
Abstract
The introduction of next-generation sequencing technology has revealed that mutations in the gene that encodes titin (TTN) are linked to multiple skeletal and cardiac myopathies. The most prominent of these myopathies is dilated cardiomyopathy (DCM). Over 60 genes are linked to the etiology of DCM, but by far, the leading cause of DCM is mutations in TTN with truncating variants in TTN (TTNtvs) associated with familial DCM in ∼ 20% of the cases. Titin is a large (3–4 MDa) and abundant protein that forms the third myofilament type of striated muscle where it spans half the sarcomere, from the Z-disk to the M-line. The underlying mechanisms by which titin mutations induce disease are poorly understood and targeted therapies are not available. Here, we review what is known about TTN mutations in muscle disease, with a major focus on DCM. We highlight that exon skipping might provide a possible therapeutic avenue to address diseases that arise from TTNtvs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Titin is a giant myofilament that extends from the Z-disk (N-terminus) to the M-band (C-terminus) region of the sarcomere and is encoded by the TTN gene [11, 37, 43, 44, 69]. Due to its enormous size, TTN has been insufficiently analyzed in the past. However, recent whole genome sequencing studies revealed that TTN is a major human disease gene [13, 26, 45, 56, 74, 75, 96, 98, 99]. Many titin mutations are also linked to neuromuscular diseases [20, 26, 87, 89, 98], but this review mainly focuses on the role of titin in cardiomyopathies where TTNtvs have been studied most.
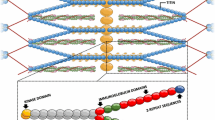
The human titin gene contains 364 exons, of which 363 exons are coding exons. Titin has a maximum molecular mass of ~ 4200 kDa [11, 69] and has a modular domain composition consisting of immunoglobulin (Ig) and fibronectin type III (FnIII) domains and unique sequences [69, 106] (see Fig. 1 supplemental Table S1). Titin provides passive stiffness to the striated muscle sarcomere and modulates active contractile force [4, 9, 16, 18, 33,34,35,36, 42, 50, 73, 79, 104]. Titin’s N-terminus is embedded in the Z-disk and acts as a mechano-sensor [65]. The I-band region of titin functions as a molecular spring and is the main determinant of cardiac myocyte elasticity in cardiac muscles [25, 42, 75, 77, 113, 117]. It comprises three distinct elements, the tandem Ig segment, the PEVK region (rich in proline, glutamic acid, valine, and lysine residues) and the N2B element, containing the extensible N2B unique sequence (N2B-Us) [11, 55, 69]. The spring elements can be posttranslational modified, altering their elastic behaviors [8, 49, 53, 54, 58, 59, 92, 121]. In addition to providing elasticity, these segments also interact with signaling proteins and have been proposed to function as mechanosensor complexes [43, 67, 77, 81, 88, 95, 114] with mouse models that genetically target individual spring elements supporting such roles [15, 23, 47, 61, 93, 94]. The A-band segment of titin contains 178 Ig and Fn3 domains and is functionally inextensible [16, 69, 106]. The A-band segment contains the so-named I/A zone, D-zone, C-zone, and M-band regions (supplemental Table S1). The IA zone is near the ends of the thick filaments and is striking in that the regular domain patterns of Ig and FnIII domains is broken with a stretch of 6 FnIII domains that is found preceding the D zone. It has been suggested that the unique domain composition of the IA zone reflects an alteration in titin–myosin interaction that is critical for the termination of the thick filament [14]. However, a mouse model in which titin’s IA junction was targeted revealed that deleting the IA junction does not alter thick filament length [48]. In the D-zone region of the A-band, Ig and FnIII domains form 6 repeats, each containing 7 domains and in the C-zone 11 Ig and FnIII domains form super-repeats, each containing 11 domains [69]. The C-zone region of titin likely plays a role in anchoring MyBP-C [31], regulating actomyosin interaction [82], and regulating the thick filament length [103]. Titin’s M-band region contains the serine/threonine kinase (TK) domain and is involved in numerous signaling pathways [19, 39, 83, 90, 91, 115, 116]. Moreover, exon 363 (Mex5), coding for is7 domain in the M-band region, is differentially spliced and gives rise to is7+ and is7- titin isoforms [21, 66].

Titin isoforms and mapped disease-associated missense mutations. Titin isoforms assembled from the metatranscript, cardiac N2BA, cardiac N2B, skeletal muscle N2A, Novex3 and Cronos transcripts (from top to bottom). See text for details. Missense mutations causing DCM, HCM, ARVC, RCM and myopathy are shown by vertical lines mapped on the protein domains where they occur. Missense mutations downloaded from the TITINdb (http://fraternalilab.kcl.ac.uk/TITINdb/), see Laddach et al. [71]. Domain colors: red: Ig domains, white: Fn domains, green: Z-repeats, yellow: PEVK sequence, blue: unique sequences
Extensive mRNA splicing results in distinct titin isoforms [11, 70] (Fig. 1). In the heart, three titin isoform classes are present: fetal cardiac titin (3.5–3.6 MDa), adult N2BA (~ 3.3 MDa), and adult N2B (~ 3.0 MDa) isoforms [11, 69, 72]. An important titin splicing factor is RBM20. Deficiency in RBM20 is leading to increased expression of large N2BA-type titin isoforms in the adult heart [50, 61, 79, 80]. In addition to full-length titins, isoforms that are not full-length also exist (Fig. 1). Novex-3 titin, a ∼ 700 kDa titin isoform is found in cardiac and skeletal muscle [11, 46, 64]. The 3′ end of novex-3 contains the stop codon polyadenylation signal and functions as an alternative C-terminus, resulting in a truncated titin isoform [11]. Unlike full-length titin isoforms, novex-3 is too short to reach the A-band region [11, 96]. Recently, an alternative start site has been identified in the titin gene that is predicted to results in expression of cronos titin, a ~ 2000 kDa isoform that lacks the Z-disk and most of the I-band domains but contains the A-band and M-line domains [123]. The functions of novex-3 and cronos titin have not been established. Due to alternative splicing, adult full-length cardiac isoforms differ in the length of their tandem and PEVK segments in the I-band and their stiffness varies accordingly [11, 17, 117] [32]. The adult full-length cardiac isoforms (N2B and N2BA) are co-expressed at the level of the half sarcomere [105]; their expression ratio is approximately 50:50 in humans [84, 85] but can vary in disease states [84, 85, 118,119,120].
