
Abstract
Transient receptor potential melastatin 4 (TRPM4) is a voltage-dependent, nonselective cation channel. Under pathological conditions, sustained activation of TRPM4 leads to oncotic cell death. Here, we report the upregulation of TRPM4 in vascular endothelium following hypoxia/ischemia in vitro and in vivo. In human umbilical vein endothelial cells, TRPM4 expression was increased at both the mRNA and protein levels following oxygen–glucose deprivation. Blocking TRPM4 with 9-phenanthrol greatly enhanced tube formation on Matrigel. In a rat permanent middle cerebral artery occlusion model, TRPM4 was upregulated in the vascular endothelium within the penumbra region after stroke. TRPM4 expression peaked 1 day post-occlusion and gradually decreased. In vivo siRNA-mediated TRPM4 silencing enhanced angiogenesis and improved capillary integrity. A twofold reduction in infarct volume and a substantial recovery of motor function were observed in animals receiving the siRNA treatment. Interestingly, the protective effect of TRPM4 suppression disappeared 5 days after stroke induction, indicating that TRPM4 upregulation is critical for cerebral damage during the acute phase of stroke. TRPM4 could be a potential therapeutic target for ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transient receptor potential melastatin 4 (TRPM4) is a voltage-dependent, nonselective cation channel. It is impermeable to Ca2+, activated by elevated cytosolic Ca2+, and modulated by ATP [29]. TRPM4 belongs to the mammalian transient receptor potential superfamily. The TRPM4 and TRPM5 are unique because they only conduct monovalent cations, whereas most other TRP channels are permeable to both monovalent and divalent ions. TRPM4 is important for the function of immune cells, including dendritic, mast, and T cells [2, 13, 30]. When activated, TRPM4 can depolarize the membrane potential and regulate Ca2+ homeostasis by decreasing the driving force for Ca2+ entry in non-excitable cells. Gain-of-function mutations in TRPM4 are associated with familial heart disease [12, 14]. TRPM4 also participates in the pathophysiology of spinal cord injury (SCI) and experimental autoimmune encephalomyelitis (EAE) [7, 20]. Ectopic expression of TRPM4 has been found in capillaries after SCI and in neurons after EAE. Activation of TRPM4 in SCI and EAE results in unchecked ion influx and subsequently leads to oncotic cell death.
Cerebral edema following brain injury is bound to cell death. Edema resulting from ischemic stroke leads to tissue damage and worsens neurological functions. Recently, upregulation of the nonselective cation channel NCCa-ATP was observed in neurovascular cells, including astrocytes, neurons, and vascular endothelia, after ischemic stroke [22]. Enhanced NCCa-ATP current can lead to unchecked Na+ entry, subsequently oncotic cell death, and is believed to cause brain edema [10]. The current exhibits many properties similar to those of TRPM4, including a smaller single-channel conductance, permeability to Na+ and Cs+, and activation by intracellular Ca2+ [22, 25]. The NCCa-ATP current is also involved in other central nervous system injuries, including traumatic brain injury, spinal cord injury, and subarachnoid hemorrhage [23, 24, 26]. However, studies of NCCa-ATP channel in stroke have mainly focused on the sulfonylurea receptor-1, an auxiliary subunit of KATP channels [22]. The role of TRPM4 after ischemic stroke is unclear. Using a rat permanent middle cerebral artery occlusion model (MCAO), we investigated the expression and functions of TRPM4 in ischemic stroke.
Materials and methods
Animal model
This study was approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the National Neuroscience Institute, Singapore. As per the approved protocol, male Wistar rats weighing approximately 300 g were subjected to MCAO. Prior to surgery, the animals were anesthetized with ketamine (75 mg/kg) and xylazine (10 mg/kg) intraperitoneally. Under an operating microscope, the left common carotid artery was exposed and temporarily ligated using a vascular clip (Aesculap, B. Braun, Germany). Next, the left external carotid artery (ECA) and internal carotid artery (ICA) were dissected from the surrounding tissues. Occipital artery and superior thyroid artery (branches of ECA) were occluded. The ECA was then ligated with a 4–0 silk suture. The ICA was also free from the adjacent vagus nerve. Subsequently, a loose knot was made at the ECA stump near the bifurcation with a 4–0 silk suture. The extracranial branch of ICA was temporarily ligated with a vascular clip as well. The distal end of the ECA was cut and a silicon-coated filament (0.37 mm, Cat #403756PK10, Doccol Corp, Redlands, CA) was introduced into the ICA through the ECA stump. Subsequently, the suture around the ECA stump was tightened around the intraluminal filament, and the microvascular clip was removed. The filament was then gently advanced from the ECA to the ICA lumen for approximately 18–20 mm. The ligation on the CCA was released after the suture on the ECA-intraluminal filament was tightened. The sham-operated rats underwent similar procedures, except for the insertion of the suture.
Infarct volume measurement
2,3,5-Triphenyltetrazolium chloride (TTC) staining relies on the ability of the dehydrogenase enzymes and cofactors present in the living tissue to react with tetrazolium salts, the main component of the TTC solution, to form a formazan pigment. After the animals were euthanized, the brains were collected, and the cerebellum and overlying membranes were removed. The brain was sectioned into 2-mm-thick coronal slices using a brain-sectioning block. The sections were stained with 0.1 % TTC (Sigma, USA) solution at 37 °C for 30 min and then preserved in 4 % formalin solution. The sections were scanned and the infarct size was analyzed using an image analyzer system (Scion image from Windows, Microsoft).
