
Abstract
Over a half of mammalian genomes is occupied by repetitive elements whose ability to provide functional sequences, move into new locations, and recombine underlies the so-called genome plasticity. At the same time, mobile elements exemplify selfish DNA, which is expanding in the genome at the expense of the host. The selfish generosity of mobile genetic elements is in the center of research interest as it offers insights into mechanisms underlying evolution and emergence of new genes. In terms of numbers, with over 20,000 in count, protein-coding genes make an outstanding >2 % minority. This number is exceeded by an ever-growing list of genes producing long non-coding RNAs (lncRNAs), which do not encode for proteins. LncRNAs are a dynamically evolving population of genes. While it is not yet clear what fraction of lncRNAs represents functionally important ones, their features imply that many lncRNAs emerge at random as new non-functional elements whose functionality is acquired through natural selection. Here, we explore the intersection of worlds of mobile genetic elements (particularly retrotransposons) and lncRNAs. In addition to summarizing essential features of mobile elements and lncRNAs, we focus on how retrotransposons contribute to lncRNA evolution, structure, and function in mammals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
At the beginning, until Darwin and Mendel created foundations for understanding evolution and heredity, our knowledge was formless void and darkness covered the face of the deep. The term gene emerged as a name for a fundamental physical and functional unit of heredity at the beginning of the twentieth century [42]. Deciphering of the genetic code half a century later [13] strongly tied the concept of a gene with protein coding. However, this is only one of the many contexts in which the term gene is being used [8] and the definition of a gene as a unit of heredity has been evolving ever since (reviewed in [29]). Importantly, genome and transcriptome sequencing during the last two decades revealed that protein-coding genes are a critical but relatively small world in the whole universe of heritable information in terms of both, the genome content and the genome fraction transcribed into RNA. For example, human genome sequencing revealed that less than 2 % of its nucleotide sequence codes for proteins, while 55 % is composed of repetitive elements [40]. Next generation sequencing (NGS) provided evidence for mobile elements being one of the major factors driving genome evolution [53]. Mobile elements have an ability to insert themselves into new genomic locations and recombine, thereby causing genetic alterations along with multiplying their numbers in the genome.
In this review, we focus on intersection of two large worlds in the universe of heritable information: mobile genetic elements (particularly retrotransposons) and long non-coding RNAs (lncRNAs). There is a plethora of literature separately covering mobile elements and lncRNAs (for example [12, 31, 36, 45, 62]). Here, we summarize essential features of mobile elements and lncRNAs and focus on how retrotransposons contribute to lncRNA evolution, structure, and function in two main mammalian model organisms—mice and humans.
Retrotransposons
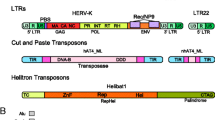
Based on the mode of transposition, mobile elements fall into two major classes: Class I includes “copy and paste” retrotransposons and Class II includes “cut and paste” DNA transposons (Fig. 1, reviewed in [36]). The latter class is characterized by terminal inverted repeats and ability to release itself with a transposase (TPase) from the genome and insert elsewhere (hence cut and paste). However, DNA transposons do not seem to be currently active in mammalian genomes and their remnants are so-called DNA transposon fossils [24]. The human genome contains >100,000 copies of short (180–1200 bp) elements with 14- to 25-bp terminal inverted repeats generated by target site duplications [10, 40, 76]. Class I mobile elements, also called retrotransposons, transpose through an RNA intermediate. Retrotransposons can be further divided into four subclasses based on their retrotransposition competence (autonomous vs. non-autonomous) and the presence/absence of long terminal repeats (LTRs) at their 5′ and 3′ ends (LTR vs. non-LTR elements).

Autonomous LTR retrotransposons evolve from retroviruses when their life cycle becomes confined into a host cell as they lose the ability to be released and infect another cell [43, 52, 80]. Accordingly, structure of LTR retrotransposons closely resembles that of retroviruses. LTR retrotransposons are ∼5–12 kb long, they have two long terminal repeats (LTRs) flanking a protein-encoding region, which carries RNA-dependent DNA polymerase (POL, reverse transcriptase) but often lacks an envelope (ENV) protein-encoding gene (reviewed in [36]). In the mouse genome, there is currently one highly active LTR retrotransposon group (Intracisternal A Particle (IAP)) and several presumable LTR retrotransposon fossils, including Mouse Endogenous Retrovirus type-L (MuERV-L) insertions, which are transcribed during early development [78]. The human genome hosts a family of actively retrotransposing Human Endogenous Retroviruses (HERVs, reviewed in [48]). Autonomous LTR-retrotransposons usually reach hundreds to several thousands of insertions before they die out because inserts accumulate mutations abolishing their coding capacity. At the same time, complementation causes that mutant transcripts compete with intact ones for retrotransposition. This eventually minimizes retrotransposon’s chance to make a copy of a functional element while random mutagenesis continues eliminating remaining functional copies.
In non-autonomous LTR elements, the sequence flanked by LTRs does not contain open-reading frames. They are significantly smaller than autonomous LTR elements, ranging usually between 1 and 1.5 kb. Because of the lack of coding capacity, their retrotransposition requires factors provided by autonomous retrotransposons. A representative example of non-autonomous LTR elements is Mammalian apparent LTR Retrotransposons (MaLR, reviewed in [75]). MaLRs include Mouse Transcript (MT) elements, which provide oocyte-specific promoters in mouse oocytes [71].
Autonomous non-LTR elements are represented by long interspersed nuclear elements (LINEs), which are among the most abundant retrotransposons in mammalian genomes (868,000 insertions in the human genome (20 %) [40] and 660,000 insertions in the mouse genome (9 %) [10]). LINE elements are 6–7 kb long and carry two open reading frames but most of the genome insertions are truncated at the 5′ end. Importantly, LINE elements are resistant to the above-mentioned problem of integration of faulty retrotransposon copies because of a strong cis-preference of the retrotransposition machinery. In other words, proteins translated from a LINE RNA preferentially associate with and retrotranspose the RNA from which they were translated [89].
Non-autonomous non-LTR short interspersed nuclear elements (SINEs) are relatively short sequences (<0.5 kb) related to RNA Polymerase (Pol III)-transcribed small RNAs and do not encode any proteins. Except of rodents and primates, animal SINEs are usually related to tRNAs (reviewed in detail in [74]). There are ∼1.5 million SINEs in human and mouse genomes, which occupy ∼11 and 8 % of the genomes, respectively [10, 11]. The most studied mammalian SINEs are human Alu elements, which are derived from the small cytoplasmic 7SL RNA and are the most abundant transposable elements in the human genome (∼1 million insertions [40]). SINE elements in mice are more heterogeneous. The most abundant murine SINE element is SINE B1, which is ∼140 bp long element derived from a portion of 7SL RNA, which has ∼560 000 copies (2.66 %) in a haploid genome [10]. Another noteworthy murine SINE element is SINE B2, which is a tRNA-derived ∼190 bp long element, which has ∼350,000 copies (2.39 %) in the genome [10].
