinitiate growth in the spring.
Proteins are generally classified as storage reserves based on their abundance and pattern of accumulation and degradation. Some have no other known biological activity, but others are enzymatically active or have other biological properties that can raise questions about what their primary function is. For example, certain lectins of bark tissue have been considered storage proteins (Greenwood et al. 1986), but they may also have other roles, such as plant defense (Peumans and Van Damme 1995). The abundant potato storage reserve patatin has lipid acyl hydrolase activity (Andrews et al. 1988) and the sporamin tuber protein of sweet potato is related to trypsin inhibitors (Yeh et al. 1997). An abundant storage protein from the Andean tuber crop oca has antimicrobial activity, which may also be important for protection against pathogens (Flores et al. 2002). A functional β-amylase also appears to serve as a storage reserve in alfalfa taproots and it is not clear that enzymatic activity has an important function (Gana et al. 1998).
There are also storage proteins that are clearly derived from active proteins based on sequence homology, but they have lost some or all of their activity. Among these is an abundant but inactive RNAse-like storage protein from rhizomes of Calystegia sepium (hedge bindweed; Van Damme et al. 2000) and bark lectin-like proteins from Cladastris lutea (yellow wood) and Sambucas nigra (black elderberry) that lack sugar-binding capacity (Van Damme et al. 1995; Chen et al. 2002). Loss of these biological properties may have occurred because they were redundant and offered no selective advantage, or because inactivation was required for the protein to function as an abundant storage reserve.
Soybean (Glycine max) accumulates a limited number of VSPs to high level in developing vegetative sink tissues and these are later preferentially degraded. The major soybean VSPs are VSPα and VSPβ, two glycoproteins of about 27 kDa that are around 80% identical in sequence (for review, see Staswick 1994). Consistent with a storage role, the corresponding VSP genes are regulated developmentally in a source/sink-dependent manner, and are induced by the removal of seed pods and by the availability of excess nitrogen (Mason and Mullet 1990; Staswick et al. 1991; Mason et al. 1992; Sadka et al. 1994). On the other hand, their induction by stresses such as drought and high salt could suggest other roles are also possible.
Soybean VSPα and β are related to tomato acid phosphatase-1 (Aarts et al. 1991; Williamson and Colwell 1991). The VSPs occur as both homo and heterodimers and have acid phosphatase activities on o-carboxyphenyl phosphate ranging from 0.3 U mg−1 protein for VSPα homodimer to 10 U mg−1 protein for the heterodimer (DeWald et al. 1992). Compared to several other plant acid phosphatases these values are somewhat low, raising the question of the relevance of VSP catalytic activity. Although VSP levels and total acid phosphatase activity increased dramatically in leaves of depodded soybean plants, VSPα and β accounted for no more than 0.1% of the total acid phosphatase activity in these leaves (Staswick et al. 1994). Rather, an unrelated 51-kDa phosphatase was responsible for most of the activity, having a specific activity of 1,353 U mg−1 protein.
Soybean root nodules also contain an acid phosphatase with 69% sequence identity with the VSPs (Penheiter et al. 1997; Penheiter 1998). Interestingly, the specific activity of recombinant nodule acid phosphatase (APase) on its optimal substrate (monophosphates) was about 30-fold higher than that previously reported for purified soybean VSPα/β, which was most active on polyphosphates (DeWald et al. 1992; Penheiter et al. 1998). The VSPs and nodule APase contain three short sequence motifs that suggest they belong to the bacterial class-B family of acid phosphatases, which are members of the haloacid dehalogenase (HAD) superfamily (Koonin and Tatusov 1994; Penheiter 1998; Morais et al. 2000; Selengut 2001). In plant and bacterial class-B APases the motif-I consensus sequence is FD[I,V]D[D,E]TXL. By analogy with the extensively characterized bacterial l-2-HAD (Liu et al. 1995) it was suggested that the first Asp in motif I is critical for enzyme activity because it makes a nucleophilic attack on the substrate phosphate (Penheiter 1998). However, the role of this Asp has not been experimentally demonstrated for plant APases. In l-2-HAD and in several related acid phosphatases, motif 1 is at the amino terminus (Selengut 2001), whereas it is near the center of the plant APases and VSPs. This suggests that the structure of the bacterial and plant proteins is somewhat different. Therefore, it is conceivable that one of the other acidic residues of motif I could play the catalytic role.
Interestingly, the putative catalytic Asp is present in nodule APase (Asp116), but it is substituted by Ser106 and Gly106 in VSPα and VSPβ, respectively. It was suggested that the relatively low enzyme activity of soybean VSPs might result from this substitution of Asp in motif I (Penheiter 1998). Recombinant VSP has not been previously reported so it has not been possible to directly compare its activity with recombinant nodule APase.