Immunofluorescent staining
The animals were euthanized and perfused with saline followed by 4 % paraformaldehyde (PFA). The brains were then collected and post-fixed with 4 % PFA for 2 h. Dehydration was subsequently carried out by immersing the brain in a 15 % sucrose solution, followed by 30 % sucrose solution. Next, the rat brain was cryosectioned at 20 μm of thickness. After washing with 0.2 % Triton X-100 phosphate buffered saline (PBST), 100 μl of the blocking serum (10 % goat serum and 1 % bovine serum albumin in 0.2 % PBST) was added to the sections for 1 h. The brain sections were then incubated with primary antibodies overnight at 4 °C. The primary antibodies used in the study are anti-TRPM4 (sc-27540, Santa Cruz, CA, USA), anti-NeuN (MAB377, Millipore), anti-GFAP (IF03L, Calbiochem, Millipore), anti-smooth muscle actin (CBL171, Millipore), and anti-von Willebrand factor (vWF) (AB7356, Millipore, MA, USA). On the following day, the tissue sections were washed three times with TNT wash buffer (0.1 M Tris–HCl buffer pH 7.5 containing 0.15 M NaCl and 0.05 % Tween 20). The slides were incubated with secondary antibodies for 1 h at room temperature. After washing three times of wash buffer, the slides were mounted with FluorSaveTM reagent (Merck, Germany). The results were visualized by a laser scanning confocal microscope (Fluoview BX61, Olympus). The negative control underwent an identical procedure with the exception of the primary antibody incubation; no positive signal was observed.
Capillary counting
Capillary counting was conducted stereologically. Every fifth 100-μm brain section across the entire region of infarction was counted. For random sampling, six fields per brain section were randomly chosen under a confocal microscope under ×40 magnification. The number of blood vessels was measured by counting the number of elongated tube-like structures with positive vWF immunoreactivity.
Western blot
Tissues from the penumbra region and the contralateral control sites were collected from the TTC-stained brain slices as described earlier [32]. The brain tissues were homogenized in 300 μl of HEPES lysis buffer (20 mM HEPES, 137 mM NaCl, 1 % Triton X-100, 10 % glycerol, 1.5 mM MgCl2, 1 mM EGTA) with freshly added protease inhibitors (1:50 dilution, Roche Diagnostics). The homogenized samples were then centrifuged at 14,000 rpm for 15 min, and the supernatants were collected. Protein concentration was determined using the Bradford assay. For Western blot analysis, 200 μg of lysate (with 2× protein loading dye) was separated on an 8 % SDS-PAGE gel. The gel was then transferred overnight at 30 V onto a PVDF membrane at 4 °C. Subsequently, the membrane was blocked with 1 % BSA in 1× PBS + 0.1 % Tween 20 for 1 h and probed with an anti-TRPM4 goat polyclonal antibody (1:2,000 dilution, SC27540, Santa Cruz Biotechnology) overnight at 4 °C. The next day, the membrane was washed hree times with 0.1 % Tween 20 in 1× PBS for 10 min. The membrane was then probed with a goat secondary antibody conjugated to HRP (1:5,000 dilution, A5420, Sigma Aldrich) for 1 h. After washing, the TRPM4 band was detected using the Amersham ECL Western Blotting Analysis System (RPN2109, GE Healthcare).
Human umbilical vein endothelial cells culture and hypoxia induction
Human umbilical vein endothelial cells (HUVECs) (Lonza, Wokingham, UK) were cultured at 37 °C with 5 % CO2. The culture medium is endothelial growth medium-2 (EGM-2) consisting of endothelial basal medium (EBM), supplemented with 2 % fetal bovine serum, hydrocortisone, hFGF, VEGF, R3-IGF-1, ascorbic acid, HEGF, GA-1000, and heparin (Lonza, Wokingham, UK). Cells at passage 6–10 were used for experiments. HUVECs were subjected to oxygen/glucose deprivation (OGD). To achieve supplement deprivation, EGM-2 was changed to EBM without fetal bovine serum or growth supplements. Hypoxia was induced by culturing the cells in a hypoxic chamber (Stem Cell Technologies, Vancouver, Canada) with 1 % O2 and 5 % CO2 at 37 °C for 24 h.
Trypan blue exclusion and MTT assay
Cell viability was determined using the Trypan blue exclusion method. The cells were trypsinized and incubated with Trypan blue. The number of cells was quantified by light microscopy using a hemocytometer chamber and is expressed as the percentage of viable cells. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay (Roche, Switzerland) was performed to assess cell viability. Cell viability was quantified by the amount of MTT reduction. The cells were incubated with anhydrous MTT for 4 h, and the product was solubilized in a DMSO solution. The optical density was measured at 540 nm. The data is expressed as the percentage of viable cells.