Titin gene mutations as a cause of cardiomyopathies
Cardiomyopathies are diseases that cause primary abnormalities in the heart muscle [57]. The most common type is dilated cardiomyopathy (DCM) with a prevalence of up to ~ 1:250 [57, 99]. DCM is characterized by left ventricular dilation and systolic dysfunction [57]. Recent landmark sequence studies in large patient cohorts revealed that mutations in the titin gene (TTN) are responsible for ~ 20% of all DCM cases [56, 96, 99]. Many of the DCM-causing TTN mutations are heterozygous truncating variants (TTNtv) that include frameshift, nonsense, and essential splice site mutations and are over-represented in the A-band segment of titin [56, 96], see Fig. 1. Moreover, TTNtvs show a high penetrance after the age of 40 years and there is a possibility that secondary stressors are needed to develop DCM phenotype [27, 56]. Titin missense mutations are also likely to contribute to a small fraction of DCM [13, 38] and they are a rare cause of hypertrophic cardiomyopathy (HCM) and of arrhythmogenic right ventricular dysplasia [10, 16, 56, 75, 102] (Fig. 1).
TTNtv-induced DCM
Epidemiology and penetrance of TTNtv
DCM is the most common indication for heart transplantation and is associated with TTNtv in ~ 20% of DCM cases [56, 57, 96, 99]. Surprisingly, 1–3% of the general population has a TTNtv but the overwhelming majority does not present a cardiac phenotype, and thus, the genotype-phenotype relationship of TTNtvs is uncertain [5,6,7, 56, 99]. Clearly, it is important to focus on the underlying mechanisms of TTNtv-induced DCM.
Localization of TTNtv
TTNtv are predominantly found in the A-band region of titin and show a position-dependent manner with increasing disease severity closer to the C-terminus [56, 60, 96, 99]. Recently, TTNtv-induced DCM has also been associated with Z-disk, I-band, and M-band exons in a small subset of patients [99]. The position-dependent effect might be explained by TTN exon usage in left ventricular tissue, characterized by the relative incorporation of exons into titin transcripts, termed proportion spliced-in (PSI) [96]. Constitutively expressed exons have high PSI values, whereas exons that are subject to alternative splicing show low PSI scores [27, 96]. Therefore, titin’s A-band exons that have high PSI scores and are incorporated in all titin isoforms are most affected by TTNtvs [27, 60, 96]. Notably, exons in the I-band region where intense alternative splicing occurs have low PSI values [96]. Consequently, I-band exons with TTNtv can be excluded from the transcript without resulting in a frameshift, acting as a natural ‘exon-skipping’ mechanism [77, 96]. Hence, it has been suggested that TTNtv can be tolerated in the healthy population because the majority of the mutations fall in I-band exons that are subject to alternative splicing [60, 96]. It has also been proposed that the upregulation of cronos titin [24], a novel titin isoform driven by an internal promoter (Fig. 1), could rescue the effects of truncating mutations that localize proximal to its internal I-band promoter [24, 123]. Truncating variants in the novex-3 exon that functions as an alternative C-terminus occur equally in patients with DCM and in healthy controls [96, 99, 110]. Although currently there is lack of evidence for pathogenicity of novex-3 titin mutations [96], whole-exome sequencing technologies are enabling the identification of novel rare cardiomyopathy-causing titin truncating variants [101] and it is possible that in future studies novex-3 titin truncating mutations will be shown to play a role in the pathomechanism of some cardiomyopathies [22, 64].
Currently, there is much uncertainty about the exact mechanism by which titin truncating mutations lead to a cardiac phenotype. Multiple mechanisms have been proposed to explain TTNtv-induced DCM: haploinsufficiency, poison-peptide/dominant-negative mechanism, and perturbation of cardiac metabolism and signaling.
Haploinsufficiency
Schafer et al. developed 2 rat strains and modeled a proximal and distal TTNtv mutation and their RNA-seq study revealed a profound nonsense mediated mRNA decay (NMD) of the allele with TTNtv, indicating haploinsufficiency [99]. However, protein gels did not reveal truncated titins, suggesting that either no truncated proteins are produced or that they are produced but rapidly degraded [99]. Moreover, total protein levels of full-length titin appear not different, suggesting an upregulation of the wild-type allele, consistent with the transcript findings of the Schafer study [99]. To study the effect of titin deficiency, Radke et al. generated a conditional KO mouse model with progressive postnatal loss of the complete titin protein achieved by removing exon 2 (E2-KO) [94]. Results showed that titin deficiency leads to sarcomere disassembly and atrophy in striated muscle and eventually DCM. Overall, these animal studies suggest a need to further investigate the haploinsufficiency mechanism in DCM patients with TTNtvs.
Poison peptide mechanism
Another possible mechanism by which TTNtv can induce DCM is the poison peptide/dominant-negative mechanism. A limited amount of truncated protein has been found in induced pluripotent stem cell (iPSC) cardiomyocytes derived from patients with TTNtv [60]. Additionally, heterozygous TTNtv mutant iPSC-s have fewer myofibrils and show sarcomere disorganization [60]. A recent study by Schick et al. also demonstrates defects in sarcomere assembly in patient-derived iPSC cardiomyocytes [100]. In a large DCM patient cohort, Roberts et al. found that TTNtv containing transcripts are not subjected to NMD and no changes in the protein expression levels of major titin isoforms are detectable, suggesting the possible role of poison peptide/dominant-negative mechanism in TTNtv-related DCM [96].
Perturbation of cardiac metabolism and signaling
It is known that mTORC1, which functions as a nutrient/energy sensor and controls protein synthesis, is activated in DCM patients [99, 122]. It is of interest therefore to determine whether distinct molecular pathways are associated with TTNtv-based DCM. Indeed, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis suggests altered cardiac metabolism in TTNtv rats, independently of the position of the truncation [99]. Furthermore, biochemical analysis revealed a shift from fatty acids toward glycolysis, similar to those seen in the failing heart that may be adaptive [99]. The levels of metabolites that can activate mTOR are also increased in TTNtv rats [99]. Interestingly, major signaling pathways, involving transforming growth factor-β, vascular endothelial growth factor, and mitogen-activated protein kinases, that are critically important to cardiomyocyte function, are diminished in iPS-derived cardiac cells containing TTNtv [60, 110]. Additionally, Verdonschot et al. found that all components of the mitochondrial electron transport chain are significantly upregulated in patients with TTNtv, leading to pronounced cardiac alterations in mitochondrial function [109]. In accordance with these alterations, Zhou et al. found decreased oxygen consumption rate, elevated reactive oxygen species (ROS) levels and increased mitochondrial protein ubiquitination in rat hearts with TTNtv, indicating mitochondrial dysfunction caused by TTNtv [2]. Additionally, TTNtv hearts show increased mTOR phosphorylation and impaired autophagy function [2]. Interestingly, mutated iPSC cardiomyocytes, derived from DCM patients with TTNtv, show attenuated response to isoproterenol, [Ca2+]out and angiotensin II. Furthermore, mutated cells display a longer recovery period after caffeine administration [100]. These changes suggest altered function of calcium-handling proteins, such as SERCA, phospholamban (PLB), and calsequestrin [100]. Overall, the importance of changes in cardiac metabolism and calcium handling in DCM caused by TTNtv warrant further investigation, including whether these changes develop directly from the truncating mutation or, more likely, are secondary effects.