The purpose of this study was to investigate the biochemical basis for the apparent low catalytic activity of soybean VSPs. Specifically, we tested whether restoring the putative catalytic Asp in motif I would elevate the activity of VSPα when expressed as a glutathione S-transferase (GST) fusion protein in Escherichia coli. We also compared the sequence of VSP cDNAs isolated from distant relatives of cultivated soybean and evaluated the phylogenetic relationship among 25 other plant proteins related to soybean VSPs.
Materials and methods
Plant and DNA material used
Perennial soybeans from the subgenus Glycine were grown in a temperature-regulated greenhouse and young leaves were collected for RNA extraction as described by Staswick (1997). cDNA libraries were constructed in lambda UNI ZAP-XR vectors (Stratagene) with reverse-transcribed poly(A)-mRNA from G. falcata and G. tomentella leaves. 32P-labeled VSPA and VSPB DNA was used as a probe to screen the cDNA libraries. Low-stringency hybridizations were done overnight at 55°C and blots were washed twice with 1× SSC, 0.1% SDS solution at 53°C. Plaques that produced positive signals were selected and re-screened to homogeneity. After confirming the clones’ relationship to the VSPA/B sequences by restriction endonuclease digestion, one cDNA from each species was sequenced.
A partial cDNA that included motif I for a VSP homologue was obtained from G. curvata. Total RNA was used with the 3′ RACE System from GIBCO–BRL according to the manufacturer’s instructions. The VSPA-related fragment was amplified with primers for sites flanking motif I that are relatively conserved in the other VSPs (Fwd: 5′ GTGGAAGCACACAACATC 3′, VSPA nucleotides 145–162; Rev: 5′ TCTTCCTGACAAGAATA 3′, VSPA nucleotides 485–502). PCR products were cloned into pGem-T-Easy vector (Promega) and putative VSP clones were sequenced, translated and aligned with other VSP sequences.
Preparation and expression of recombinant APase and VSPA
The soybean [Glycine max (L.) Merr.] root nodule APase and VSPA cDNAs were amplified by PCR with primers designed to eliminate the amino terminal signal peptide and incorporate XhoI sites compatible with the GST fusion expression vector. The primers used for PCR of APase were:
-
Fwd, 5′ GATCTCGAGATTCCGGAGGTATCATGC 3′;
-
Rev, 5′ TCACTCGAGTCAACTAATGTAGTACATGGGATCAGG 3′.
For VSPA the primers were:
-
Fwd, 5′ CCACTCGAGAACACTGGCTATGGTG 3′;
-
Rev, 5′ GATCTCGAGCTACTGAATGTAGTACAG 3′.
The PCR products were cloned into pGem-T-Easy vector, sequenced to verify their integrity, and then fused into the XhoI site of pGEX-4T-1. The APase:pGEX-4T-1 and VSPA:pGEX-4T-1 were transformed into E. coli strain BL21. For expression, cultures in Luria-Bertani (LB) medium containing 100 μg ml−1 ampicillin were grown to OD600=0.6 and then induced at room temperature with 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG) for 4–6 h. Cells were harvested and washed once in Mes–NaOH (pH 6.0), and were stored as pellets at −80°C if not used immediately. Proteins were evaluated by SDS–PAGE and the molecular weights of the GST–APase and GST–VSPα fusions were approximately 52 kDa, as expected.
PCR was also used to introduce a site-specific mutation in VSPα converting Ser106 to Asp using the wild-type VSPA as template. Because the targeted region was in the internal region of the cDNA, four oligonucleotides were used to generate two PCR fragments that could be joined by ligation at a common ClaI restriction endonuclease site. Primers for the 5′-end cDNA fragment were:
-
Fwd: 5′ GATCTCGAGAACACTGGCTATGGTG 3′;
-
Rev: 5′ ATCGATATCGAACACAAATGTGTCCTTGGG 3′.
Primers for the 3′-end cDNA fragment were:
-
Fwd: 5′ GTGTTCAGTATCGATGGCACCG 3′;
-
Rev: 5′ GATCTCGAGCTACTGAATGTAGTACAG 3′.
The two PCR products were ligated into pGem-T-Easy, verified by sequencing and the reconstructed insert was then cloned into the XhoI site of pGEX4-T-1
Purification of GST fusion proteins
The GST–APase and GST–VSPα were partially purified from sonicated E. coli extracts by ammonium sulfate precipitation and ion-exchange chromatography. Ammonium sulfate was added to 60% saturation and the extract left on ice for 30 min. The precipitate was collected by centrifugation (12,000 rpm, 20 min) and resuspended in 4 ml of 50 mM Mes–NaOH (pH 6.0). The suspension was desalted by dialysis at 4°C against 20 mM Tris–HCl, pH 8.0 (two exchanges, 6–12 h). The dialyzed sample was then centrifuged at 15,000 rpm for 20 min. The supernatant was loaded onto a DEAE-50 cellulose column pre-equilibrated with 50 mM Mes–NaOH (pH 6.0). The eluted sample was concentrated osmotically and assayed for protein and APase activity. Fractions were analyzed by SDS–PAGE on 12% minigels (Bio-Rad) following the manufacturer’s procedures. Protein concentrations were determined with the Bio-Rad DC protein assay according to the protocol supplied by the company. Activity of acid phosphatase was examined by monitoring phosphate released from p-nitrophenol phosphate (pNPP) at 405 nm. Activity determinations were routinely performed in triplicate at room temperature in 0.05 M Mes–NaOH (pH 6.0) containing 1 mM MgCl2.