Since 1980’s, complex eukaryotic genomes were considered loaded with selfish DNA (or “junk” DNA), which expands in a genome without contributing to (or even at the expense of) the fitness of the organism [17, 68]. Accordingly, retrotransposons were seen as harmful genomic parasites causing mutations and threatening the genome integrity. This view was reinforced by their retroviral origin and identification of disease-causing mutations [34, 45]. However, retrotransposons were also proposed to be one of the major contributors to genome evolution [15]. More detailed analyses of eukaryotic genome fueled by NGS surge painted a colorful picture where mutated transpositionally incompetent elements can still provide functional cis-elements regulating adjacent genes such as alternative promoters, transcription factor binding sites, enhancers, exons, terminators, and splice junctions (Fig. 2a) [1, 22, 27, 33, 35, 44, 51, 71]. It has been estimated that 16 % of eutherian-specific conserved non-coding elements are derived from mobile elements, implicating their major contribution to mammalian evolution [23, 63]. Furthermore, transposable elements are evolutionarily among the most lineage-specific sequence elements, especially in mammals [59, 82]. Mouse- and human-specific retrotransposons constitute 87.0 and 51.9 % of all mouse and human retrotransposons, respectively [10].

Contribution of retrotransposon sequences to lncRNAs. a Retrotransposons can provide different functional sequences to lncRNAs. Retrotransposon sequences in a lncRNA gene scheme are indicated by black color. pA, polyadenylation signal. The arrow depicts the transcription start site and direction of transcription. b Heureka, a kinetic statue by Jean Tinguely in Zurich, symbolizes her stochastic building of a new functional structure from used material. Photo by Martin Moravec, 2016
Long non-coding RNAs
Large-scale genome analyses brought surprising findings, which changed perspective on “junk DNA”, a traditional label for a part of a genome that did not encode proteins. Transcriptome and chromatin analyses revealed complex RNA production and typical gene-like chromatin signatures outside of protein-coding gene loci (areas traditionally and doubtfully termed intergenic regions) as well as much more complex RNA synthesis within protein-coding gene loci than previously appreciated [16, 32, 81]. Importantly, in the current view of genome organization, the term “junk DNA” does not need to be replaced; it is only the interpretation that should shift from “useless waste” towards “material for recycling”. When one explores RNA expression in intergenic regions and compares same loci in different species, an idea comes to mind of a large scrap yard, where an old material is being recycled in a TinuelyFootnote 1-like fashion (Fig. 2b).
Large-scale genome analyses also revealed large numbers of lncRNAs that do not have an apparent protein-coding potential. A handful of lncRNAs such as Xist, H19, and few others were known already before the NGS era [46]. However, NGS began pouring lncRNAs by thousands [5, 16, 32, 85]. LncRNAs are generally >200 nt, have a bias towards two-exon transcripts, and have predominantly nuclear localization [16]. Their biogenesis resembles that of mRNAs—they are transcribed by Pol II, capped, usually spliced with a high degree of alternative splicing, and frequently polyadenylated, but they are not translated into proteins [32, 67, 81].
LncRNAs generally lack sequence conservation. In fact, lncRNA promoters are more conserved than lncRNA exons [16]. While lncRNA exons are more conserved than neutrally evolving sequences, their conservation is lower than that of untranslated regions in mRNAs but higher than introns of protein-coding genes [44]. DNA sequence conservation in genes is linked to non-coding features important for gene structure and expression (e.g., promoters, enhances, intron boundaries, or polyadenylation sites) and to the functionally important information stored in the encoded RNA (e.g., encoded protein). However, a non-coding RNA function often depends more on the secondary structure rather than on the primary nucleotide sequence. Thus, a conserved secondary RNA structure of a functionally important module in a lncRNA can be maintained via compensating mutations while a common primary sequence analysis of an entire lncRNA might show only a weak conservation. For example, an imprinting-regulating lncRNA Airn exhibits low expression, conservation, and stability, yet it is involved in silencing Igf2r, as the process of transcription is more important than stable transcripts accumulation [73]. Taken together, while conserved regions are assumed to have a function, it should not be assumed that function needs to be associated with sequence conservation [69].
LncRNAs are usually categorized based on their localization relative to the nearest protein-coding gene (Fig. 3a). Categorization by genomic position and exonic structure is the most widely used method because current bioinformatics expertise is not sufficient to perform reasonable function prediction and classification based on lncRNA exonic sequences. This contrasts with protein-coding genes where one can classify protein-coding RNAs and make functional predictions based on identification of annotated functional domains encoded in nucleic acid sequences.

LncRNA classification. a LncRNA can be classified based on their position relative to the nearest protein-coding genes. The scheme depicts diversity of lncRNAs in terms of possible overlaps with exons and introns of the nearest protein-coding gene. b Four major effects mediated by lncRNAs. Shown are schemes of basic mechanisms by which lncRNAs act
LncRNAs have been linked to transcriptional regulation and chromatin modification, especially during pluripotency and differentiation [3, 16, 27, 47, 92]. However, their range of roles is much broader. In terms of functions, lncRNAs are a heterogeneous group that can be classified in many ways. According to the place of action relative to the encoding locus, lncRNAs are classified as cis- or trans-acting lncRNA. Another criterion can be binding partners (protein, DNA, RNA, or a combination) or cellular localization (nuclear/cytoplasmic). Here, we decided to combine classification of lncRNA effects described in the literature [31, 86] into four categories reflecting distinct modes of action: (i) signaling/allosteric effects, (ii) decoying, (iii) scaffolding, and (iv) guiding and tethering (Fig. 3b). Importantly, a specific lncRNA can exert a combination of these effects as its sequence can carry functionally different modules.
Retrotransposon sequences in lncRNAs
Retrotransposons make a strong contribution to lncRNA sequences. Over two thirds of mature lncRNA sequences (75 and 68 % of human and mouse, respectively) have at least a partial retrotransposon insertion in their sequence, which is more than other type of RNA sequences, such as protein-coding sequences, small RNAs, or untranslated regions [44]. The high content of retrotransposon sequences is likely a contributing factor to sequence diversification and high complexity of lncRNAs. At the same time, it was found that human lncRNAs rarely have extensive sequence similarity to each other outside of shared repetitive elements [16].