Tube formation
HUVECs were divided into the following groups: normoxia, normoxia + 5 μM 9-phenanthrol (Sigma), hypoxia, and hypoxia + 5 μM 9-phenanthrol. A 24-well culture plate was precoated with 250 μl of growth factor-reduced Matrigel (Sigma, USA) at 37 °C for 30 min. Twenty-four hours after OGD, 4 × 104 cells (in 300 μl of EGM-2) from each group were seeded onto the Matrigel-coated plates. The formation of capillary structures was examined under a light microscope after 4 h. This study was replicated more than four times.
Reverse transcriptase PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen, Life Technologies Corporation, USA) according to the manufacturer's protocol. Superscript III (Invitrogen, Life Technologies Corporation, USA) was used to generate first strand cDNA. The PCR was performed as follows: a denaturation step at 95 °C for 5 min; 35 cycles of 94 °C for 30 s, 60 °C for 45 s, and 72 °C for 30 s; and a final extension step at 72 °C for 10 min. GAPDH was used as the endogenous control. The primers used for TRPM4 were as follows: 5′-CTGGTTCTCGCCTTCTTTTG-3′ (forward) and 5′-CATGAAGTCGATGCAGAGGA-3′ (reverse); and GAPDH: 5′-GAAGGTGAAGGTCGGAGTCAACG-3′ (forward) and 5′-TGCCATGGGTGGAATCATATTGG-3′ (reverse).
Patch clamp measurements
Whole-cell currents were recorded under voltage clamp with Axopatch 200B amplifier (Molecular Devices Corp. Sunnyvale, CA, USA). Data were digitized at 10 kHz and filtered at 1 kHz. The pClamp 10.2 software was used for data acquisition and analysis. The internal pipette solution contained (in millimole/liter): CsCl 140, NaCl 5, MgCl2 1, BAPTA 1, CaCl2 0.83, and pH 7.2 adjusted with CsOH. The bath solution contained (in millimole/liter): NaCl 140, CsCl 5, CaCl2 2, MgCl2 1, glucose 10, HEPES 10, and pH 7.4 adjusted with NaOH.
In vivo siRNA delivery
TRPM4 in vivo Ready siRNA was purchased from Ambion, Life Technologies Corporation, USA. The sequences were as follows: sense 5′-CGCUAGUAGCAGCAAAUCUtt-3′ and antisense 5′-AGAUUUGCUGCUACUAGCGtg-3′. Immediately after operation, the rats received a loading dose of 19 nmol siRNA intravenously. Subsequently, 6 nmol siRNA was given via the jugular vein through an implanted mini-osmotic pump (Alzet, Durect Corporation). The infusion rate was 7.8 μl/h for 24 h.
Behavioral analysis
Motor function after MCAO was evaluated using a rotarod apparatus (Ugo Basile, Italy). The performance of the rats was measured by observing the latency with which the rats fell off the rotarod. Three days before the surgery, the rats received three training trials each day with 15-min intervals. The accelerating rotarod was set from 4 to 80 rpm within 10 min. The mean duration of time that the animals remained on the device was recorded 1 day before MCAO as an internal baseline control. At different time points following surgery, the mean duration of latency was recorded and compared to the internal baseline control.
Statistics
All of the results are presented as the mean ± S.E.M. Data were graphed using Prism, version 4 (GraphPad Software, CA). Student's t test was used to compare two sample means and one-way ANOVA followed by Dunnett's post hoc tests was used to compare the means of data from three or more groups. The results were considered significant if P < 0.05.
Results
Upregulation of TRPM4 in the vascular endothelium after MCAO
To investigate the role of TRPM4 in ischemic stroke, we permanently occluded the middle cerebral artery in rats. The infarction was located in ipsilateral cortex and striatum (left hemisphere in this study). DAB staining of the capillaries with the endothelial marker vWF in the contralateral and ipsilateral regions indicated prominent angiogenesis in the penumbra region 1 day post-stroke (Fig. 1a). Endothelial vWF staining was stronger in the penumbra region than in the contralateral brain tissues possibly due to endothelial activation and/or dysfunction after stroke [5]. Using a TRPM4-specific antibody, we detected strong staining of TRPM4 in the penumbra region that was co-labeled with vWF 1 day post-MCAO (Fig. 1b). TRPM4 was almost undetectable in the vascular endothelium in the uninjured contralateral hemisphere (Fig. 1b) and sham-operated brain (Supplementary Fig. 1a). Another TRPM channel, TRPM5, which is structurally similar to TRPM4, was not expressed in the endothelium (Supplementary Fig. 1b).

Upregulation of TRPM4 in the vascular endothelium within the penumbra region 1 day after MCAO. a DAB staining of the capillaries with vWF in the contralateral and ipsilateral regions. Scale bar, 200 μm. Capillary counting showed significant angiogenesis in the ipsilateral region. ***P < 0.0001, n = 25. b Representative staining of TRPM4 and colocalization with vWF in the contralateral and ipsilateral regions. Scale bar, 50 μm
Western blot analysis revealed a twofold upregulation of TRPM4 in the penumbra region (Fig. 4a) as compared to the contralateral region. The rat TRPM4 is approximately 135 kDa, suggesting that this protein represents the longer TRPM4b isoform [17]. The same antibody was able to detect the mouse TRPM4 protein which was expressed in HEK cells as a positive control, confirming the specificity of the antibody for TRPM4. We consistently observed that the size of rat TRPM4 was slightly smaller than the mouse TRPM4.