Enzymes for the analysis of mutated VSPα were affinity-purified with glutathione agarose. Induced cells were pelleted, resuspended in 10 mM Tris–HCl, 5 mM NaCl, and 3 mM MgCl2 at pH 7.5, and then sonicated. Insoluble material was precipitated by centrifugation at 10,000 rpm for 10 min, and the supernatant was combined with glutathione agarose beads for 30 min at 4°C with continuous rotation. Beads were prepared in the same buffer as used for cell lysis. Recombinant proteins bound to the beads were washed 5 times with the same buffer and then digested with Thrombin Protease (Amersham) at 4°C for 6 h to release each protein from the GST fusion. The protease-digested supernatant was collected after centrifugation at 300 rpm for a few seconds. Protein amount was estimated using the Bio-Rad DC assay according to the manufacturer’s instructions; bovine serum albumin was used as a standard. Proteins were analyzed by SDS–PAGE on 12% minigels (Bio-Rad) according to the manufacturer’s procedure.
Assays for enzyme activity
Phosphatase activity was determined by the method of Fiske and Subbarow (1925) using a kit from Sigma following the manufacturer’s instructions. A variety of phosphorylated compounds were used as substrates: ADP, GMP, FMN, pNPP, P-tyrosine, and tripolyphosphate, each at 3 mM. Acid phosphatase reactions were performed in triplicate at 37°C in 0.05 M Mes–NaOH containing 1 mM MgCl2, pH 6.0. Kinetic measurements were determined by activity assays at pH 6.0 with pNPP and GMP substrate concentrations ranging from 0 to 6 mM. The kinetic parameters V max and K m were evaluated by the Michaelis–Menten method.
Analysis of DNA and protein sequence
DNA sequencing was done by the University of Nebraska Genome Core Research Facility. DNA and protein sequence was analyzed with SeqWeb v. 2 (Accelrys). For phylogenetic analysis VSP-related plant sequences were identified by blastP searches of the non-redundant translated database using soybean VSPα (AAA34020) and tomato APase-1 (AAA34135) as the query sequences. Non-redundant sequences were judged to be full length based on comparison with those known to be complete (e.g. soybean VSPs, soybean nodule APase, tomato APase-1). Phylogenetic relationships were analyzed with Grow Tree using the default settings (Kimura distance, neighbor distance, blossum62 scoring matrix, gap penalty 8). The tree was displayed using the Nexus output with Tree View 1.6.6 (http://taxonomy.zoology.gla.ac.uk/rod/rod.html). Sequences for the motif-I region included those used for tree construction, three partial sequences from the blastP search (PPI309082, AY106317, CAB71336) and the partial sequence we derived for G. curvata.
Results
The putative nucleophilic Asp is substituted in VSPs from perennial soybeans
We first isolated cDNA clones from wild perennial relatives of cultivated soybean in order to determine whether the absence of the putative catalytic Asp was unique to the two G. max VSPs, or whether this was more generally the case in the Glycine genus. Full-length clones from G. falcata and G. tomentella had predicted open reading frames encoding 253 amino acids, compared with 254 for VSPα and VSPβ from G. max. Analysis with Grow Tree indicated the perennial proteins were more closely related to VSPα than VSPβ. The proteins from G. falcata and G. tomentella shared 85% sequence identity with each other and were each 81 and 76% identical with VSPα and VSPβ, respectively. Based on this evidence we have classified these as VSPα proteins. Sequence identity with soybean nodule APase was 64 and 62% for the G. falcata and G. tomentella proteins, respectively.