Retrotransposons overlap with various lncRNA elements—an internal part of an exon, a transcription start site (TSS), a polyadenylation (polyA) site, a splice donor or acceptor (Fig. 4). The contribution of retrotransposons to functional features of lncRNA is much more than protein-coding loci. Approximately, 23 and 30 % of non-redundant TSS and polyA sites, respectively, used by lncRNA transcripts in the human GENCODE v13 set, were found to be provided by retrotransposons [44]. This strongly contrasts with retrotransposon association with 1.7 % of TSS and 7.9 % of polyA sites of protein-coding genes. In total, 29,519 transposable-element derived functional features (TSS, polyA and splice sites) were identified in GENCODE v13 [16, 44].

Four mechanisms of a new lncRNA evolution. While (i) requires duplication of an entire locus, (ii) and (iii) involve insertions, deletions. All three mechanisms rely on pre-existing transcriptional units. (iv) entails a sequence change (mutation, retrotransposon insertion) leading to a formation of a new promoter in a previously untranscribed region. The newly emerged lncRNA is depicted in black. Gray boxes connected with a dashed line represent exons. White rectangles depict protein-coding sequences. Dark gray rectangles represent two LTRs of a retrotransposon
Apart from mutated retrotransposons, which produce non-coding RNAs themselves, some lncRNAs are almost completely made of several different retrotransposon sequences. An example of such a lncRNA is UCA1. Its expression is enriched in bladder carcinomas and it conserved only in a few primate species [87]. In addition, many annotated lncRNAs share a significant proportion of their sequence with retrotransposons, for example, XIST [19], lincRNA-RoR [56], BORG, UCA1 [87], HULC [70], SLC7A2-IT1A/B [7] etc. Some of these mature lncRNA transcripts are almost entirely composed of transposable elements sequences. For example, the first three exons of the mature transcript of human LncRNA BANCR, which is involved in melanoma cell migration [25], are derived from a MER41 retrotransposon of ERV1 LTR retrotransposon family [44]. Mouse lncRNA Borg, which is proposed to have a role in bone morphogenesis [79], has three of its splice site overlapping with B4 SINE elements and MaLR family LTR elements while its second exon is completely composed of an LTR sequence of EVRL-MaLR family retrotransposon. A unique case of retrotransposon sequence-enriched lncRNAs is precursors for small PIWI-associated RNAs (piRNAs), in which accumulation of retrotransposon sequences is functionally desirable (discussed further in the section Retrotransposons and lncRNA functions).
All four major retrotransposon types (Fig. 1) contribute to lncRNA exons approximately proportionally to their occurrence in the genome [44]. Relative to protein-coding genes, LTR/ERV elements were found to be the most enriched retrotransposon families in mouse and human lncRNAs, especially in the lncRNA exons and proximal to lncRNA genes [44]. Moreover, over 40 % of retrotransposon-derived TSSs in the GENCODEv13 map within ERVs [16, 44]. In embryonic stem cells (ESCs), the class of non-coding ESC-specific non-annotated stem transcripts (NASTs) was strongly associated with LTR retrotransposons, particularly with the ERVK and MaLR LTR subfamilies in mice and with ERV1 in humans [27]. Consistent with this, ERVK and MaLR families appeared to be significantly more highly expressed in mouse ESCs; ERV1 and ERVKs showed similar trends in human undifferentiated ESCs [22].
Retrotransposon contribution to tissue-specific lncRNA expression
Tissue-specific expression is one of the characteristic features of lncRNAs. According to the GENCODE v7 data, majority of human protein-coding genes are expressed in multiple tissues whereas expression of majority of lncRNAs is restricted to single tissues [16]. Certain tissues also exhibit enriched lncRNA expression [38, 65]. In this context, it is worth of noting that retrotransposons (especially LTRs) contain regulatory cis-acting elements, which may function as promoters or enhancers [27, 44, 51].
Retrotransposon expression is naturally selected for germline cells because somatic retrotransposition in a sexually reproducing organism is not transmitted into the next generation. Therefore, to increase their copy number in the genome, retrotransposons must direct their activity into the germline. This rationale is consistent with the observation that testes exhibit higher expression of lncRNAs among different organs, with stronger specificity for young than for old lncRNAs [16, 65]. It is believed that chromatin remodeling during male germ cell development provides window of opportunity for this extensive transcription and higher expression of lncRNA [77]. This window is also explored by retrotransposons, which may lead to the birth of new or younger retrotransposon-driven lncRNA transcripts. Contribution of retrotransposons to tissue specific expression has also been well documented for mouse oocytes, where several non-autonomous LTR retrotransposons drive expression of oocyte-specific mRNAs [71]. Accordingly, a recent study reported high expression of lncRNAs in oocytes, from MaLR and EVRK family retrotransposons [84].
Retrotransposons do not support expression only in the germline. There are multiple examples showing that LTR sequences function as enhancers/promoters also in somatic cells. To name a few: Cap analysis of gene expression (CAGE) method revealed that MaLR elements provide promoters in murine adipose tissue, hippocampus, neuroblastoma, and hepatoma cells [22]. Murine VL30 retrotransposon LTRs were shown to function as promoter and enhancer elements in hepatocytes in vivo [37]. LTRs of the human ERV-9 endogenous retrovirus (2-4000 copies/genome) possess enhancer activities in embryonic and hematopoietic cells [54]. Importantly, whether a retrotransposon sequence would function as a promoter or enhancer depends on the chromatin context while the bulk of retrotransposon sequences is silenced by heterochromatin formation during cell differentiation [60, 93]. This implies that expression of retrotransposon-driven lncRNAs would emerge under conditions favoring loss of heterochromatin marks.
LncRNAs in ESCs
A large volume of lncRNA data comes from ESCs, which are an artificial undifferentiated cell type derived from an early embryo, which can be propagated in cell culture while retaining pluripotency. Retrotransposons significantly contribute to ESC-specific expression of lncRNAs, which is conceivable given the reduced heterochromatin at repetitive elements observed in undifferentiated ESCs [60]. Human and mouse ESC lncRNA promoters are located more often in specific LTR retrotransposon families than in the differentiated cells [27]. Approximately 30 % of transcripts (CAGE tags) derived from human embryonic tissues were found to be associated with repetitive elements (16 % retrotransposon, 10 % satellite, 5 % simple repeat), particularly in LINE subfamilies [22]. Among the above-mentioned NASTs (lncRNAs), those associated with LTR-associated promoters accumulate to higher levels than those expressed from promoters not associated with repeats [27]. A quarter of POU5F1, NANOG, and CTCF-bound regions in humans and mouse were found to be within transposable elements [51]. In addition, enrichment for stem cell transcription factors bound at lncRNA (NASTs) loci associated with mouse ERVK and MaLR and human ERV1 elements was greater than for the non-expressed elements [27, 51]. Several HERVH lncRNAs were found expressed at higher levels in ESCs than in any other tissue or cell line [47]. Likewise, the mouse EVRK family also manifested this kind of stem cell-specific expression [47].