We further studied the expression of TRPM4 in the vascular endothelium within the penumbra at the following time points post-MCAO: 0 h, 6 h, 1 day, 3 days, and 7 days (Fig. 2). Strong TRPM4 staining was observed in the endothelium as early as 6 h. The expression levels remained prominent at 1 day post-operation. The almost complete colocalization of TRPM4 with vWF indicated that the majority of the endothelial cells within the penumbra region expressed high levels of TRPM4 following stroke. However, by day 3, TRPM4 expression began to decrease gradually. Many vWF-positive cells did not express TRPM4. Furthermore, we observed vascular fragmentation within penumbra region after stroke induction, a typical sign of the loss of vascular integrity. This fragmentation was prominent by day 3 and correlated with the reduced expression of TRPM4. Thus, we postulate that TRPM4 upregulation in the vascular endothelium may contribute to capillary death post-stroke.

Time-dependent expression of TRPM4 in the endothelium within the penumbra region after MCAO. Co-staining for TRPM4 and vWF was performed at 0 h, 6 h, 1 day, 3 days, and 7 days post-operation. Scale bar, 50 μm
In vitro blockade of TRPM4 in human umbilical vein endothelial cells induces tube formation
We tested this hypothesis in HUVECs under OGD. The cells were exposed to hypoxic conditions and starved of glucose and serum/growth factors. After 24 h of OGD, significant cell death occurred. A twofold increase of TRPM4 mRNA by RT-PCR (Fig. 3a) and a 1.7-fold increase of TRPM4 protein by Western blot (Fig. 3b) were observed after OGD treatment. Such increase was further confirmed by immunofluorescence staining (Fig. 3c).



The expression and function of TRPM4 in HUVECs. a RT-PCR for TRPM4 in HUVECs 1 day after OGD. GAPDH was used as a loading control. N, normoxia; H, hypoxia. After normalization to GAPDH, TRPM4 was significant increased in HUVECs after ODG treatment. ***P < 0.001, n = 4. b Western blot for TRPM4 in HUVECs 1 day after OGD. β-Actin was used as a loading control. N, normoxia; H, hypoxia. After normalization to β-actin, TRPM4 was significant increased in HUVECs after ODG treatment. *P < 0.05, n = 4. c Immunofluorescent staining of TRPM4 (green) in HUVECs 1 day after hypoxic induction. The nuclei were labeled with DAPI (blue). Scale bar, 20 μm. d The effects of 5 μM of 9-phenanthrol on tube formation in HUVECs after OGD. 9-Phenanthrol treatment significantly increased tube branches after OGD (n = 4). N-C: normoxia control; N-P, normoxia with 9-phenanthrol; H-C, hypoxia control; H-P, hypoxia with 9-phenanthrol. *P < 0.05; ***P < 0.001. e Sample TRPM4 currents from HUVEC cells after 1 day hypoxic treatment. The holding potential was 0 mV. A 400-ms ramp protocol from −100 to +100 mV was applied to the cells followed by a 20-ms step at +100 mV. A new ramp protocol was performed every 2 s. The TRPM4 current was inhibited by 100 μM 9-phenanthrol. f Summary of peak currents at +100 mV. *P < 0.05. n control = 10, n 1d hypoxia = 4. g Summary of peak current densities at +100 mV. n control = 7, n 1d hypoxia = 6
To test the functional impact of TRPM4 on HUVECs, we incubated the cells with the TRPM4-specific blocker 9-phenanthrol. This blocker does not affect the TRPM5 channel [8], another TRPM channel with similar electrophysiological properties to TRPM4 [16, 29]. A study was performed in which 9-phenantrol with the concentration ranged from 0.1 to 30 μM was added to HUVECs. No difference was observed in cell death between 0.1 and 5 μM. Cell death became prominent when 10 μM 9-phenanthrol was added to the culture medium. Very few cells survived treatment with 30 μM 9-phenanthrol (Supplementary Fig. 2a). Thus, 5 μM 9-phenanthrol was used to treat HUVECs. Unexpectedly, 9-phenanthrol treatment did not reduce cell death after OGD (Supplementary Fig. 2b, c). However, we observed a large enhancement of tube formation on Matrigel by 9-phenanthrol treatment after OGD (Fig. 3d). This in vitro study proves that blocking TRPM4 improves endothelial functions under hypoxic conditions.
TRPM4 currents are increased in HUVEC cells after hypoxia
To evaluate the functions of upregulation of TRPM4 in cultured HUVEC cells after hypoxia, we used whole-cell patch clamp method to measure the TRPM4 currents. A typical ramp protocol was applied to the cells under normoxia or hypoxia treatment for 1 day. An increased outward rectifying current sensitive to 9-phenanthrol (Fig. 3e) was recorded from the cells after hypoxia treatment (Fig. 3f) correlating with the increased expression of TRPM4 channels. Although the current density was higher in hypoxic cells, no significance was observed when compared to the control cells (Fig. 3g), P = 0.33.