The sequence of 22 amino acids surrounding motif I for each of these proteins is shown in Fig. 1, along with the same region from previously characterized G. max VSPs and from the VSP-like pod storage protein (PSP) from Phaseolus vulgaris (Zhong et al. 1997). In addition, a translated sequence from a partial cDNA from G. curvata is also shown. Like the G. max VSPs, those from all three perennial species lacked the putative catalytic Asp, which in each case was substituted by a Ser residue. The Asp is also substituted in PSP by Asn. Together these are denoted as Group-I proteins. The sequence from this region was also compared to that of 25 other plant VSP/APase-like proteins. Included in this second group are two enzymes that are known to have acid phosphatase activity, the soybean nodule APase and tomato Apase-1 (Aarts et al. 1991; Williamson and Colwell 1991; Penheiter et al. 1997). All of the Group-II proteins from eight different plant species contained the Asp that is predicted to have a catalytic role. Two arabidopsis genes have been called VSP1 and VSP2 based on predicted amino acid sequence similarity with the soybean VSPs (Utsugi et al. 1998), but the proteins they encode have not been evaluated for their potential enzymatic activity. The remainder are putative proteins based on complete or partial database nucleotide sequences. In addition to the putative nucleophilic Asp, two other residues conserved in Group-II proteins are absent in Group I. These are Trp three residues upstream and Asp or Glu three residues downstream of the putative nucleophilic Asp

Comparison of conserved motif I from soybean VSPs and related plant APases. The invariant nucleophilic Asp of Group-II proteins is substituted by the boxed residue indicated for each Group-I protein (Ser106 in G. max VSPα). Invariant or highly conserved Group-II residues are shaded and represented similarly if present in Group I. Species designations are At, Arabidopsis thaliana; Gc, Glycine curvata; Gf, Glycine falcata; Gt, Glycine tomentella; Gm, Glycine max; Hv, Hordeum vulgare; Le, Lycopersicon esculentum; Os, Oryza sativa; Pv, Phaseolus vulgaris; Pp, Pinus pinaster; Zm, Zea maize
Nodule APase expressed in E. coli is catalytically active
Recovery of soybean VSPs and nodule APase in active form from E. coli had not previously been reported. In order to test the feasibility of obtaining these enzymes from bacteria, VSPα and nodule APase were each expressed as GST fusion proteins. The constructs excluded the signal peptide found in each full-length cDNA. GST–APase and GST–VSPα were partially purified by DEAE ion-exchange chromatography following precipitation with 60% ammonium sulfate (Table 1). The specific activity of GST–APase was about 72 μmol min−1 mg−1 using 3 mM pNPP as the substrate. This was similar to the activity reported previously for this enzyme expressed in yeast and purified in a similar manner (66.7 μmol min−1 mg−1 with 5 mM pNPP; Penheiter 1998). This result indicated that APase could be recovered from E. coli in active form.
GST–APase was assayed for its pH optimum with pNPP as substrate. Figure 2 shows that the enzyme had a broad pH activity profile typical of acid phosphatases, with a maximum occurring around pH 6.0. This is similar to the pH optimum found for APase expressed in yeast (Penheiter 1998). Therefore, all further kinetic studies were carried out at this pH. To evaluate the substrate specificity for GST–APase, several phosphorylated substrates were tested (Table 2). The highest activity was observed with the monophosphorylated substrate 5′-GMP, which was in agreement with previous results for native and recombinant APase from yeast (Penheiter 1998). Several other compounds (FMN, ADP, and P-tyrosine) were also dephosphorylated, but at a much lower rate than 5′-GMP. In contrast, we found no evidence that tripolyphosphate was hydrolyzed. Based on these data, the kinetics for the two most active substrates was determined and is shown in Table 3. Highest affinity was observed for 5′-GMP (K m=0.9 mM). The observed K m value for pNPP was somewhat higher at 8.8 mM. The V max value for pNPP was greater than that calculated for 5′-GMP and the V max/K m values for pNPP and 5′-GMP were 279 and 1,926, respectively.

The effect of pH on soybean (Glycine max) nodule APase activity. Enzyme purified from Escherichia coli was assayed with pNPP as a substrate at the indicated pH values. Activity is expressed as percent of the value at pH 6. Mes–NaOH was the buffer for pH 5 and 6, and Tris–HCl for pH 7–9
In contrast to GST–APase the VSPα fusion protein possessed little phosphatase activity. The only significant activity among the substrates tested was with GMP and pNPP (22.6 and 3.2 U mg−1, respectively). This was about 20-fold lower than for GST–APase with the same substrates. Extracts from E. coli expressing GST alone exhibited no detectable activity with any of these substrates (data not shown), indicating that the low activity observed for GST–VSPα was in fact due to the recombinant enzyme and not endogenous phosphatases co-purified from E. coli. As observed for nodule GST–APase, P-tyrosine, ADP and tripolyphosphate, were poor substrates for GST–VSPα.
Previous studies showed that cyclic nucleotides inhibited purified nodule APase (Penheiter 1998). To determine the effect of this inhibitor on GST–APase, enzyme activity was assayed in the presence of cAMP. Substrate pNPP concentrations ranging from 2.5 to 20 mM, and two concentrations of inhibitor, 5 and 50 μM cAMP, were used. The K i value of GST–APase was about 15 μM (not shown), only slightly higher than that determined for APase from nodule extract (12 μM; Penheiter 1998). Collectively, these results indicate that in all regards tested nodule GST–APase from E. coli behaves similarly to native APase.