Interestingly, ten human lncRNAs significantly upregulated in induced pluripotent stem cells (iPSCs) relative to ESCs were identified [56]. Among them, linc-RoR, which acts as an important modulator of iPSC reprogramming, is almost entirely composed of retrotransposon-derived sequence from seven different retrotransposon families and has an ERV1 LTR at its TSS [47, 56]. Accordingly, it was suggested that endogenous retroviruses shape pluripotency networks via lncRNA regulation in mammals [27, 47]. This notion was corroborated by another study, where 9241 human and 981 mouse lncRNAs that were found to be strongly associated with LTR elements; expressed lncRNAs (NASTs) were associated with mouse ERVK, mouse MaLR and human ERV1 elements, which become silenced by heterochromatin upon differentiation [27].
LncRNA evolution and retrotransposon contributions
LncRNAs are poorly conserved through evolution. The primary lncRNA sequence is loosely connected with functional conservation and importance, as exemplified by XIST, a lncRNA controlling X chromosome inactivation in mammals (reviewed for example in [28]). Mouse and human Xist/XIST transcripts show 49 % sequence identity, which is lower than 5′ and 3′ UTR regions but slightly higher than introns. The homology is not continuous but represents alternating totally unrelated sequences and seven gap-free regions (90–160 bp) of relatively high homology (68–86 %) [4, 66]. Mammalian Xist is also a good example of complex lncRNA evolution with a strong contribution of retrotransposons. It has been proposed that Xist evolved in early eutherians from a protein-coding gene Lnx3 by integration of transposable elements [19]. The Xist gene promoter region and 4/10 exons retain homology to Lnx3 exons. The remaining six Xist exons including those with simple tandem repeats have similarity to different transposable elements. Furthermore, transposable elements in Xist exons are species-specific hence contributing to diversification of Xist transcripts during eutherian evolution [18, 19].
Four possible mechanisms were proposed for new lncRNA origins (Fig. 4): (i) genomic duplication of another lncRNA—this mechanism is also common for protein-coding genes, (ii) birth of a long non-coding RNA from a pseudogene or a protein coding gene, which loses its coding potential, (iii) derivation a new lncRNA from retrotransposon sequences, and (iv) de novo emergence from a previously untranscribed genomic location [44, 72, 83]. Retrotransposons can contribute towards birth of lncRNAs from protein-coding genes by either disrupting the gene or producing a processed pseudogene by reverse transcribing and integrating its mRNA. De novo emergence of lncRNA in a previously untranscribed location can be induced by a novel retrotransposon insertion, which will provide a promoter. Thus, retrotransposons can play a major role in origin and diversification of lncRNAs. This notion is supported by analysis of lineage specific lncRNAs in mammals whose emergence can be mainly credited to retrotransposon sequences (especially LTRs) [27, 59].
Retrotransposons and lncRNA function
As mentioned above, four basic mechanisms of action were proposed for lncRNAs: (i) signaling/allosteric effects, (ii) decoying, (iii) scaffolding, and (iv) guiding and tethering. LncRNAs have various biological functions, including regulation of chromatin structure and transcription where lncRNA can attract silencing or activating complexes to the locus. For example, lncRNA Air and KCNQ1ot1, recruit a chromatin modifying complex to the site of their transcription and silence the locus [50, 64]. Although molecular mechanisms through which lncRNAs act are still only partially understood, a few interesting examples emerged concerning contribution of retrotransposons (particularly of the SINE class) to lncRNA function.
In the first example, a non-coding RNA from a specific retrotransposon regulates spatiotemporal control of gene expression. A specific SINE B2 element functions as a boundary element and its transcription is implicated in the control of growth hormone gene (GH) activity during embryonic development. Pituitary gland-specific expression of GH is repressed until the embryonic day E17.5. A repressive H3K9me3 mark observed at the GH promoter until the E12.5 is replaced by an H3K9me2 mark by E14.5, which is completely lost by E17.5 [57]. A specific SINE B2 element located ∼14 kb upstream of the promoter appears to regulate the temporal activation of GH gene by bidirectional Pol II and Pol III-transcribed non-coding RNAs, which are necessary and sufficient to enable repositioning of the GH locus between nuclear compartments. According to the model, Pol III transcription is implicated in the maintenance of the H3K9me3 repressive mark while Pol II transcription correlates with the loss of heterochromatin and gene activation [57].
Nuclear SINE B2 RNA was also implicated in transcriptional repression under stress conditions. Pol III-transcribed SINE B2 RNA forms secondary structures that can bind Pol II and interfere with polymerase binding, hence causing transcriptional block [21]. Consequently, non-coding RNA transcripts from mouse SINE B2 lead to transcriptional repression during heat shock response [20]. A similar mechanism was reported for Alu RNA, which forms secondary structures similar to B2 SINE RNA, directly binds Pol II, and inhibits transcription during heat shock response in humans [59].
Alu elements are one of the most abundant (1.3 million copies) primate-specific repetitive elements in the human genome [2, 40]. Alu elements regulate gene expression by acting as silencers, promoters, or enhancers [35, 58]. They can also provide templates for A-to-I editing by adenosine deaminase acting on RNA (ADAR) enzyme family. An Alu sequence in lncRNAs can function as a guide and induce Staufen 1 (STAU1)-mediated mRNA decay (SMD) by base pairing with a complementary Alu sequence harbored in the cognate mRNA [30]. STAU1 is a double-stranded RNA binding protein, which was shown to target mRNAs to SMD through binding a STAU1-binding site (SBS), a 3′ UTR 19 nt stem loop structure [49]. However, some SMD-targeted mRNAs, such as Serpine1 and Ankrd57 transcripts, lack SBS. Instead, their 3′ UTRs contain an Alu element sequence. This sequence can then base pair with an Alu-containing lncRNA, forming an imperfect double stranded stem structure mimicking SBS, which in turn leads to SMD. This mechanism might be much more common as many mRNAs carry Alu elements in their 3′ UTRs and 23 % of lncRNAs carry Alu sequence [30]. A similar mechanism of lncRNA and mRNA base-pairing was also shown in mice for SINE elements [88].