In vivo TRPM4 knockdown enhances angiogenesis and reduces infarction after MCAO
To demonstrate that TRPM4 is critical for vascular endothelial functions in vivo, we used an siRNA against rat TRPM4 in our animal model of MCAO. A total of 25 nmol of siRNA was delivered into rats with a body weight of 300 g. A single dose of 19 nmol was injected intravenously post-operation and the remaining 6 nmol was delivered into the jugular vein with an osmotic pump for 24 h. Intravenous delivery reduces the loss of siRNA during absorption via other routes and maximizes contact with the endothelium.
Western blot analysis revealed that in the saline-treated animals there was an upregulation of TRPM4 in the ipsilateral region and very low expression of TRPM4 in the contralateral region. Knockdown of TRPM4 via siRNA successfully prevented the upregulation of TRPM4 in the ipsilateral hemisphere 1 day post-MCAO (Fig. 4a). TRPM4 protein levels were reduced to a level similar to that in the contralateral region. De novo expression of TRPM4 was previously shown to contribute to the damage of capillary integrity following SCI [7]. We studied the capillary structure in the rat brains 3 days post-MCAO. In the saline-treated animals, the capillaries were generally short, and fragmentation was present in almost all capillaries (Fig. 4b). The capillaries within the penumbra region of the siRNA-treated animals were elongated without segmentation, indicating greater structural integrity. In addition, the number of capillaries in the penumbra region was increased 2.5-fold relative to the saline-treated animals (saline 8.8 ± 0.8 vs. siRNA 21.4 ± 0.5, P < 0.0001, Student's t test). Thus, TRPM4 inhibition can promote angiogenesis in a rat model of stroke.


Knockdown of TRPM4 in vivo enhances angiogenesis and reduces infarct volume after MCAO in rats. a Representative Western blot of TRPM4 in rat brains 1 day post-MCAO. HEK 293 cells transfected with mouse TRPM4 (mTRPM4) were used as the positive controls and HEK 293 cells transfected with GFP vector alone (vc) were used as the negative control. Samples from ipsilateral penumbra region (L) and corresponding contralateral region (R) with saline (− siRNA) or siRNA treatment (+ siRNA) were probed with an antibody against TRPM4. The protein size of the rat TRPM4 (indicated by the arrow) was slightly smaller than the control mouse TRPM4. Without siRNA treatment, a twofold increase of TRPM4 expression was found in the penumbra region (L). *P < 0.05; n = 4. In vivo siRNA application successfully inhibited TRPM4 upregulation within the penumbra region. b Staining of the penumbra region with vWF showed the fragmentation of capillaries (indicated by the arrows) in the saline-treated rats 3 days post-MCAO. Elongated intact capillaries were identified in the siRNA-treated rats. Scale bar, 50 μm. The number of capillaries was increased by twofold with the siRNA treatment. #P < 0.0001; n = 7. c TTC staining of the rat brains 1 day post-MCAO. There was a significant reduction of infarct volume by siRNA treatment. **P < 0.01; n = 5
Next, we examined whether TRPM4 deletion affects brain tissue loss after stroke. In the saline-treated animals 1 day post-MCAO, large infarction was observed in the ipsilateral hemisphere according to TTC staining (Fig. 4c). Both the cortex and striatum were affected. By contrast, siRNA treatment greatly reduced infarction. The cortex was preserved almost completely, while a smaller infarction occurred mainly in the striatum. The infarction volume was greatly reduced from 24 ± 3.2 to 8.8 ± 1 % (P = 0.0017, Student's t test) (Fig. 4c).
In vivo TRPM4 knockdown improves motor function after MCAO
We used the rotarod test to evaluate the motor function of the rats receiving siRNA treatment. The sham-operated animals performed well on the test (Fig. 5a). The performance of MCAO rats (n = 8) receiving saline treatment decreased to 28 ± 4 % 1 day post-operation and gradually improved in subsequent days. The performance of the 21 siRNA-treated MCAO rats greatly improved to 57 ± 3.7 % 1 day post-MCAO, which is significantly higher than the saline group but lower than the sham-operated group (P < 0.0001, one-way ANOVA followed by Dunnett's post hoc analysis). The effects of siRNA were most prominent 3 days post-MCAO. There was no difference between the siRNA-treated animals and sham-operated animals (Fig. 5b). The protective effect lasted until day 5 post-operation. By day 7, the motor function deficit in the siRNA-treated animals decreased to levels that were similar to those in the saline-treated animals (Fig. 5a).

TRPM4 knockdown via siRNA improves motor function during the acute phase of stroke. a Rotarod performance of the sham-operated, saline-treated, and TRPM4-siRNA-treated MCAO rats. The data represent the percentage of the mean duration from three trials at each time point after normalization to the baseline control prior to the operation. The performance of the siRNA-treated rats was better than that of the saline-treated rats at days 1, 3, and 5. siRNA vs saline: #P < 0.0001; n sham = 5; n saline = 6–7; n siRNA = 18–21. b Rotarod performance at day 3 post-MCAO. Both sham-operated and siRNA-treated rats performed better than the saline-treated rats. #P < 0.0001. There was no difference between the sham-operated and siRNA-treated animals
Transient protective effect of TRPM4 knockdown
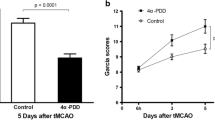
The results from the rotarod test suggest that TRPM4 knockdown does not maintain functional improvement after the acute phase of stroke. To study the effects of TRPM4 knockdown on tissue loss after the acute phase, we performed TTC staining to measure the infarction volume at different time points (Fig. 6a). In the saline-treated rats, the infarct volume decreased gradually by day 7, indicating that the process of reabsorption was occurring. By contrast, the protective effects of siRNA treatment disappeared by day 5. Although the mean infarct volumes were smaller than those in the saline-treated animals, there was no difference between the two groups (Fig. 6b). Thus, we believe that the protective effect of TRPM4 knockdown is transient and occurs during the acute phase of stroke. These results support that brain tissue loss correlates with functional changes after MCAO.