Mutation of VSPα increases its acid phosphatase activity
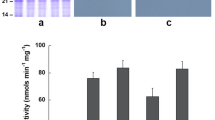
We next examined whether conversion of Ser106 to Asp in VSPα would alter its phosphatase activity relative to the wild-type protein. For this assay, highly purified enzymes were recovered by isolation on glutathione agarose followed by cleavage with thrombin to isolate the enzyme from GST. Analysis of wild-type and mutant VSPα, as well as nodule APase, by SDS–PAGE indicated the major protein bands in each case had the expected molecular weights of approximately 27 kDa after thrombin cleavage (Fig. 3). For each protein only minor contaminants were evident.

Analysis of glutathione agarose-purified GST fusion proteins. Major bands at about 27 kDa are wild-type nodule APase, VSPα and mutated VSPαAsp106, as indicated. Position of molecular markers is indicated to the left in kDa. Purified proteins were loaded on 12% SDS–PAGE gels
The result of the assay of purified enzyme with five phosphorylated substrates is shown in Table 4. The activity for thrombin-cleaved APase on GMP was 845 U mg−1. This was the most active substrate, as noted earlier for the partially purified GST–APase. Wild-type VSPα cleaved from GST yielded 30- to 40-fold lower activity on both pNPP and GMP. In contrast, the conversion of Ser106 to Asp dramatically increased the enzyme activity of VSPα on GMP to 439 U mg−1, almost 20-fold higher than for wild-type VSPα and about half that of nodule APase. VSPα Asp106 also hydrolyzed pNPP at 35 U mg−1 and P-tyrosine at 20 U mg−1. This was about one-third of the level found for nodule APase with pNPP as a substrate and about 10-fold higher than for wild-type VSPα on these substrates. As for nodule APase, the mutant VSPα had no detectable activity towards FMN or tripolyphosphate. These results establish that the major reason for the low catalytic activity of VSPα is the single amino acid substitution of Ser106 for the catalytic Asp found in this family of APases.
Discussion
Penheiter et al. (1998) previously reported that nodule APase expressed in E. coli as a His tag fusion was found in inclusion bodies rather than as a soluble protein. Our results show that expression of both VSPα and nodule APase as GST fusions is a viable means to obtain these enzymes in soluble and active form from bacteria. This expression system permitted the rapid affinity purification of the enzymes, which facilitated the study of VSPα by mutational analysis. The specific activity of the glutathione-purified and thrombin-cleaved APase on GMP was about 2-fold higher than that purified by ion exchange. The substrate specificity was also similar to that found for the native enzyme, confirming that purification by this method yields enzyme that is representative of the natural enzyme. We were also able to directly compare the activity of recombinant nodule APase with that of VSPα, because the two enzymes were produced and purified under identical conditions. Wild-type VSPα had about 40-fold lower activity than nodule APase, confirming previous indications that soybean VSP is a relatively weak acid phosphatase. A recent study showed that a marked down-regulation of the VSP genes, resulting in only about 2% of the normal VSP protein, had no adverse effect on plant growth or productivity (Staswick et al. 2001). While this does not necessarily mean that VSPs have no catalytic role, at least the role is not essential and is possibly made redundant by other enzymes. The same is apparently true for the storage role that has been assumed for these VSPs.
Site-specific mutation of the first Asp of motif I in bacterial l-2-HAD and in magnesium-dependent acid phosphatase-1 from mouse has shown this residue is critical for catalytic activity (Liu et al. 1995; Selengut 2001). The functional relevance of this residue had not previously been determined in plant enzymes. This was important to establish because all Group-II plant proteins, including two demonstrated to be acid phosphatases, contain two additional acidic residues just down stream of the proposed catalytic Asp in motif I (DxD[D/E]). It was conceivable that one of these, rather than the first Asp, might be structurally positioned to act as a nucleophile in these enzymes. We found that substitution of Ser106 withAsp at the position corresponding with the proposed catalytic Asp increased VSPα activity nearly 20-fold, confirming its essential role in catalysis in the plant enzymes. Interestingly, all Group-I proteins also lacked the invariant Trp and the conserved acidic residue found in Group-II proteins corresponding to positions 103 and 109 in VSPα, respectively. It is possible that these residues also enhance acid phosphatase activity, perhaps affecting the substrate-binding site.
Although Asp106 is clearly of major importance, wild-type recombinant VSPα did have low activity, as was previously reported for native VSPs (DeWald et al. 1992). It is not clear why this is so if Asp106 is essential for catalysis. All Group-I VSPs retain the Asp corresponding to position 108 in VSPα. It would be of interest to determine whether Asp108 can also act as the nucleophile for catalysis in VSPs, if only inefficiently. VSPαAsp106 had only half the activity of nodule APase. It is possible that other residues in VSPα are also suboptimal for activity. The fact that the VSPβ homodimer purified from plants had a specific activity about 10-fold higher than the VSPα homodimer (DeWald et al. 1992) supports this possibility.