Is a TTNtv sufficient to induce a phenotype or are modifying effects required?
Not all individuals that carry a TTNtv develop DCM and a multifactorial disease model has been proposed where multiple factors contribute to the development of a TTNtv-based phenotype [27, 99]. In this model, a second genetic variant and/or environmental stressor is needed, as a ‘second or third hit’, to uncover the effects of the TTNtv.
Sex differences
Interestingly, the onset of DCM is ∼ 40 years and the penetrance of TTNtv is sex dependent [30, 56]. The median age of onset in males is estimated to be 28 years and 56 years in females [30]. In addition, women carrying TTNtv mutations have a better prognosis than men [30, 56]. Further studies are needed to establish whether the sex dependence might be more related to the link between titin phosphorylation and increased oxidative stress [12, 30] and whether the cardioprotective effects of estrogen in premenopausal women contribute to sex-related differences [62, 76].
Peripartum cardiomyopathy
TTNtv have also been linked to peripartum cardiomyopathy (PPCM) where the distribution of truncating variants in PPCM is similar to that found in DCM [108, 112]. PPCM can also be a manifestation of familial DCM and TTNtv in PPCM patients is a possible prognostic factor for low recovery rate [108, 112].
Second mutation
Often additional rare truncating variants or other pathogenic cardiomyopathy genes are present in TTNtv carriers that can increase the severity of DCM or can be associated with an earlier onset of the disease [51, 56, 86, 97].
Environmental factors
Although the onset of TTNtv-induced DCM is ∼ 40 years [56], environmental insults, such as chemotherapy, can induce pediatric-onset DCM cases [28]. Furthermore, TTNtv can be associated with a more severe form of chemotherapy-induced cardiomyopathy (CCMP). Recently, it has been reported that patients with TTNtv have a prevalent genetic predisposition for alcoholic cardiomyopathy and an even more impaired ejection fraction can be observed in TTNtv-induced DCM patients with alcohol abuse [110]. Therefore, alcohol is an additional environmental risk that can contribute to a more severe outcome of TTNtv-associated DCM. Finally, Gramlich et al. showed that hemodynamic stress caused by angiotensin II or isoproterenol can induce a more severe phenotype in heterozygous TTNtv mice compared to control litter mates [40]. This finding suggests that hypertension, a common risk factor for heart disease and stroke [52], results in a more severe form of DCM in patients with TTNtv [40].
In summary, many additional genetic and environmental factors can influence the outcome of an existing TTNtv. In most of the cases these stressors can unmask the effects of TTNtv or induce an even more severe DCM phenotype.
Comparison between TTNtv- and TTNtv+ DCM
To date, there are contradictory observations in patient populations about the symptoms and differences between DCM patients with (TTNtv+) or without (TTNtv-) mutations. Furthermore, as discussed above, there is much debate about the genotype-phenotype relationship of TTNtv in DCM, as truncating titin mutations can be found in 1–3% of the general population [5, 7, 56, 99]. Herman et al. showed no significant differences in clinical manifestations between TTNtv+ and TTNtv- subjects, including the risk of major cardiac events [56]. Similarly, others reported that TTNtv+ does not appear to be associated with worse prognosis and DCM patients with TTNtv are unaccompanied by conduction disease [30]. Although, Verdonschot et al. found more life-threatening arrhythmias in TTNtv+ patients associated with enhanced interstitial myocardial fibrosis, the survival rate was similar between TTNtv+ and TTNtv- patients at long-term follow-up [109]. Interestingly, recent whole-exome sequencing studies by Ahlberg et al. identified TTNtv as a major genetic contributor to atrial fibrillation [3]. A new zebrafish model that contains a TTNtv mutation displays increased fibrosis and altered sarcomere structure in the atria. Moreover, TTNtv+ zebrafish show electrophysiological defects that could potentially develop into arrhythmia [3]. Comparing TTNtv+ and TTNtv- DCM patients, Roberts et al. observed more severely impaired left ventricular (LV) function, lower stroke volumes, and more sustained ventricular tachycardia in TTNtv+ patients [96]. Furthermore, patients with TTNtv are at higher risk to more adverse cardiac events, as death, cardiac transplant, or LV assist device [96]. Overall, it is still uncertain whether or not patients with TTNtv have more severe symptoms compared to TTNtv- DCM patients.
Patients with DCM caused by TTNtv respond to standard DCM therapies [63] and long-term prognosis is similar to that of patients without TTNtvs [29, 109]. Recovery from TTNtv-associated PPCM is also possible with proper and careful medical assistance [68]. Based on the metabolic changes in TTNtv+ humans and animal models, mTOR pathway modulation with metformin or ‘rapalogues’ (rapamycin analogues) could serve as a potential treatment for TTNtv-induced DCM [2, 110]. Zhou et al. observed that the mTORC1 inhibitor rapamycin is able to rescue the attenuated autophagy in rat hearts containing TTNtv mutations [2]. Additionally, research groups are focusing on exon-skipping approaches to cure TTNtv-associated DCM. Most TTN exons can be deleted while keeping the reading frame intact. The deletion of a large TTN exon induced by antisense oligonucleotides has been accomplished [41], but it is currently uncertain how well the absence of exons is tolerated or whether it might lead to a cardiac phenotype at some stage of life. An exon-skipping therapeutic strategy has already been approved by the Food and Drug Administration (FDA) for use in Duchenne muscular dystrophy [1, 110], and the hope is that similar exon-skipping approaches are feasible and be beneficial in TTNtv patients as well. In summary, exon skipping has the potential to cure TTNtv-induced DCM but much research is required first, particularly focused on possible off-target effects that might occur.
Conclusions and perspectives
It is now well established that TTN is a major human disease gene that causes multiple neuromuscular and cardiac diseases [13, 20, 26, 56, 74, 75, 89, 96, 98, 99]. Most studies are currently focused on TTNtv that cause dilated cardiomyopathy [56, 96, 99]. Even though TTNtv mutations are likely to affect ribosome activity [99], sarcomeric organization [40, 60], and alter cardiac metabolism [99, 109], a clear genotype-phenotype correlation is often lacking. Indeed, 1–3% of the general population has a TTNtv, and it has been proposed that additional genetic and/or environmental stressors might be needed to unmask the effects of TTNtv [40, 78, 97, 108, 110, 111]. Although TTNtv+ patients present more life-threatening arrhythmias associated with enhanced interstitial myocardial fibrosis, the survival rate is similar between TTNtv+ and TTNtv- patients at long-term follow-up [29, 109]. In addition, TTNtv-associated DCM patients respond well to standard DCM therapies [63]. Mimicking natural skipping of exons with low PSI scores [77, 96], exon skipping with antisense oligonucleotides could provide a more specific treatment option for patients with DCM caused by TTNtv. Clearly, more research is required into the pathomechanism by which TTNtv mutations induce DCM and into the possibility of exon skipping as a therapy.