Transient effect of TRPM4-siRNA treatment post-stroke. a TTC staining at day 1 and day 5 after MCAO. b Infarct volume measurement at days 1, 5, and 7 after MCAO. **P < 0.01; n = 5 for both saline and siRNA groups
Extended siRNA treatment does not improve motor functions
To examine the effects of extended siRNA treatment, we gave a second dose of siRNA to MCAO rats intravenously on day 4 after operation. Day 4 was chosen for the boosting dose because the motor functions were well preserved on days 3 and 5 in rats receiving single-dose siRNA. Motor functions of the rats with the double-doses siRNA treatment were similar from day 5 until day 21 except for day 7 to those with single-dose treatment (Fig. 7a). However, the improved motor functions observed at day 7 by the boosting siRNA were not different from those with single-dose siRNA (Fig. 7b).

Motor functions of MCAO rats after prolonged treatment of siRNA. a Motor functions of MCAO rats received single dose or double doses of siRNA treatment. The boosting dose of 19 nmol of siRNA was given to five rats at day 4 after stroke induction. b Motor functions at day 7. P = 0.18
Discussion
The ectopic expression and activation of TRPM4 is generally harmful to cells. Under pathological conditions, an increase in intracellular Ca2+ concentrations and the depletion of ATP leads to TRPM4 activation [29], resulting in oncotic cell death due to unchecked Na+ influx [7]. The cessation of the blood supply to part of the brain after ischemic stroke can increase intracellular Ca2+ levels and lower ATP concentrations within the affected area [19]; both events can enhance TRPM4 activities.
An increase in TRPM4 current has been observed in cardiomyocytes from spontaneous hypertensive rats [9]. In the central nervous system, de novo expression of TRPM4 has also been identified in the capillaries following SCI [7]. We observed a similar upregulation of TRPM4 protein in the capillary endothelia after stroke. In cultured HUVECs, TRPM4 was expressed at basal levels under normoxic conditions, similar to a previous report [3]. In this study, TRPM4 expression is low in the healthy rat brain. The culture conditions may have caused the increased TRPM4 expression in HUVECs. OGD treatment increased TRPM4 expression at both the transcriptional and translational levels. As a result, increased TRPM4 currents were observed in HUVECs after 1 day OGD. Interestingly, there was no significance for the current density. This is possibly due to an enlarged cell volume caused by hypoxia-induced cellular edema. Blocking TRPM4 channels with 9-phenanthrol enhanced tube formation, a sign of angiogenesis after hypoxia. Thus, the upregulation of TRPM4 is harmful to endothelial cells, and TRPM4 inhibition can protect HUVECs from hypoxic insult. This is supported by another study indicating that blocking TRPM4 can prevent HUVECs from lipopolysaccharide-induced cell death [3].
In SCI, de novo expression of TRPM4 in the endothelium caused capillary fragmentation, and deletion of TRPM4 greatly enhanced recovery by promoting angiogenesis [7]. The loss of vascular integrity was also apparent within the penumbra region in our MCAO model. The disrupted vascular structure was likely partially caused by the upregulation of TRPM4 that leads to oncotic cell death. TRPM4 knockdown via siRNA greatly promoted angiogenesis and improved the motor functions of the rats. This indicates that the upregulation of TRPM4 in the endothelium following stroke plays a similar pathological role as in traumatic SCI. The increased expression of TRPM4 in the capillaries after SCI led to secondary hemorrhage. It is possible that the upregulation of TRPM4 is a cause of hemorrhage transformation observed in many patients with ischemic stroke.
In the MCAO model, the animals that were treated with the siRNA displayed not only intact capillaries but also an increased number of capillaries compared to the saline-treated rats. However, blocking TRPM4 in HUVECs only improved tube formation without increasing the cell number. This could be due to the differences in the levels of growth factors in the two studies. In the animal model, VEGF and other growth factors generated after the onset of stroke could promote capillary proliferation, whereas in HUVECs under OGD, we completely removed the serum and growth factors. Thus, no cell proliferation was observed.
The ectopic expression of TRPM4 within the axonal processes contributes to the tissue damage induced by EAE [20]. TRPM4 was also found in cerebral vascular smooth muscle cells. Blocking TRPM4 with antisense oligonucleotides greatly reduced arterial constriction [6, 18]. However, TRPM4 knockout mice are hypertensive due to elevated circulating catecholamines, which is independent of the TRPM4 deletion in endothelial and smooth muscle cells [15]. The reasons for the difference are unknown. Our initial staining result did not support an alteration of TRPM4 expression in vascular smooth muscles after stroke (data not shown). The Western blot used for detecting TRPM4 expression was done using brain homogenates. The beneficial effect of blocking TRPM4 after stroke could be partially due to the inhibition of TRPM4 in other cell types in the brain. More experiments are needed to clarify the role of TRPM4 in these cells after stroke.