In contrast to the earlier finding of highest activity on polyphosphates for native VSPα/β (DeWald et al. 1992), we found no detectable activity on this substrate. One possible explanation is that we evaluated only VSPα, which presumably forms a homodimer in E. coli. The native VSPα homodimer from plant extracts had much lower activity overall than VSPα/β and was relatively less active on polyphosphates than was the heterodimer. We chose to investigate VSPα because it is the more abundant of the two subunits in soybean during normal plant growth. The previous analysis of native VSP (DeWald et al. 1992) also did not include GMP, which was the substrate giving the highest activity in our analysis. It will be important in future studies to determine the level of activity and substrate specificity for the β VSP that has had Asp106 restored. VSPβAsp106 could be expressed alone in E. coli and along with VSPαAsp106, in which case it could presumably form the heterodimer as in soybean.
The sequence of VSPs from perennial soybeans had not previously been reported. We characterized two new VSP cDNAs and found they encode proteins with high sequence identity to the G. max VSPs. The VSPs from the perennial Glycine spp. are nearly identical in size to the G. max VSPs and are expected to have signal peptides of 29 amino residues. Previous analysis indicated there was considerable apparent size heterogeneity among proteins from the perennials that cross-reacted with VSP antisera (Staswick 1997). Our results here suggest that part of the heterogeneity on SDS–PAGE is due to variable mobility caused by charge differences or glycosylation, rather than differences in polypeptide length. This is also the basis for the apparent size difference between G. max VSPα and VSPβ. In the earlier study, leaf extracts from G. falcata produced a single VSP immuno cross-reacting band while G. tomentella yielded two distinct bands. This suggests there may be a second gene in G. tomentella, possibly a VSPβ-type protein as in G. max. Like the G. max VSPs, those from the perennial species also lacked the first Asp residue in motif I, suggesting that they are probably catalytically inefficient as well. It appears that the loss of the catalytic Asp originated before the domestication of Glycine max. We would also predict that the enzyme activity of P. vulgaris PSP would be low due to the absence of the catalytic Asp in this protein as well.
A phylogenetic analysis of the full-length sequences of 30 VSP and VSP-like plant proteins suggests that catalytic inactivation of VSPs may not be a recent event (Fig. 4). Of the currently known VSP-like plant proteins the one most closely related to the Glycine VSPs is PSP from P. vulgaris. Together, these constitute the Group-I proteins described in Fig. 1. All of the Group-II proteins have the nucleophilic Asp, including the G. max nodule and pathogenesis-associated APases (Acc. No. BAB86895), and are more distantly related to the VSPs than is PSP. These relationships suggest that enzymatic inactivation of an ancestral APase may have occurred after the divergence of VSPs from soybean root nodule APase, but before the divergence of Phaseolus from Glycine. We cannot, however, rule out the possibility that PSP lost the nucleophilic Asp independently of the soybean VSPs.

Phylogenetic analysis of plant VSPs and VSP-like APases. Group-II proteins lacking the nucleophilic Asp residue are boxed. Proteins are designated by a two-letter genus/species designation (see Fig. 1), followed by the database accession number
Our results document clearly that inactivity in VSPα is due primarily to a single amino acid substitution in an APase active site, because we have been able to restore activity to this protein by mutation. It is somewhat surprising that other residues necessary for catalysis had not also diverged, since there would not seem to be a reason to retain them in this inactive enzyme. It is possible that some of these must be maintained for proper folding and protein stability. However, the low overall level of sequence conservation among VSPs comprising Group-I proteins, as well as in the Group-II VSP-like acid phosphatases, indicates there is wide room for sequence variation in this family of proteins.
Other storage proteins also appear to be inactivated by simple mutations, although in each case mentioned below it has not been experimentally demonstrated that repairing the proposed defect alone is sufficient to restore activity. For example, sequence comparison suggested the RNase-like rhizome storage protein of Calystegia sepium is inactive due to substitution of a conserved His that is involved in catalysis in related plant RNAses (Van Damme et al. 2000). Type-2 ribosome-inactivating proteins (RIP) can also function as storage reserves. A critical component of their activity is a B-chain polypeptide with agglutinating activity. Sambucas nigra contains an abundant RIP-like protein that is inactive apparently because its B-chain lacks critical residues for agglutinating activity. Substitution of one of these in a closely related active B-chain reduced binding activity 50%, and a second mutation further lowered activity (Chen et al. 2002). Structural modeling of a lectin-like storage protein from Cladastris lutea also suggested its inactivity is due to three extra amino acids in the presumed carbohydrate-binding site (Van Damme et al. 1995). In each of these cases it would be of interest to correct the apparent defects in the inactive proteins and determine whether this alone would restore activity, or if additional residues must also be changed.