References
Aartsma-Rus A, Krieg AM (2017) FDA approves eteplirsen for duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther 27:1–3. https://doi.org/10.1089/nat.2016.0657
Adams M, Fleming JR, Riehle E, Zhou T, Zacharchenko T, Markovic M, Mayans O (2019) Scalable, non-denaturing purification of phosphoproteins using Ga(3+)-IMAC: N2A and M1M2 titin components as study case. Protein J. https://doi.org/10.1007/s10930-019-09815-w
Ahlberg G, Refsgaard L, Lundegaard PR, Andreasen L, Ranthe MF, Linscheid N, Nielsen JB, Melbye M, Haunso S, Sajadieh A, Camp L, Olesen SP, Rasmussen S, Lundby A, Ellinor PT, Holst AG, Svendsen JH, Olesen MS (2018) Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun 9:4316. https://doi.org/10.1038/s41467-018-06618-y
Ait-Mou Y, Hsu K, Farman GP, Kumar M, Greaser ML, Irving TC, de Tombe PP (2016) Titin strain contributes to the frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc Natl Acad Sci U S A 113:2306–2311. https://doi.org/10.1073/pnas.1516732113
Akinrinade O, Koskenvuo JW, Alastalo TP (2015) Prevalence of titin truncating variants in general population. PLoS One 10:e0145284. https://doi.org/10.1371/journal.pone.0145284
Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, Koillinen H, Kaartinen M, Nieminen MS, Myllykangas S, Alastalo TP, Koskenvuo JW, Helio T (2015) Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J 36:2327–2337. https://doi.org/10.1093/eurheartj/ehv253
Akinrinade O, Alastalo TP, Koskenvuo JW (2016) Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet 90:49–54. https://doi.org/10.1111/cge.12741
Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernandez JM (2014) S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 156:1235–1246. https://doi.org/10.1016/j.cell.2014.01.056
Anderson BR, Granzier HL (2012) Titin-based tension in the cardiac sarcomere: molecular origin and physiological adaptations. Prog Biophys Mol Biol 110:204–217. https://doi.org/10.1016/j.pbiomolbio.2012.08.003
Anderson BR, Bogomolovas J, Labeit S, Granzier H (2013) Single molecule force spectroscopy on titin implicates immunoglobulin domain stability as a cardiac disease mechanism. J Biol Chem 288:5303–5315. https://doi.org/10.1074/jbc.M112.401372
Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S (2001) The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 89:1065–1072
Beckendorf L, Linke WA (2015) Emerging importance of oxidative stress in regulating striated muscle elasticity. J Muscle Res Cell Motil 36:25–36. https://doi.org/10.1007/s10974-014-9392-y
Begay RL, Graw S, Sinagra G, Merlo M, Slavov D, Gowan K, Jones KL, Barbati G, Spezzacatene A, Brun F, Di Lenarda A, Smith JE, Granzier HL, Mestroni L, Taylor M, Familial Cardiomyopathy R (2015) Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc 4. https://doi.org/10.1161/JAHA.115.002645
Bennett PM, Gautel M (1996) Titin domain patterns correlate with the axial disposition of myosin at the end of the thick filament. J Mol Biol 259:896–903. https://doi.org/10.1006/jmbi.1996.0367
Brynnel A, Hernandez Y, Kiss B, Lindqvist J, Adler M, Kolb J, van der Pijl R, Gohlke J, Strom J, Smith J, Ottenheijm C, Granzier HL (2018) Downsizing the molecular spring of the giant protein titin reveals that skeletal muscle titin determines passive stiffness and drives longitudinal hypertrophy. Elife 7. https://doi.org/10.7554/eLife.40532
Burke MA, Cook SA, Seidman JG, Seidman CE (2016) Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol 68:2871–2886. https://doi.org/10.1016/j.jacc.2016.08.079
Cazorla O, Freiburg A, Helmes M, Centner T, McNabb M, Wu Y, Trombitas K, Labeit S, Granzier H (2000) Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ Res 86:59–67
Cazorla O, Wu Y, Irving TC, Granzier H (2001) Titin-based modulation of calcium sensitivity of active tension in mouse skinned cardiac myocytes. Circ Res 88:1028–1035
Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S (2001) Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 306:717–726. https://doi.org/10.1006/jmbi.2001.4448
Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, Swanson LC, Soemedi R, Vasli N, Iannaccone ST, Shieh PB, Shur N, Dennison JM, Lawlor MW, Laporte J, Markianos K, Fairbrother WG, Granzier H, Beggs AH (2013) Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 81:1205–1214. https://doi.org/10.1212/WNL.0b013e3182a6ca62
Charton K, Suel L, Henriques SF, Moussu JP, Bovolenta M, Taillepierre M, Becker C, Lipson K, Richard I (2016) Exploiting the CRISPR/Cas9 system to study alternative splicing in vivo: application to titin. Hum Mol Genet 25:4518–4532. https://doi.org/10.1093/hmg/ddw280
Chen K, Song J, Wang Z, Rao M, Chen L, Hu S (2018) Absence of a primary role for TTN missense variants in arrhythmogenic cardiomyopathy: from a clinical and pathological perspective. Clin Cardiol 41:615–622. https://doi.org/10.1002/clc.22906
Chung CS, Hutchinson KR, Methawasin M, Saripalli C, Smith JE 3rd, Hidalgo CG, Luo X, Labeit S, Guo C, Granzier HL (2013) Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation 128:19–28. https://doi.org/10.1161/CIRCULATIONAHA.112.001268
Deo RC (2016) Alternative splicing, internal promoter, nonsense-mediated decay, or all three: explaining the distribution of truncation variants in titin. Circ Cardiovasc Genet 9:419–425. https://doi.org/10.1161/CIRCGENETICS.116.001513
Elhamine F, Radke MH, Pfitzer G, Granzier H, Gotthardt M, Stehle R (2014) Deletion of the titin N2B region accelerates myofibrillar force development but does not alter relaxation kinetics. J Cell Sci 127:3666–3674. https://doi.org/10.1242/jcs.141796
Evila A, Palmio J, Vihola A, Savarese M, Tasca G, Penttila S, Lehtinen S, Jonson PH, De Bleecker J, Rainer P, Auer-Grumbach M, Pouget J, Salort-Campana E, Vilchez JJ, Muelas N, Olive M, Hackman P, Udd B (2017) Targeted next-generation sequencing reveals novel TTN mutations causing recessive distal titinopathy. Mol Neurobiol 54:7212–7223. https://doi.org/10.1007/s12035-016-0242-3
Fatkin D, Huttner IG (2017) Titin-truncating mutations in dilated cardiomyopathy: the long and short of it. Curr Opin Cardiol 32:232–238. https://doi.org/10.1097/HCO.0000000000000382