Retrotransposons can also contribute to post-transcriptional control in the cytoplasm by selectively stimulating proteosynthesis, as it was demonstrated for Uchl1-AS, a lncRNA antisense to ubiquitin carboxy-terminal hydrolase L1 (Uchl1) gene. UCHL1 is a dual function protein with deubiquitinating and ubiquityl ligase activities expressed mainly in neuronal cells [55]. UCHL1 has been associated with brain function and neurodegenerative diseases such as Parkinson’s and Alzheimer’s disease [9, 55]. Uchl1-AS transcripts, which overlap with the 5′ end of Uchl1 mRNAs, are initially retained in the nucleus, while Uchl1 mRNAs translocate to cytoplasm. Upon cellular stress, Uchl1-AS transcripts move to cytoplasm, which in turn accelerates Uchl1 mRNA translation. This stress-induced proteosynthesis stimulation requires a particular SINE B2 element at the 3′ end of Uchl1-AS along with the 5′ overlap region [6]. The same antisense and SINEB2 dependence was also reported for UXT chaperon protein [6]. The mechanism by which SINE B2 element exerts post-transcriptional regulation is not known. It is conceivable that it, like the mechanisms above, involves a secondary structure, which is a signaling cue for the assembly of translation enhancers or directly binds them.
A distinct case of guiding function of lncRNA-embedded retrotransposon sequences is lncRNA substrates processed into piRNAs, small RNAs (24-32 nucleotides) guiding repressive ribonucleoprotein complexes. The piRNA pathways (reviewed in detail in [90]) suppress retrotransposons and protect the genome integrity in the germline at both, transcriptional and post-transcriptional levels. A complex biogenesis of piRNAs from long lncRNA precursors involves a concerted action of PIWI proteins and other RNA nucleases. Precursor lncRNAs originate from distinct genomic regions (piRNA cluster regions), which harbor retrotransposon sequences and can be seen as checkpoints for screening retrotransposons expanding through the genome. Once a retrotransposon expanding in the genome integrates into such a checkpoint locus, it will be recognized by the piRNA system and all transcripts of that retrotransposon will be recognized and targeted in the germline by complementary piRNAs [41, 90].
Finally, there are also examples linking retrotransposon-lncRNA function to pathophysiology. For example, a trans-acting lncRNA ANRIL, which maps to the atherosclerosis locus on the chromosome 9p21 locus [26, 61, 91], contains an Alu motif implicated in binding genes with a similar Alu motif. The Alu motif mutations in ANRIL reverse trans-effects and pro-atherogenic cellular properties [39]. A single point mutation, which has been linked to an encephalopathy, was found in LINE1 sequence in lncRNA SLC7A2-IT1A/B. This mutation results eightfold downregulation of SLC7A2 intronic lncRNA transcripts in patients brain tissue and increased apoptosis [7]. Finally, high expression of LINE-1 chimeric non-coding transcripts has been observed in breast and colon cancers, which contribute to tumor invasion and metastasis through antisense-mediated downregulation of TFPI2 gene [14].
Summary
Retrotransposons are closely associated with birth, evolution, expression, and function of lncRNAs. Retrotransposons provide mobile platforms giving a rise to novel lncRNAs from protein-coding genes as well as from previously untranscribed regions. Thus, retrotransposons serve as a recycling system probing at random potential of “junk DNA” and creating novel functions through lncRNA. Retrotransposon-derived promoter and enhancer platforms offer synchronization and coordination of lncRNA expression. Retrotransposons also distribute complementary sequences across the genome, providing opportunities for guiding and tethering functions of lncRNAs. At the same time, lncRNAs are employed by the genome defense where they allow for surveying the retrotransposon content and mediating their silencing.
Notes
Jean Tinguely (1925-1991) Swiss sculptor known for kinetic art using scrape yard metal material.
References
Babushok DV, Ostertag EM, Kazazian HH Jr (2007) Current topics in genome evolution: molecular mechanisms of new gene formation. Cell Mol Life Sci 64:542–554. doi:10.1007/s00018-006-6453-4
Berger A, Strub K (2011) Multiple roles of Alu-related noncoding RNAs. Prog Mol Subcell Biol 51:119–146. doi:10.1007/978-3-642-16502-3_6
Bohmdorfer G, Wierzbicki AT (2015) Control of chromatin structure by long noncoding RNA. Trends Cell Biol 25:623–632. doi:10.1016/j.tcb.2015.07.002
Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF (1992) The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71:527–542
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Gene Dev 25:1915–1927. doi:10.1101/gad.17446611
Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, Pesce E, Ferrer I, Collavin L, Santoro C, Forrest AR, Carninci P, Biffo S, Stupka E, Gustincich S (2012) Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491:454–457. doi:10.1038/nature11508
Cartault F, Munier P, Benko E, Desguerre I, Hanein S, Boddaert N, Bandiera S, Vellayoudom J, Krejbich-Trotot P, Bintner M, Hoarau JJ, Girard M, Genin E, de Lonlay P, Fourmaintraux A, Naville M, Rodriguez D, Feingold J, Renouil M, Munnich A, Westhof E, Fahling M, Lyonnet S, Henrion-Caude A (2012) Mutation in a primate-conserved retrotransposon reveals a noncoding RNA as a mediator of infantile encephalopathy. Proc Natl Acad Sci U S A 109:4980–4985. doi:10.1073/pnas.1111596109
Carver R, Waldahl R, Breivik J (2008) Frame that gene. A tool for analysing and classifying the communication of genetics to the public. EMBO Rep 9:943–947. doi:10.1038/embor.2008.176
Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L (2004) Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem 279:13256–13264. doi:10.1074/jbc.M314124200
Consortium MGS, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O’Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562. doi:10.1038/nature01262
Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10:691–703. doi:10.1038/nrg2640
Craig NL, Chandler M, Gellert M, Lambowitz AM, Rice PA, Sandmeyer SB (2015) Mobile DNA III. ASM press, Washington