The expression of TRPM4 was transient after ischemic stroke. It peaked within 1 day and then gradually decreased. As angiogenesis occurs soon after the onset of stroke, TRPM4 is likely to affect the regeneration of capillaries. In fact, TRPM4 knockdown immediately after MCAO preserved vascular integrity, enhanced angiogenesis, and as a result, reduced infarction, and promoted functional recovery. However, the effect of siRNA treatment was transient. By day 5, the rotarod performances of the animals were similar to those of the saline-treated rats. When the rats received a boosting siRNA on day 4 post-MCAO, motor functions were only slightly improved on day 7 but with no statistical difference. These results suggest that TRPM4 upregulation only participates in endothelial damage during the acute phase of stroke. This is supported by the observation that by day 7, many vascular endothelial cells did not express TRPM4 channels. The mechanism for this unique expression pattern is not known. Acute stroke reperfusion treatments are currently restricted by the very narrow time window (<4.5 h for intravenous thrombolysis), limiting this treatment to only a minority of patients. This is due to the progression of the ischemic penumbra to infarction over the first few hours following cerebral arterial occlusion [1]. According to our animal study, blocking TRPM4 during the acute phase protects the brain tissue for as long as 5 days. Thus, blocking TRPM4 could potentially extend the therapeutic time window of acute reperfusion treatments.
The role that TRPM4 plays in the immune response after stroke is unknown. Functional TRPM4 is required for dendritic cell (DC) migration and the release of cytokines in mast and T cells [2, 13, 30]. Although these cells are not present or activated in the healthy brain, they can be recruited into the infarct and activated to participate in the immune response due to the disruption of blood–brain barrier (BBB) after stroke [11, 28, 31]. Enhanced endothelial TRPM4 expression may contribute to the BBB disruption and facilitate the migration of immune cells to the damaged brain tissues. TRPM4 may have opposite effects in these cells. DCs invading the brain after stroke activate T cells, inducing a long-lasting immune response [11]. Blocking TRPM4 could reduce DC migration and ameliorate neurological deficits. By contrast, the release of cytokines from mast cells contributes to cell death after stroke. TRPM4 blockade can induce mast cell degranulation [30] and potentially increases BBB disruption and edema [27]. However, TRPM4 expression is also critical for mast cell migration [21]. TRPM4 inhibition may reduce mast cell recruitment and cytokine-induced tissue damage. In T cells, blocking TRPM4 increases IL-2 production [13]. Inhibiting TRPM4 after stroke can enhance cytokine production in T lymphocytes. Suppressing the immune response often ameliorates acute ischemic damage [4]. Thus, blocking TRPM4 in T cells may enhance IL-2 secretion and lead to further tissue damage. To the best of our knowledge, there have been no reports studying the expression and functions of TRPM4 in immune cells after ischemic stroke. The functional role of TRPM4 warrants further investigation.
In summary, the transient expression of TRPM4 during the acute phase following ischemic stroke is critical for capillary integrity. Blocking TRPM4 can protect brain tissue by promoting angiogenesis and could be a potential drug target for stroke therapy.
References
Astrup J, Siesjo BK, Symon L (1981) Thresholds in cerebral ischemia—the ischemic penumbra. Stroke 12:723–5
Barbet G, Demion M, Moura IC, Serafini N, Leger T, Vrtovsnik F, Monteiro RC, Guinamard R, Kinet JP, Launay P (2008) The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat Immunol 9:1148–56. doi:10.1038/ni.1648
Becerra A, Echeverria C, Varela D, Sarmiento D, Armisen R, Nunez-Villena F, Montecinos M, Simon F (2011) Transient receptor potential melastatin 4 inhibition prevents lipopolysaccharide-induced endothelial cell death. Cardiovasc Res 91:677–84. doi:10.1093/cvr/cvr135
Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R (2012) The immunology of acute stroke. Nat Rev Neurol 8:401–10. doi:10.1038/nrneurol.2012.98
De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C (2012) von Willebrand factor: an emerging target in stroke therapy. Stroke 43:599–606. doi:10.1161/STROKEAHA.111.628867
Earley S, Waldron BJ, Brayden JE (2004) Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res 95:922–9. doi:10.1161/01.RES.0000147311.54833.03
Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, Ivanov A, Geng Z, Chen Z, Nilius B, Flockerzi V, Freichel M, Simard JM (2009) De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med 15:185–91. doi:10.1038/nm.1899
Grand T, Demion M, Norez C, Mettey Y, Launay P, Becq F, Bois P, Guinamard R (2008) 9-Phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br J Pharmacol 153:1697–705. doi:10.1038/bjp.2008.38
Guinamard R, Demion M, Magaud C, Potreau D, Bois P (2006) Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension 48:587–94. doi:10.1161/01.HYP.0000237864.65019.a5