Fatkin D, Lam L, Herman DS, Benson CC, Felkin LE, Barton PJR, Walsh R, Candan S, Ware JS, Roberts AM, Chung WK, Smoot L, Bornaun H, Keogh AM, Macdonald PS, Hayward CS, Seidman JG, Roberts AE, Cook SA, Seidman CE (2016) Titin truncating mutations: a rare cause of dilated cardiomyopathy in the young. Prog Pediatr Cardiol 40:41–45. https://doi.org/10.1016/j.ppedcard.2016.01.003
Felkin LE, Walsh R, Ware JS, Yacoub MH, Birks EJ, Barton PJ, Cook SA (2016) Recovery of cardiac function in cardiomyopathy caused by titin truncation. JAMA Cardiol 1:234–235. https://doi.org/10.1001/jamacardio.2016.0208
Franaszczyk M, Chmielewski P, Truszkowska G, Stawinski P, Michalak E, Rydzanicz M, Sobieszczanska-Malek M, Pollak A, Szczygiel J, Kosinska J, Parulski A, Stoklosa T, Tarnowska A, Machnicki MM, Foss-Nieradko B, Szperl M, Sioma A, Kusmierczyk M, Grzybowski J, Zielinski T, Ploski R, Bilinska ZT (2017) Titin truncating variants in dilated cardiomyopathy—prevalence and genotype-phenotype correlations. PLoS One 12:e0169007. https://doi.org/10.1371/journal.pone.0169007
Freiburg A, Gautel M (1996) A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem 235:317–323
Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, Granzier H, Labeit S (2000) Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res 86:1114–1121
Fukuda N, Granzier H (2004) Role of the giant elastic protein titin in the frank-Starling mechanism of the heart. Curr Vasc Pharmacol 2:135–139
Fukuda N, Granzier HL (2005) Titin/connectin-based modulation of the Frank-Starling mechanism of the heart. J Muscle Res Cell Motil 26:319–323. https://doi.org/10.1007/s10974-005-9038-1
Fukuda N, Wu Y, Farman G, Irving TC, Granzier H (2003) Titin isoform variance and length dependence of activation in skinned bovine cardiac muscle. J Physiol 553:147–154. https://doi.org/10.1113/jphysiol.2003.049759
Fukuda N, Wu Y, Farman G, Irving TC, Granzier H (2005) Titin-based modulation of active tension and interfilament lattice spacing in skinned rat cardiac muscle. Pflugers Arch 449:449–457. https://doi.org/10.1007/s00424-004-1354-6
Furst DO, Osborn M, Nave R, Weber K (1988) The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J Cell Biol 106:1563–1572
Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, Granzier H, Mestroni L (2016) A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front Cardiovasc Med 3:21. https://doi.org/10.3389/fcvm.2016.00021
Gotthardt M, Hammer RE, Hubner N, Monti J, Witt CC, McNabb M, Richardson JA, Granzier H, Labeit S, Herz J (2003) Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J Biol Chem 278:6059–6065. https://doi.org/10.1074/jbc.M211723200
Gramlich M, Michely B, Krohne C, Heuser A, Erdmann B, Klaassen S, Hudson B, Magarin M, Kirchner F, Todiras M, Granzier H, Labeit S, Thierfelder L, Gerull B (2009) Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J Mol Cell Cardiol 47:352–358. https://doi.org/10.1016/j.yjmcc.2009.04.014
Gramlich M, Pane LS, Zhou Q, Chen Z, Murgia M, Schotterl S, Goedel A, Metzger K, Brade T, Parrotta E, Schaller M, Gerull B, Thierfelder L, Aartsma-Rus A, Labeit S, Atherton JJ, McGaughran J, Harvey RP, Sinnecker D, Mann M, Laugwitz KL, Gawaz MP, Moretti A (2015) Antisense-mediated exon skipping: a therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol Med 7:562–576. https://doi.org/10.15252/emmm.201505047
Granzier HL, Irving TC (1995) Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68:1027–1044. https://doi.org/10.1016/S0006-3495(95)80278-X
Granzier HL, Labeit S (2005) Titin and its associated proteins: the third myofilament system of the sarcomere. Adv Protein Chem 71:89–119. https://doi.org/10.1016/S0065-3233(04)71003-7
Granzier HL, Labeit S (2006) The giant muscle protein titin is an adjustable molecular spring. Exerc Sport Sci Rev 34:50–53
Granzier H, Wu Y, Siegfried L, LeWinter M (2005) Titin: physiological function and role in cardiomyopathy and failure. Heart Fail Rev 10:211–223. https://doi.org/10.1007/s10741-005-5251-7
Granzier H, Radke M, Royal J, Wu Y, Irving TC, Gotthardt M, Labeit S (2007) Functional genomics of chicken, mouse, and human titin supports splice diversity as an important mechanism for regulating biomechanics of striated muscle. Am J Physiol Regul Integr Comp Physiol 293:R557–R567. https://doi.org/10.1152/ajpregu.00001.2007
Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschope C, McNabb M, Larson DF, Labeit S, Gotthardt M (2009) Truncation of titin’s elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ Res 105:557–564. https://doi.org/10.1161/CIRCRESAHA.109.200964
Granzier HL, Hutchinson KR, Tonino P, Methawasin M, Li FW, Slater RE, Bull MM, Saripalli C, Pappas CT, Gregorio CC, Smith JE 3rd (2014) Deleting titin's I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci U S A 111:14589–14594. https://doi.org/10.1073/pnas.1411493111
Grutzner A, Garcia-Manyes S, Kotter S, Badilla CL, Fernandez JM, Linke WA (2009) Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys J 97:825–834. https://doi.org/10.1016/j.bpj.2009.05.037
Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, Dauksaite V, Vakeel P, Klaassen S, Gerull B, Thierfelder L, Regitz-Zagrosek V, Hacker TA, Saupe KW, Dec GW, Ellinor PT, MacRae CA, Spallek B, Fischer R, Perrot A, Ozcelik C, Saar K, Hubner N, Gotthardt M (2012) RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 18:766–773. https://doi.org/10.1038/nm.2693
Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Muller S, Kayvanpour E, Vogel B, Sedaghat-Hamedani F, Lim WK, Zhao X, Fradkin D, Kohler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjaer H, Jorgensen M, Garcia-Giustiniani D, Ortiz-Genga M, Crespo-Leiro MG, Deprez RH, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Morner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA, Meder B (2015) Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 36:1123–1135a. https://doi.org/10.1093/eurheartj/ehu301
Hales CM, Carroll MD, Simon PA, Kuo T, Ogden CL (2017) Hypertension prevalence, awareness, treatment, and control among adults aged >/=18 years—Los Angeles County, 1999-2006 and 2007-2014. MMWR Morb Mortal Wkly Rep 66:846–849. https://doi.org/10.15585/mmwr.mm6632a3
Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Kruger M, Backs J, Linke WA (2013) Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 112:664–674. https://doi.org/10.1161/CIRCRESAHA.111.300105
Hamdani N, Herwig M, Linke WA (2017) Tampering with springs: phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys Rev 9:225–237. https://doi.org/10.1007/s12551-017-0263-9
Helmes M, Trombitas K, Centner T, Kellermayer M, Labeit S, Linke WA, Granzier H (1999) Mechanically driven contour-length adjustment in rat cardiac titin’s unique N2B sequence: titin is an adjustable spring. Circ Res 84:1339–1352
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE (2012) Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366:619–628. https://doi.org/10.1056/NEJMoa1110186
Hershberger RE, Hedges DJ, Morales A (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10:531–547. https://doi.org/10.1038/nrcardio.2013.105
Hidalgo C, Granzier H (2013) Tuning the molecular giant titin through phosphorylation: role in health and disease. Trends Cardiovasc Med 23:165–171. https://doi.org/10.1016/j.tcm.2012.10.005
Hidalgo CG, Chung CS, Saripalli C, Methawasin M, Hutchinson KR, Tsaprailis G, Labeit S, Mattiazzi A, Granzier HL (2013) The multifunctional Ca(2+)/calmodulin-dependent protein kinase II delta (CaMKIIdelta) phosphorylates cardiac titin's spring elements. J Mol Cell Cardiol 54:90–97. https://doi.org/10.1016/j.yjmcc.2012.11.012
Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, Haghighi A, Homsy J, Hubner N, Church G, Cook SA, Linke WA, Chen CS, Seidman JG, Seidman CE (2015) HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349:982–986. https://doi.org/10.1126/science.aaa5458
Hinze F, Dieterich C, Radke MH, Granzier H, Gotthardt M (2016) Reducing RBM20 activity improves diastolic dysfunction and cardiac atrophy. J Mol Med (Berl) 94:1349–1358. https://doi.org/10.1007/s00109-016-1483-3
Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M (2017) The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ 8:33. https://doi.org/10.1186/s13293-017-0152-8
Jansweijer JA, Nieuwhof K, Russo F, Hoorntje ET, Jongbloed JD, Lekanne Deprez RH, Postma AV, Bronk M, van Rijsingen IA, de Haij S, Biagini E, van Haelst PL, van Wijngaarden J, van den Berg MP, Wilde AA, Mannens MM, de Boer RA, van Spaendonck-Zwarts KY, van Tintelen JP, Pinto YM (2017) Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail 19:512–521. https://doi.org/10.1002/ejhf.673
Kellermayer D, Smith JE 3rd, Granzier H (2017) Novex-3, the tiny titin of muscle. Biophys Rev 9:201–206. https://doi.org/10.1007/s12551-017-0261-y
Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR (2002) The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111:943–955
Kolmerer B, Olivieri N, Witt CC, Herrmann BG, Labeit S (1996) Genomic organization of M line titin and its tissue-specific expression in two distinct isoforms. J Mol Biol 256:556–563. https://doi.org/10.1006/jmbi.1996.0108
Kramerova I, Kudryashova E, Wu B, Ottenheijm C, Granzier H, Spencer MJ (2008) Novel role of calpain-3 in the triad-associated protein complex regulating calcium release in skeletal muscle. Hum Mol Genet 17:3271–3280. https://doi.org/10.1093/hmg/ddn223
Kryczka KE, Dzielinska Z, Franaszczyk M, Wojtkowska I, Henzel J, Spiewak M, Stepinska J, Bilinska ZT, Ploski R, Demkow M (2018) Severe course of peripartum cardiomyopathy and subsequent recovery in a patient with a novel TTN gene-truncating mutation. Am J Case Rep 19:820–824. https://doi.org/10.12659/AJCR.909601
Labeit S, Kolmerer B (1995) Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270:293–296
Labeit S, Lahmers S, Burkart C, Fong C, McNabb M, Witt S, Witt C, Labeit D, Granzier H (2006) Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. J Mol Biol 362:664–681. https://doi.org/10.1016/j.jmb.2006.07.077
Laddach A, Gautel M, Fraternali F (2017) TITINdb-a computational tool to assess titin’s role as a disease gene. Bioinformatics 33:3482–3485. https://doi.org/10.1093/bioinformatics/btx424
Lahmers S, Wu Y, Call DR, Labeit S, Granzier H (2004) Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 94:505–513. https://doi.org/10.1161/01.RES.0000115522.52554.86
Lee EJ, Nedrud J, Schemmel P, Gotthardt M, Irving TC, Granzier HL (2013) Calcium sensitivity and myofilament lattice structure in titin N2B KO mice. Arch Biochem Biophys 535:76–83. https://doi.org/10.1016/j.abb.2012.12.004
LeWinter MM, Granzier HL (2013) Titin is a major human disease gene. Circulation 127:938–944. https://doi.org/10.1161/CIRCULATIONAHA.112.139717
LeWinter MM, Granzier HL (2014) Cardiac titin and heart disease. J Cardiovasc Pharmacol 63:207–212. https://doi.org/10.1097/FJC.0000000000000007
Li S, Gupte AA (2017) The role of estrogen in cardiac metabolism and diastolic function. Methodist Debakey Cardiovasc J 13:4–8. https://doi.org/10.14797/mdcj-13-1-4
Linke WA (2018) Titin gene and protein functions in passive and active muscle. Annu Rev Physiol 80:389–411. https://doi.org/10.1146/annurev-physiol-021317-121234
Linschoten M, Teske AJ, Baas AF, Vink A, Dooijes D, Baars HF, Asselbergs FW (2017) Truncating titin (TTN) variants in chemotherapy-induced cardiomyopathy. J Card Fail 23:476–479. https://doi.org/10.1016/j.cardfail.2017.03.003
Methawasin M, Hutchinson KR, Lee EJ, Smith JE 3rd, Saripalli C, Hidalgo CG, Ottenheijm CA, Granzier H (2014) Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 129:1924–1936. https://doi.org/10.1161/CIRCULATIONAHA.113.005610
Methawasin M, Strom JG, Slater RE, Fernandez V, Saripalli C, Granzier H (2016) Experimentally increasing the compliance of titin through RNA binding Motif-20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation 134:1085–1099. https://doi.org/10.1161/CIRCULATIONAHA.116.023003
Moriscot AS, Baptista IL, Bogomolovas J, Witt C, Hirner S, Granzier H, Labeit S (2010) MuRF1 is a muscle fiber-type II associated factor and together with MuRF2 regulates type-II fiber trophicity and maintenance. J Struct Biol 170:344–353. https://doi.org/10.1016/j.jsb.2010.02.001