Crick FH, Barnett L, Brenner S, Watts-Tobin RJ (1961) General nature of the genetic code for proteins. Nature 192:1227–1232
Cruickshanks HA, Vafadar-Isfahani N, Dunican DS, Lee A, Sproul D, Lund JN, Meehan RR, Tufarelli C (2013) Expression of a large LINE-1-driven antisense RNA is linked to epigenetic silencing of the metastasis suppressor gene TFPI-2 in cancer. Nucleic Acids Res 41:6857–6869. doi:10.1093/nar/gkt438
Deininger PL, Moran JV, Batzer MA, Kazazian HH Jr (2003) Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 13:651–658
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22:1775–1789. doi:10.1101/gr.132159.111
Doolittle WF, Sapienza C (1980) Selfish genes, the phenotype paradigm and genome evolution. Nature 284:601–603
Duret L, Chureau C, Samain S, Weissenbach J, Avner P (2006) The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312:1653–1655. doi:10.1126/science.1126316
Elisaphenko EA, Kolesnikov NN, Shevchenko AI, Rogozin IB, Nesterova TB, Brockdorff N, Zakian SM (2008) A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PloS One 3, e2521. doi:10.1371/journal.pone.0002521
Espinoza CA, Allen TA, Hieb AR, Kugel JF, Goodrich JA (2004) B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat Struct Mol Biol 11:822–829. doi:10.1038/nsmb812
Espinoza CA, Goodrich JA, Kugel JF (2007) Characterization of the structure, function, and mechanism of B2 RNA, an ncRNA repressor of RNA polymerase II transcription. RNA 13:583–596. doi:10.1261/rna.310307
Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, Waki K, Hornig N, Arakawa T, Takahashi H, Kawai J, Forrest AR, Suzuki H, Hayashizaki Y, Hume DA, Orlando V, Grimmond SM, Carninci P (2009) The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41:563–571. doi:10.1038/ng.368
Feschotte C (2008) Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9:397–405. doi:10.1038/nrg2337
Feschotte C, Pritham EJ (2007) DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41:331–368. doi:10.1146/annurev.genet.40.110405.090448
Flockhart RJ, Webster DE, Qu K, Mascarenhas N, Kovalski J, Kretz M, Khavari PA (2012) BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res 22:1006–1014. doi:10.1101/gr.140061.112
Folkersen L, Kyriakou T, Goel A, Peden J, Malarstig A, Paulsson-Berne G, Hamsten A, Hugh W, Franco-Cereceda A, Gabrielsen A, Eriksson P, Consortia P (2009) Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PloS One 4:e7677. doi:10.1371/journal.pone.0007677
Fort A, Hashimoto K, Yamada D, Salimullah M, Keya CA, Saxena A, Bonetti A, Voineagu I, Bertin N, Kratz A, Noro Y, Wong CH, de Hoon M, Andersson R, Sandelin A, Suzuki H, Wei CL, Koseki H, Consortium F, Hasegawa Y, Forrest AR, Carninci P (2014) Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet 46:558–566. doi:10.1038/ng.2965
Galupa R, Heard E (2015) X-chromosome inactivation: new insights into cis and trans regulation. Curr Opin Genet Dev 31:57–66. doi:10.1016/j.gde.2015.04.002
Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO, Emanuelsson O, Zhang ZD, Weissman S, Snyder M (2007) What is a gene, post-ENCODE? History and updated definition. Genome Res 17:669–681. doi:10.1101/gr.6339607
Gong C, Maquat LE (2011) lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature 470:284–288. doi:10.1038/nature09701
Guttman M, Rinn JL (2012) Modular regulatory principles of large non-coding RNAs. Nature 482:339–346. doi:10.1038/nature10887
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227. doi:10.1038/nature07672
Han JS, Szak ST, Boeke JD (2004) Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 429:268–274. doi:10.1038/nature02536
Hancks DC, Kazazian HH Jr (2012) Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22:191–203. doi:10.1016/j.gde.2012.02.006
Hasler J, Strub K (2006) Alu elements as regulators of gene expression. Nucleic Acids Res 34:5491–5497. doi:10.1093/nar/gkl706
Havecker ER, Gao X, Voytas DF (2004) The diversity of LTR retrotransposons. Genome Biol 5:225. doi:10.1186/gb-2004-5-6-225
Herquel B, Ouararhni K, Martianov I, Le Gras S, Ye T, Keime C, Lerouge T, Jost B, Cammas F, Losson R, Davidson I (2013) Trim24-repressed VL30 retrotransposons regulate gene expression by producing noncoding RNA. Nat Struct Mol Biol 20:339–346. doi:10.1038/nsmb.2496
Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I (2015) Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep 11:1110–1122. doi:10.1016/j.celrep.2015.04.023
Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, Finstermeier K, Stahringer A, Wilfert W, Beutner F, Gielen S, Schuler G, Gabel G, Bergert H, Bechmann I, Stadler PF, Thiery J, Teupser D (2013) Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet 9, e1003588. doi:10.1371/journal.pgen.1003588
International Human Genome Sequencing Consortium, Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Szustakowki J, International Human Genome Sequencing C (2001) Initial sequencing and analysis of the human genome. Nature 409:860-921. doi:10.1038/35057062
Iwasaki YW, Siomi MC, Siomi H (2015) PIWI-interacting RNA: its biogenesis and functions. Ann Rev Biochem 84:405–433. doi:10.1146/annurev-biochem-060614-034258
Johannsen W (1909) Elemente der exakten erblichkeitslehre. Deutsche wesentlich erweiterte ausgabe in fünfundzwanzig vorlesungen. G. Fischer Verlag, Jena
Kaneko-Ishino T, Ishino F (2012) The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front Microbiol 3:262. doi:10.3389/fmicb.2012.00262
Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C (2013) Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet 9, e1003470. doi:10.1371/journal.pgen.1003470
Kazazian HH Jr (2004) Mobile elements: drivers of genome evolution. Science 303:1626–1632. doi:10.1126/science.1089670
Kelley RL, Kuroda MI (2000) Noncoding RNA genes in dosage compensation and imprinting. Cell 103:9–12
Kelley D, Rinn J (2012) Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 13:R107. doi:10.1186/gb-2012-13-11-r107
Khodosevich K, Lebedev Y, Sverdlov E (2002) Endogenous retroviruses and human evolution. Comp Funct Genom 3:494–498. doi:10.1002/cfg.216
Kim YK, Furic L, Parisien M, Major F, DesGroseillers L, Maquat LE (2007) Staufen1 regulates diverse classes of mammalian transcripts. EMBO J 26:2670–2681. doi:10.1038/sj.emboj.7601712