An intriguing question is whether loss of acid phosphatase activity was required for the VSPs to function as storage proteins, or whether they were recruited for storage proteins because they were redundant and not serving another critical role. Storage proteins by definition accumulate to a high level. It may be that over accumulation of an active phosphatase would raise the cellular phosphate pool to toxic levels. It is also possible that in their role as storage proteins, legume VSPs have simply lost a non-essential function, but that loss is not required. One way to address this question would be to express the gene for activated VSPαAsp106 in transgenic soybean under its native promoter to determine whether VSPs with high catalytic activity would be tolerated.
Because the complete genome is available we should have identified all putative VSP-like proteins from arabidopsis. None of the nine proteins that were found lacked the nucleophilic Asp. This suggests that if any of these are storage proteins their enzymatic inactivation is not required or involves other amino acids not yet identified. It has not been conclusively established that arabidopsis contains proteins that function specifically as storage reserves. Two genes have been called VSP1 and VSP2 based on sequence homology and some regulatory similarity with the soybean VSP genes. However, their relative abundance has not been well characterized, so it is not clear whether they could have a significant storage role. Retention of the catalytic Asp in these proteins suggests they may be active enzymes. It should also be noted that regulatory similarity with the soybean VSP genes does necessarily imply they encode storage proteins. For example, soybean VSPs respond to several stress factors that may also trigger the production of APases that function in stress response. Depodding of soybean plants dramatically elevated acid phosphatase activity from a 51-kDa protein, along with the rise in VSP level. But the low abundance of the 51-kDa protein indicates it is not a significant storage reserve (Staswick et al. 1994).
In summary, the five soybean VSPs from four distinct species of Glycine sequenced to date, as well as a PSP from P. vulgaris, all lack an Asp residue that we have demonstrated is critical for high catalytic function in VSPα. A single amino acid substitution replacing the native Ser with Asp in VSPα increased its acid phosphatase activity about 20-fold, to a level similar to that of nodule APase. This establishes that the major biochemical basis for the low enzymatic activity of VSPα is the substitution of this single amino acid residue, and that other residues necessary for high enzyme activity are retained in VSPα.
Abbreviations
- APase :
-
Acid phosphatase
- GST :
-
Glutathione S-transferase
- HAD :
-
Haloacid dehalogenase
- pNPP :
-
Para-nitrophenol phosphate
- PSP :
-
Pod storage protein
- RIP :
-
Ribosome inactivating protein
- VSP :
-
Vegetative storage protein
References
Aarts JMMJG, Hontelez GJ, Fischer P, Verkerk R, Kammen A, Zabel P (1991) Acid phosphatase-1, a tightly linked molecular marker for root-knot nematode resistance in tomato: from protein to gene, using PCR and degenerate primers containing deoxyinosine. Plant Mol Biol 16:647–661
Andrews DL, Beams B, Summers MD, Park WD (1988) Characterization of the lipid acyl hydrolase activity of the major potato (Solanum tuberosum) tuber protein, patatin, cloning and abundant expression in a baculovirus vector. Biochem J 252:199–206
Bewley JD (2002) Root storage proteins, with particular reference to taproots. Can J 80:321–329
Chen Y, Rouge P, Peumans WJ, van Damme EJ (2002) Mutational analysis of the carbohydrate-binding activity of the NeuAc(alpha-2,6)Gal/GalNAc-specific type 2 ribosome-inactivating protein from elderberry (Sambucus nigra) fruits. Biochem J 364:587–592
Derbyshire E, Writght DJ, Boulter D (1976) Legumin and vicilin storage proteins of legume seeds. Phytochemistry 15:3–24
DeWald DB, Mason HS, Mullet JE (1992) The soybean vegetative storage proteins VSPα and VSPβ are acid phosphatases active on polyphosphates. J Biol Chem 267:15658–15964
Fiske CH, Subbarow Y (1925) The colorometric detection of phosphorous. J Biol Chem 66:375–400
Flores T, Alape-Girón A, Flores-Díaz M, Flores HE (2002) Ocatin. A novel tuber storage protein from the andean tuber crop oca with antibacterial and antifungal activities. Plant Physiol 128:1291–1302
Gana JA, Kalengamaliro NE, Cunningham SM, Volenec JJ (1998) Expression of β-amylase from alfalfa taproots. Plant Physiol 118:1495–1506
Greenwood JS, Stinissen HM, Peumans WJ, Chrispeels MJ (1986) Sambucus nigra agglutinin is located in proteins bodies in the phloem parenchyma of the bark. Planta 167:275–278
Koonin EV, Tatusov RL (1994) Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity. J Mol Biol 244:125–132