Muhle-Goll C, Habeck M, Cazorla O, Nilges M, Labeit S, Granzier H (2001) Structural and functional studies of titin's fn3 modules reveal conserved surface patterns and binding to myosin S1—a possible role in the Frank-Starling mechanism of the heart. J Mol Biol 313:431–447. https://doi.org/10.1006/jmbi.2001.5017
Musa H, Meek S, Gautel M, Peddie D, Smith AJ, Peckham M (2006) Targeted homozygous deletion of M-band titin in cardiomyocytes prevents sarcomere formation. J Cell Sci 119:4322–4331. https://doi.org/10.1242/jcs.03198
Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL (2004) Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110:155–162. https://doi.org/10.1161/01.CIR.0000135591.37759.AF
Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA (2002) Titin isoform switch in ischemic human heart disease. Circulation 106:1333–1341
Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, Guo S, Gonzalez M, Hedges DJ, Robertson PD, Krumm N, Nickerson DA, Hershberger RE, National Heart L, Blood Institute GOESP, the Exome Sequencing Project Family Studies Project T (2013) Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet 6:144–153. https://doi.org/10.1161/CIRCGENETICS.111.000062
Oates EC, Jones KJ, Donkervoort S, Charlton A, Brammah S, Smith JE 3rd, Ware JS, Yau KS, Swanson LC, Whiffin N, Peduto AJ, Bournazos A, Waddell LB, Farrar MA, Sampaio HA, Teoh HL, Lamont PJ, Mowat D, Fitzsimons RB, Corbett AJ, Ryan MM, O’Grady GL, Sandaradura SA, Ghaoui R, Joshi H, Marshall JL, Nolan MA, Kaur S, Punetha J, Topf A, Harris E, Bakshi M, Genetti CA, Marttila M, Werlauff U, Streichenberger N, Pestronk A, Mazanti I, Pinner JR, Vuillerot C, Grosmann C, Camacho A, Mohassel P, Leach ME, Foley AR, Bharucha-Goebel D, Collins J, Connolly AM, Gilbreath HR, Iannaccone ST, Castro D, Cummings BB, Webster RI, Lazaro L, Vissing J, Coppens S, Deconinck N, Luk HM, Thomas NH, Foulds NC, Illingworth MA, Ellard S, McLean CA, Phadke R, Ravenscroft G, Witting N, Hackman P, Richard I, Cooper ST, Kamsteeg EJ, Hoffman EP, Bushby K, Straub V, Udd B, Ferreiro A, North KN, Clarke NF, Lek M, Beggs AH, Bonnemann CG, MacArthur DG, Granzier H, Davis MR, Laing NG (2018) Congenital titinopathy: comprehensive characterization and pathogenic insights. Ann Neurol 83:1105–1124. https://doi.org/10.1002/ana.25241
Ojima K, Kawabata Y, Nakao H, Nakao K, Doi N, Kitamura F, Ono Y, Hata S, Suzuki H, Kawahara H, Bogomolovas J, Witt C, Ottenheijm C, Labeit S, Granzier H, Toyama-Sorimachi N, Sorimachi M, Suzuki K, Maeda T, Abe K, Aiba A, Sorimachi H (2010) Dynamic distribution of muscle-specific calpain in mice has a key role in physical-stress adaptation and is impaired in muscular dystrophy. J Clin Invest 120:2672–2683. https://doi.org/10.1172/JCI40658
Ottenheijm CA, Granzier H (2010) Role of titin in skeletal muscle function and disease. Adv Exp Med Biol 682:105–122. https://doi.org/10.1007/978-1-4419-6366-6_6
Peng J, Raddatz K, Labeit S, Granzier H, Gotthardt M (2005) Muscle atrophy in titin M-line deficient mice. J Muscle Res Cell Motil 26:381–388. https://doi.org/10.1007/s10974-005-9020-y
Peng J, Raddatz K, Molkentin JD, Wu Y, Labeit S, Granzier H, Gotthardt M (2007) Cardiac hypertrophy and reduced contractility in hearts deficient in the titin kinase region. Circulation 115:743–751. https://doi.org/10.1161/CIRCULATIONAHA.106.645499
Perkin J, Slater R, Del Favero G, Lanzicher T, Hidalgo C, Anderson B, Smith JE 3rd, Sbaizero O, Labeit S, Granzier H (2015) Phosphorylating titin’s cardiac N2B element by ERK2 or CaMKIIdelta lowers the single molecule and cardiac muscle force. Biophys J 109:2592–2601. https://doi.org/10.1016/j.bpj.2015.11.002
Radke MH, Peng J, Wu Y, McNabb M, Nelson OL, Granzier H, Gotthardt M (2007) Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc Natl Acad Sci U S A 104:3444–3449. https://doi.org/10.1073/pnas.0608543104
Radke MH, Polack C, Methawasin M, Fink C, Granzier HL, Gotthardt M (2019) Deleting full length titin versus the titin M-band region leads to differential mechanosignaling and cardiac phenotypes. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.118.037588
Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F (2012) A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J Biol Chem 287:29273–29284. https://doi.org/10.1074/jbc.M112.372839
Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O’Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O'Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA (2015) Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 7:270ra276. https://doi.org/10.1126/scitranslmed.3010134
Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, di Pasquale E, Papa L, Portararo P, Columbaro M, Forni A, Faggian G, Condorelli G, Robinson PN (2013) Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet 21:1105–1111. https://doi.org/10.1038/ejhg.2013.16
Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P (2016) Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis 3:293–308. https://doi.org/10.3233/JND-160158
Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJ, van Heesch S, Pua CJ, Kui M, Walsh R, Tayal U, Prasad SK, Dawes TJ, Ko NS, Sim D, Chan LL, Chin CW, Mazzarotto F, Barton PJ, Kreuchwig F, de Kleijn DP, Totman T, Biffi C, Tee N, Rueckert D, Schneider V, Faber A, Regitz-Zagrosek V, Seidman JG, Seidman CE, Linke WA, Kovalik JP, O'Regan D, Ware JS, Hubner N, Cook SA (2017) Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 49:46–53. https://doi.org/10.1038/ng.3719
Schick R, Mekies LN, Shemer Y, Eisen B, Hallas T, Ben Jehuda R, Ben-Ari M, Szantai A, Willi L, Shulman R, Gramlich M, Pane LS, My I, Freimark D, Murgia M, Santamaria G, Gherghiceanu M, Arad M, Moretti A, Binah O (2018) Functional abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS One 13:e0205719. https://doi.org/10.1371/journal.pone.0205719
Siegfried JD, Morales A, Kushner JD, Burkett E, Cowan J, Mauro AC, Huggins GS, Li D, Norton N, Hershberger RE (2013) Return of genetic results in the familial dilated cardiomyopathy research project. J Genet Couns 22:164–174. https://doi.org/10.1007/s10897-012-9532-8
Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, Anderson B, Simon B, Bogomolovas J, Labeit S, Granzier H, Mestroni L (2011) Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 124:876–885. https://doi.org/10.1161/CIRCULATIONAHA.110.005405