Korostowski L, Sedlak N, Engel N (2012) The Kcnq1ot1 long non-coding RNA affects chromatin conformation and expression of Kcnq1, but does not regulate its imprinting in the developing heart. PLoS Genet 8, e1002956. doi:10.1371/journal.pgen.1002956
Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, Ng HH, Bourque G (2010) Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42:631–634. doi:10.1038/ng.600
Lavillette D, Kabat D (2004) Porcine endogenous retroviruses infect cells lacking cognate receptors by an alternative pathway: implications for retrovirus evolution and xenotransplantation. J Virol 78:8868–8877. doi:10.1128/JVI.78.16.8868-8877.2004
Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AW, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL, Venter JC (2007) The diploid genome sequence of an individual human. PLoS Biol 5, e254. doi:10.1371/journal.pbio.0050254
Ling J, Pi W, Bollag R, Zeng S, Keskintepe M, Saliman H, Krantz S, Whitney B, Tuan D (2002) The solitary long terminal repeats of ERV-9 endogenous retrovirus are conserved during primate evolution and possess enhancer activities in embryonic and hematopoietic cells. J Virol 76:2410–2423
Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT Jr (2002) The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 111:209–218
Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, Manos PD, Datta S, Lander ES, Schlaeger TM, Daley GQ, Rinn JL (2010) Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 42:1113–1117. doi:10.1038/ng.710
Lunyak VV, Prefontaine GG, Nunez E, Cramer T, Ju BG, Ohgi KA, Hutt K, Roy R, Garcia-Diaz A, Zhu X, Yung Y, Montoliu L, Glass CK, Rosenfeld MG (2007) Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science 317:248–251. doi:10.1126/science.1140871
Mandal AK, Pandey R, Jha V, Mukerji M (2013) Transcriptome-wide expansion of non-coding regulatory switches: evidence from co-occurrence of Alu exonization, antisense and editing. Nucleic Acids Res 41:2121–2137. doi:10.1093/nar/gks1457
Marino-Ramirez L, Lewis KC, Landsman D, Jordan IK (2005) Transposable elements donate lineage-specific regulatory sequences to host genomes. Cytogenet Genome Res 110:333–341. doi:10.1159/000084965
Martens JH, O’Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, Jenuwein T (2005) The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J 24:800–812. doi:10.1038/sj.emboj.7600545
McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316:1488–1491. doi:10.1126/science.1142447
Mercer TR, Mattick JS (2013) Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 20:300–307. doi:10.1038/nsmb.2480
Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, Jurka J, Kamal M, Mauceli E, Searle SM, Sharpe T, Baker ML, Batzer MA, Benos PV, Belov K, Clamp M, Cook A, Cuff J, Das R, Davidow L, Deakin JE, Fazzari MJ, Glass JL, Grabherr M, Greally JM, Gu W, Hore TA, Huttley GA, Kleber M, Jirtle RL, Koina E, Lee JT, Mahony S, Marra MA, Miller RD, Nicholls RD, Oda M, Papenfuss AT, Parra ZE, Pollock DD, Ray DA, Schein JE, Speed TP, Thompson K, VandeBerg JL, Wade CM, Walker JA, Waters PD, Webber C, Weidman JR, Xie X, Zody MC, Broad Institute Genome Sequencing P, Broad Institute Whole Genome Assembly T, Graves JA, Ponting CP, Breen M, Samollow PB, Lander ES, Lindblad-Toh K (2007) Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447:167–177. doi:10.1038/nature05805
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P (2008) The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322:1717–1720. doi:10.1126/science.1163802
Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, Kaessmann H (2014) The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505:635–640. doi:10.1038/nature12943
Nesterova TB, Slobodyanyuk SY, Elisaphenko EA, Shevchenko AI, Johnston C, Pavlova ME, Rogozin IB, Kolesnikov NN, Brockdorff N, Zakian SM (2001) Characterization of the genomic Xist locus in rodents reveals conservation of overall gene structure and tandem repeats but rapid evolution of unique sequence. Genome Res 11:833–849. doi:10.1101/gr.174901
Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schonbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake JA, Bradt D, Brusic V, Chothia C, Corbani LE, Cousins S, Dalla E, Dragani TA, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons PA, Maglott DR, Maltais L, Marchionni L, McKenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple CA, Setou M, Shimada K, Sultana R, Takenaka Y, Taylor MS, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M, Yang I, Yang L, Yuan Z, Zavolan M, Zhu Y, Zimmer A, Carninci P, Hayatsu N, Hirozane-Kishikawa T, Konno H, Nakamura M, Sakazume N, Sato K, Shiraki T, Waki K, Kawai J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Imotani K, Ishii Y, Itoh M, Kagawa I, Miyazaki A, Sakai K, Sasaki D, Shibata K, Shinagawa A, Yasunishi A, Yoshino M, Waterston R, Lander ES, Rogers J, Birney E, Hayashizaki Y, Consortium F, Team IRGERGP II (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420:563–573. doi:10.1038/nature01266
Orgel LE, Crick FHC (1980) Selfish DNA: the ultimate parasite. Nature 284:604–607
Pang KC, Frith MC, Mattick JS (2006) Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22:1–5. doi:10.1016/j.tig.2005.10.003
Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M, Zatloukal K (2007) Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132:330–342. doi:10.1053/j.gastro.2006.08.026
Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, Knowles BB (2004) Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell 7:597–606. doi:10.1016/j.devcel.2004.09.004
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136:629–641. doi:10.1016/j.cell.2009.02.006
Santoro F, Mayer D, Klement RM, Warczok KE, Stukalov A, Barlow DP, Pauler FM (2013) Imprinted Igf2r silencing depends on continuous Airn lncRNA expression and is not restricted to a developmental window. Development 140:1184–1195. doi:10.1242/dev.088849
Singer MF (1982) SINEs and LINEs: highly repeated short and long interspersed sequences in mammalian genomes. Cell 28:433–434
Smit AF (1993) Identification of a new, abundant superfamily of mammalian LTR-transposons. Nucleic Acids Res 21:1863–1872
Smit AF, Riggs AD (1996) Tiggers and DNA transposon fossils in the human genome. Proc Natl Acad Sci U S A 93:1443–1448
Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, Dym M, de Massy B, Mikkelsen TS, Kaessmann H (2013) Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 3:2179–2190. doi:10.1016/j.celrep.2013.05.031
Svoboda P, Stein P, Anger M, Bernstein E, Hannon GJ, Schultz RM (2004) RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev Biol 269:276–285. doi:10.1016/j.ydbio.2004.01.028
Takeda K, Ichijo H, Fujii M, Mochida Y, Saitoh M, Nishitoh H, Sampath TK, Miyazono K (1998) Identification of a novel bone morphogenetic protein-responsive gene that may function as a noncoding RNA. J Biol Chem 273:17079–17085
Tarlinton RE, Meers J, Young PR (2006) Retroviral invasion of the koala genome. Nature 442:79–81. doi:10.1038/nature04841
The FANTOM Consortium, Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest ARR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, Chalk AM, Chiu KP, Choudhary V, Christoffels A, Clutterbuck DR, Crowe ML, Dalla E, Dalrymple BP, de Bono B, Gatta GD, di Bernardo D, Down T, Engstrom P, Fagiolini M, Faulkner G, Fletcher CF, Fukushima T, Furuno M, Futaki S, Gariboldi M, Georgii-Hemming P, Gingeras TR, Gojobori T, Green RE, Gustincich S, Harbers M, Hayashi Y, Hensch TK, Hirokawa N, Hill D, Huminiecki L, Iacono M, Ikeo K, Iwama A, Ishikawa T, Jakt M, Kanapin A, Katoh M, Kawasawa Y, Kelso J, Kitamura H, Kitano H, Kollias G, Krishnan SPT, Kruger A, Kummerfeld SK, Kurochkin IV, Lareau LF, Lazarevic D, Lipovich L, Liu J, Liuni S, McWilliam S, Babu MM, Madera M, Marchionni L, Matsuda H, Matsuzawa S, Miki H, Mignone F, Miyake S, Morris K, Mottagui-Tabar S, Mulder N, Nakano N, Nakauchi H, Ng P, Nilsson R, Nishiguchi S, Nishikawa S, Nori F, Ohara O, Okazaki Y, Orlando V, Pang KC, Pavan WJ, Pavesi G, Pesole G, Petrovsky N, Piazza S, Reed J, Reid JF, Ring BZ, Ringwald M, Rost B, Ruan Y, Salzberg SL, Sandelin A, Schneider C, Schönbach C, Sekiguchi K, Semple CAM, Seno S, Sessa L, Sheng Y, Shibata Y, Shimada H, Shimada K, Silva D, Sinclair B, Sperling S, Stupka E, Sugiura K, Sultana R, Takenaka Y, Taki K, Tammoja K, Tan SL, Tang S, Taylor MS, Tegner J, Teichmann SA, Ueda HR, van Nimwegen E, Verardo R, Wei CL, Yagi K, Yamanishi H, Zabarovsky E, Zhu S, Zimmer A, Hide W, Bult C, Grimmond SM, Teasdale RD, Liu ET, Brusic V, Quackenbush J, Wahlestedt C, Mattick JS, Hume DA, Group RGER, Group GS, Kai C, Sasaki D, Tomaru Y, Fukuda S, Kanamori-Katayama M, Suzuki M, Aoki J, Arakawa T, Iida J, Imamura K, Itoh M, Kato T, Kawaji H, Kawagashira N, Kawashima T, Kojima M, Kondo S, Konno H, Nakano K, Ninomiya N, Nishio T, Okada M, Plessy C, Shibata K, Shiraki T, Suzuki S, Tagami M, Waki K, Watahiki A, Okamura-Oho Y, Suzuki H, Kawai J, Hayashizaki Y (2005) The transcriptional landscape of the mammalian genome. Science 309:1559–1563. doi:10.1126/science.1112014
Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, Margulies EH, Blanchette M, Siepel AC, Thomas PJ, McDowell JC, Maskeri B, Hansen NF, Schwartz MS, Weber RJ, Kent WJ, Karolchik D, Bruen TC, Bevan R, Cutler DJ, Schwartz S, Elnitski L, Idol JR, Prasad AB, Lee-Lin SQ, Maduro VV, Summers TJ, Portnoy ME, Dietrich NL, Akhter N, Ayele K, Benjamin B, Cariaga K, Brinkley CP, Brooks SY, Granite S, Guan X, Gupta J, Haghighi P, Ho SL, Huang MC, Karlins E, Laric PL, Legaspi R, Lim MJ, Maduro QL, Masiello CA, Mastrian SD, McCloskey JC, Pearson R, Stantripop S, Tiongson EE, Tran JT, Tsurgeon C, Vogt JL, Walker MA, Wetherby KD, Wiggins LS, Young AC, Zhang LH, Osoegawa K, Zhu B, Zhao B, Shu CL, De Jong PJ, Lawrence CE, Smit AF, Chakravarti A, Haussler D, Green P, Miller W, Green ED (2003) Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424:788–793. doi:10.1038/nature01858
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147:1537–1550. doi:10.1016/j.cell.2011.11.055
Veselovska L, Smallwood SA, Saadeh H, Stewart KR, Krueger F, Maupetit-Mehouas S, Arnaud P, Tomizawa S, Andrews S, Kelsey G (2015) Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol 16:209. doi:10.1186/s13059-015-0769-z
Volders PJ, Verheggen K, Menschaert G, Vandepoele K, Martens L, Vandesompele J, Mestdagh P (2015) An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res 43:D174–180. doi:10.1093/nar/gku1060
Wang KC, Chang HY (2011) Molecular mechanisms of long noncoding RNAs. Mol Cell 43:904–914. doi:10.1016/j.molcel.2011.08.018
Wang F, Li X, Xie X, Zhao L, Chen W (2008) UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma and embryo, influencing cell growth and promoting invasion. FEBS Lett 582:1919–1927. doi:10.1016/j.febslet.2008.05.012
Wang J, Gong C, Maquat LE (2013) Control of myogenesis by rodent SINE-containing lncRNAs. Gene Dev 27:793–804. doi:10.1101/gad.212639.112
Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV (2001) Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol 21:1429–1439. doi:10.1128/MCB.21.4.1429-1439.2001
Weick EM, Miska EA (2014) piRNAs: from biogenesis to function. Development 141:3458–3471. doi:10.1242/dev.094037
Wellcome Trust Case Control C (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678. doi:10.1038/nature05911
Wilusz JE, Sunwoo H, Spector DL (2009) Long noncoding RNAs: functional surprises from the RNA world. Gene Dev 23:1494–1504. doi:10.1101/gad.1800909
Xi S, Geiman TM, Briones V, Guang Tao Y, Xu H, Muegge K (2009) Lsh participates in DNA methylation and silencing of stem cell genes. Stem Cells 27:2691–2702. doi:10.1002/stem.183
Acknowledgments
We thank Radek Malik (Institute of Molecular Genetics of the ASCR, Prague) for help with manuscript preparation and Martin Moravec (ETH, Zurich) for providing Fig. 2b photo. The main support for research of P.S. on RNA is provided by the European Research Council grant ERC-2014-CoG-647403 (D-FENS), the Czech Science Foundation grant GACR P305/12/G034, and Ministry of Education, Youth, and Sports project NPU1 LO1419. S.G. is supported through a Marie Curie Initial Training Network (project #607720, RNATRAIN). The institutional support is provided by RVO: 68378050.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ganesh, S., Svoboda, P. Retrotransposon-associated long non-coding RNAs in mice and men. Pflugers Arch - Eur J Physiol 468, 1049–1060 (2016). https://doi.org/10.1007/s00424-016-1818-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1818-5