Kahle KT, Simard JM, Staley KJ, Nahed BV, Jones PS, Sun D (2009) Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology (Bethesda) 24:257–65. doi:10.1152/physiol.00015.2009
Kostulas N, Li HL, Xiao BG, Huang YM, Kostulas V, Link H (2002) Dendritic cells are present in ischemic brain after permanent middle cerebral artery occlusion in the rat. Stroke 33:1129–34
Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, Ohmert I, Brink P, Pongs O (2009) Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest 119:2737–44. doi:10.1172/JCI38292
Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet JP (2004) TRPM4 regulates calcium oscillations after T cell activation. Science 306:1374–7. doi:10.1126/science.1098845
Liu H, El Zein L, Kruse M, Guinamard R, Beckmann A, Bozio A, Kurtbay G, Megarbane A, Ohmert I, Blaysat G, Villain E, Pongs O, Bouvagnet P (2010) Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ Cardiovasc Genet 3:374–85. doi:10.1161/CIRCGENETICS.109.930867
Mathar I, Vennekens R, Meissner M, Kees F, Van der Mieren G, Camacho Londono JE, Uhl S, Voets T, Hummel B, van den Bergh A, Herijgers P, Nilius B, Flockerzi V, Schweda F, Freichel M (2010) Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. J Clin Invest 120:3267–79. doi:10.1172/JCI41348
Nilius B, Owsianik G, Voets T, Peters JA (2007) Transient receptor potential cation channels in disease. Physiol Rev 87:165–217. doi:10.1152/physrev.00021.2006
Nilius B, Prenen J, Droogmans G, Voets T, Vennekens R, Freichel M, Wissenbach U, Flockerzi V (2003) Voltage dependence of the Ca2+−activated cation channel TRPM4. J Biol Chem 278:30813–20. doi:10.1074/jbc.M305127200
Reading SA, Brayden JE (2007) Central role of TRPM4 channels in cerebral blood flow regulation. Stroke 38:2322–8. doi:10.1161/STROKEAHA.107.483404
Rossi DJ, Brady JD, Mohr C (2007) Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 10:1377–86. doi:10.1038/nn2004
Schattling B, Steinbach K, Thies E, Kruse M, Menigoz A, Ufer F, Flockerzi V, Bruck W, Pongs O, Vennekens R, Kneussel M, Freichel M, Merkler D, Friese MA (2012) TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 18:1805–11. doi:10.1038/nm.3015
Shimizu T, Owsianik G, Freichel M, Flockerzi V, Nilius B, Vennekens R (2009) TRPM4 regulates migration of mast cells in mice. Cell Calcium 45:226–32. doi:10.1016/j.ceca.2008.10.005
Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V (2006) Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med 12:433–40. doi:10.1038/nm1390
Simard JM, Geng Z, Woo SK, Ivanova S, Tosun C, Melnichenko L, Gerzanich V (2009) Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab 29:317–30. doi:10.1038/jcbfm.2008.120
Simard JM, Kahle KT, Gerzanich V (2010) Molecular mechanisms of microvascular failure in central nervous system injury—synergistic roles of NKCC1 and SUR1/TRPM4. J Neurosurg 113:622–9. doi:10.3171/2009.11.JNS081052
Simard JM, Tarasov KV, Gerzanich V (2007) Non-selective cation channels, transient receptor potential channels and ischemic stroke. Biochim Biophys Acta 1772:947–57. doi:10.1016/j.bbadis.2007.03.004
Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Geng Z, Woo SK, Gerzanich V (2007) Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest 117:2105–13. doi:10.1172/JCI32041
Strbian D, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ (2006) Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J Cereb Blood Flow Metab 26:605–12. doi:10.1038/sj.jcbfm.9600228
Strbian D, Kovanen PT, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ (2009) An emerging role of mast cells in cerebral ischemia and hemorrhage. Ann Med 41:438–50. doi:10.1080/07853890902887303
Vennekens R, Nilius B (2007) Insights into TRPM4 function, regulation and physiological role. Handb Exp Pharmacol 269–85. doi: 10.1007/978-3-540-34891-7_16
Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, Schmitz F, Weissgerber P, Nilius B, Flockerzi V, Freichel M (2007) Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol 8:312–20. doi:10.1038/ni1441
Yilmaz A, Fuchs T, Dietel B, Altendorf R, Cicha I, Stumpf C, Schellinger PD, Blumcke I, Schwab S, Daniel WG, Garlichs CD, Kollmar R (2010) Transient decrease in circulating dendritic cell precursors after acute stroke: potential recruitment into the brain. Clin Sci (Lond) 118:147–57. doi:10.1042/CS20090154
Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK (2005) Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci 25:9794–806. doi:10.1523/JNEUROSCI.3163-05.2005
Acknowledgments
We thank Dr. Deidre Anne De Silva for her valuable comment on the manuscript. The research was supported by grants from the Singapore Ministry of Health's National Medical Research Council to TWS [grant number NMRC/1128/2007] and PL [grant number NMRC/1283/2011].
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 64 kb)
Rights and permissions
About this article
Cite this article
Loh, K.P., Ng, G., Yu, C.Y. et al. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch - Eur J Physiol 466, 563–576 (2014). https://doi.org/10.1007/s00424-013-1347-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-013-1347-4