Tonino P, Kiss B, Strom J, Methawasin M, Smith JE 3rd, Kolb J, Labeit S, Granzier H (2017) The giant protein titin regulates the length of the striated muscle thick filament. Nat Commun 8:1041. https://doi.org/10.1038/s41467-017-01144-9
Trombitas K, Jin JP, Granzier H (1995) The mechanically active domain of titin in cardiac muscle. Circ Res 77:856–861
Trombitas K, Wu Y, Labeit D, Labeit S, Granzier H (2001) Cardiac titin isoforms are coexpressed in the half-sarcomere and extend independently. Am J Physiol Heart Circ Physiol 281:H1793–H1799. https://doi.org/10.1152/ajpheart.2001.281.4.H1793
Tskhovrebova L, Trinick J (2004) Properties of titin immunoglobulin and fibronectin-3 domains. J Biol Chem 279:46351–46354. https://doi.org/10.1074/jbc.R400023200
UniProt C (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515. https://doi.org/10.1093/nar/gky1049
van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, Hilfiker-Kleiner D, Bollen IA, Sliwa K, Alders M, Almomani R, van Langen IM, van der Meer P, Sinke RJ, van der Velden J, Van Veldhuisen DJ, van Tintelen JP, Jongbloed JD (2014) Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J 35:2165–2173. https://doi.org/10.1093/eurheartj/ehu050
Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiaran Aizpurua A, Merken JJ, Wang P, Bierau J, van den Wijngaard A, Schalla SM, Abdul Hamid MA, van Bilsen M, van Empel VPM, Knackstedt C, Brunner-La Rocca HP, Brunner HG, Krapels IPC, Heymans SRB (2018) Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J 39:864–873. https://doi.org/10.1093/eurheartj/ehx808
Ware JS, Cook SA (2018) Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat Rev Cardiol 15:241–252. https://doi.org/10.1038/nrcardio.2017.190
Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, Tsai EJ, Hilfiker-Kleiner D, Kamiya CA, Mazzarotto F, Cook SA, Halder I, Prasad SK, Pisarcik J, Hanley-Yanez K, Alharethi R, Damp J, Hsich E, Elkayam U, Sheppard R, Kealey A, Alexis J, Ramani G, Safirstein J, Boehmer J, Pauly DF, Wittstein IS, Thohan V, Zucker MJ, Liu P, Gorcsan J 3rd, McNamara DM, Seidman CE, Seidman JG, Arany Z, Imac, Investigators I (2016) Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 374:233–241. https://doi.org/10.1056/NEJMoa1505517
Ware JS, Seidman JG, Arany Z (2016) Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 374:2601–2602. https://doi.org/10.1056/NEJMc1602671
Watanabe K, Muhle-Goll C, Kellermayer MS, Labeit S, Granzier H (2002) Different molecular mechanics displayed by titin’s constitutively and differentially expressed tandem Ig segments. J Struct Biol 137:248–258. https://doi.org/10.1006/jsbi.2002.4458
Witt CC, Ono Y, Puschmann E, McNabb M, Wu Y, Gotthardt M, Witt SH, Haak M, Labeit D, Gregorio CC, Sorimachi H, Granzier H, Labeit S (2004) Induction and myofibrillar targeting of CARP, and suppression of the Nkx2.5 pathway in the MDM mouse with impaired titin-based signaling. J Mol Biol 336:145–154
Witt SH, Granzier H, Witt CC, Labeit S (2005) MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol 350:713–722. https://doi.org/10.1016/j.jmb.2005.05.021
Witt SH, Labeit D, Granzier H, Labeit S, Witt CC (2005) Dimerization of the cardiac ankyrin protein CARP: implications for MARP titin-based signaling. J Muscle Res Cell Motil 26:401–408. https://doi.org/10.1007/s10974-005-9022-9
Wu Y, Cazorla O, Labeit D, Labeit S, Granzier H (2000) Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J Mol Cell Cardiol 32:2151–2162. https://doi.org/10.1006/jmcc.2000.1281
Wu Y, Bell SP, Trombitas K, Witt CC, Labeit S, LeWinter MM, Granzier H (2002) Changes in titin isoform expression in pacing-induced cardiac failure give rise to increased passive muscle stiffness. Circulation 106:1384–1389
Wu Y, Labeit S, Lewinter MM, Granzier H (2002) Titin: an endosarcomeric protein that modulates myocardial stiffness in DCM. J Card Fail 8:S276–S286. https://doi.org/10.1054/jcaf.2002.129278
Wu Y, Peng J, Campbell KB, Labeit S, Granzier H (2007) Hypothyroidism leads to increased collagen-based stiffness and re-expression of large cardiac titin isoforms with high compliance. J Mol Cell Cardiol 42:186–195. https://doi.org/10.1016/j.yjmcc.2006.09.017
Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H (2002) Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res 90:1181–1188
Yano T, Shimoshige S, Miki T, Tanno M, Mochizuki A, Fujito T, Yuda S, Muranaka A, Ogasawara M, Hashimoto A, Tsuchihashi K, Miura T (2016) Clinical impact of myocardial mTORC1 activation in nonischemic dilated cardiomyopathy. J Mol Cell Cardiol 91:6–9. https://doi.org/10.1016/j.yjmcc.2015.12.022
Zou J, Tran D, Baalbaki M, Tang LF, Poon A, Pelonero A, Titus EW, Yuan C, Shi C, Patchava S, Halper E, Garg J, Movsesyan I, Yin C, Wu R, Wilsbacher LD, Liu J, Hager RL, Coughlin SR, Jinek M, Pullinger CR, Kane JP, Hart DO, Kwok PY, Deo RC (2015) An internal promoter underlies the difference in disease severity between N- and C-terminal truncation mutations of titin in zebrafish. Elife 4:e09406. https://doi.org/10.7554/eLife.09406
Funding
This study was funded by National Institutes of Health grants R35HL144998, R01AR073179 and Interdisciplinary Training in Cardiovascular Research T32 HL007249.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any primary studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Sarcomeric Mutations in Pflügers Archiv—European Journal of Physiology
Electronic supplementary material
Supplemental Table S1
Domain composition of the metatranscript of titin and Novex-3 titin. Indicated are conventional names for domains based on Bang et al. [11]. Alternative domain names based on TITINdb (http://fraternalilab.kcl.ac.uk/TITINdb/), see Laddach et al. [71], and UniProt (https://www.uniprot.org/uniprot/Q8WZ42) [107]. Accession numbers for the Metatranscript and Novex-3 proteins are NP_001254479 and NP_596870. (PDF 1737 kb)
Rights and permissions
About this article

Cite this article
Kellermayer, D., Smith, J.E. & Granzier, H. Titin mutations and muscle disease. Pflugers Arch - Eur J Physiol 471, 673–682 (2019). https://doi.org/10.1007/s00424-019-02272-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-019-02272-5