Liu JQ, Kurihara T, Miyagi M, Esaki N, Soda K (1995) Reaction mechanism of L-2-haloacid dehalogenase of Pseudomonas sp YL. Identification of Asp10 as the active site nucleophile by 180 incorporation experiments. J Biol Chem 270:18309–18312
Mason HS, Mullet JE (1990) Expression of two vegetative storage protein genes during development and in response to water deficit, wounding and jasmonic acid. Plant Cell 2:569–579
Mason HS, DeWald DB, Creelman RA, Mullet JE (1992) Co-regulation of soybean vegetative storage protein gene expression by methyl jasmonate and soluble sugars. Plant Physiol 98:859–867
Morais MC, Zhang W, Baker AS, Zhang G, Dunaway-Mariano D, Allen KN (2000) The crystal structure of Bacillus cereus phosphonoacetaldehyde hydrolase: insight into catalysis of phosphorus bond cleavage and catalytic diversification within the HAD enzyme superfamily. Biochemistry 39:10385–10396
O’Kennedy BT, Titus JS (1979) Isolation and mobilization of storage proteins from apple shoot bark. Physiol Plant 45:419–424
Penheiter AR (1998) Biochemical and genetic characterization of an acid phosphatase/5′-nucleotidase from soybean root nodules. Dissertation, The University of Nebraska-Lincoln
Penheiter AR, Duff SMG, Sarath G (1997) Soybean root nodule and acid phosphatase. Plant Physiol 114:597–604
Penheiter AR, Klucas RV, Sarath G (1998) Purification and characterization of a soybean root nodule phosphatase expressed in Pichia pastoris. Protein Expr Purif 14:125–130
Peumans WJ, Van Damme EJM (1995) Lectins as plant defense proteins. Plant Physiol 109:347–352
Sadka A, DeWald DB, May GD, Park WD, Mullet JE (1994) Phosphate modulates transcription of soybean VSPB and other sugar-inducible genes. Plant Cell 6:739–749
Selengut J (2001) MDP-1 is a new and distinct member of the haloacid dehalogenase family of aspartate-dependent phosphohydrolases. Biochemistry 40:12704–12711
Staswick PE (1994) Storage proteins of vegetative plant tissue. Annu Rev Plant Physiol Plant Mol Biol 45:303–322
Staswick PE (1997) The occurrence and gene expression of vegetative storage proteins and a Rubisco Complex Protein in several perennial soybean species. J Exp Bot 48:1–7
Staswick PE, Huang J, Rhee Y (1991) Nitrogen and methyl jasmonate induction of soybean vegetative storage protein genes. Plant Physiol 96:130–136
Staswick PE, Papa C, Huang JF, Rhee Y (1994) Purification of the major soybean leaf acid phosphatase that is increased by seed-pod removal. Plant Physiol 104:49–57
Staswick PE, Zhang Z, Clemente TE, Specht JE (2001) Efficient down regulation of the major vegetative storage protein genes in transgenic soybean does not compromise plant productivity. Plant Physiol 127:1–8
Utsugi S, Sakamoto W, Murata M, Motoyoshi F (1998) Arabidopsis thaliana vegetative storage protein (VSP) genes: gene organization and tissue-specific expression. Plant Mol Biol 38:565–576
Van Damme EJ, Barre A, Bemer V, Rouge P, Van Leuven F, Peumans WJ (1995) A lectin and a lectin-related protein are the two most prominent proteins in the bark of yellow wood (Cladrastis lutea). Plant Mol Biol 29:579–598
Van Damme EJ, Hao Q, Barre A, Rouge P, Van Leuven F, Peumans WJ. (2000) Major protein of resting rhizomes of Calystegia sepium (hedge bindweed) closely resembles plant RNases but has no enzymatic activity. Plant Physiol 122:433–446
Williamson VM, Colwell G (1991) Acid phosphatase-1 from nematode resistant tomato. Plant Physiol 70:1544–1548
Wittenbach VA (1983) Effect of pod removal on leaf photosynthesis and soluble protein composition of field-grown soybeans. Plant Physiol 73:121–124
Yeh KW, Chen JC, Lin MI, Chen YM, Lin CY (1997) Functional activity of sporamin from sweet potato (Ipomoea batatas Lam.): a tuber storage protein with trypsin inhibitory activity. Plant Mol Biol 33:565–570
Zhong PY, Tanaka T, Yamauchi D, Minamikawa T (1997) A 28-kilodalton pod storage protein of French bean plants: purification, characterization, and primary structure. Plant Physiol 113:479–485
Acknowledgements
We thank Martha Rowe for her technical assistance in carrying out this study. This is Journal Series Paper No. 14,443. A contribution of the University of Nebraska Agricultural Research Division, Lincoln, NE 68583. This work was supported in part by NIH Grant Number 1 P20 RR16469 from the BRIN Program of the National Center for Research Resources (G.S.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Accession numbers for the VSPα sequences reported in this paper are from G. falcata, AY523602; G. tomentella, AY523603; G. curvata, AY523604
Rights and permissions
About this article
Cite this article
Leelapon, O., Sarath, G. & Staswick, P.E. A single amino acid substitution in soybean VSPα increases its acid phosphatase activity nearly 20-fold. Planta 219, 1071–1079 (2004). https://doi.org/10.1007/s00425-004-1294-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-004-1294-6