
Abstract
Voltage-gated sodium (Na+) and potassium (K+) channels have been found to be regulated by Src family kinases (SFKs). However, how these channels are regulated by SFKs in cochlear spiral ganglion neurons (SGNs) remains unknown. Here, we report that altering the activity of endogenous SFKs modulated voltage-gated Na+, but not K+, currents recorded in embryonic SGNs in culture. Voltage-gated Na+ current was suppressed by inhibition of endogenous SFKs or just Src and potentiated by the activation of these enzymes. Detailed investigations showed that under basal conditions, SFK inhibitor application did not significantly affect the voltage-dependent activation, but shifted the steady-state inactivation curves of Na+ currents and delayed the recovery of Na+ currents from inactivation. Application of Src specific inhibitor, Src40–58, not only shifted the inactivation curve but also delayed the recovery of Na+ currents and moved the voltage-dependent activation curve towards the left. The pre-inhibition of SFKs occluded all the effects induced by Src40–58 application, except the left shift of the activation curve. The activation of SFKs did not change either steady-state inactivation or recovery of Na+ currents, but caused the left shift of the activation curve. SFK inhibitor application effectively prevented all the effects induced by SFK activation, suggesting that both the voltage-dependent activation and steady-state inactivation of Na+ current are subjects of SFK regulation. The different effects induced by activation versus inhibition of SFKs implied that under basal conditions, endogenously active and inactive SFKs might be differentially involved in the regulation of voltage-gated Na+ channels in SGNs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Src family kinases (SFKs) are protein tyrosine kinases. The SFK family is comprised of nine members, including Src, Fyn, Lck, Lyn, and Yes, which are highly expressed in the nervous system and play important roles in the regulation of neural development and synaptic transmission [9, 22, 28, 53, 56]. Since SFKs are involved in the regulation of many physiological and pathophysiological processes, they have become important targets for developing therapeutic approaches [6, 14, 25, 39, 44].
The regulation of cation currents mediated by voltage-gated Na+ and K+ channels is a key mechanism underlying the regulation of neuronal excitability and activity. A large amount of data have shown that voltage-gated Na+ [1, 2, 5, 27, 51] and K+ [33, 36, 37, 46, 48] channels are regulated by SFKs. Depending upon expressed channel subtypes and intracellular signaling, some Na+ or K+ channels may be up-regulated, while some of them may be down-regulated by SFKs.
Previous studies [6, 25] have suggested a role of SFKs in noise-induced hearing loss. Although the mechanisms underlying the regulation of auditory functions by SFKs still require further exploration, intervention with SFK inhibitors has been suggested as a novel approach in the treatment of auditory dysfunctions [6, 25]. Cochlear spiral ganglion neurons (SGNs) provide the afferent innervation of the hair cells in the organ of Corti and are classified into two types according to their anatomical connections to inner or outer hair cells [42, 47]. Cation currents mediated by voltage-gated Na+ and K+ channels in SGNs are responsible for the generation and propagation of afferent signals along the auditory nerve. Many diseases in the auditory system are associated with dysfunction of SGNs [34, 35]. In this work, we investigated the regulation of voltage-gated Na+ and K+ currents by endogenous SFKs in SGNs. We demonstrate for the first time that voltage-gated Na+, but not K+, currents in SGNs may be regulated by endogenous SFKs. Our data also suggest that endogenously active versus inactive SFKs may be differently involved in the processes of the voltage-dependent activation, steady-state inactivation, and recovery of Na+ channels.
Methods
Cell culture and immunofluorescence staining
SGN culture was conducted as described previously [19, 50]. In brief, cochlear spiral ganglion tissue was dissected from Wistar rat embryos (18 or 19 days gestation) at 4°C. Following cervical decapitation, the cochleas were quickly removed under a stereomicroscope and placed in cold dissection medium [Hanks balanced salt solution (HBSS, VWR, Suwanee, GA) supplemented with 7.5-mM HEPES (VWR), 28-mM glucose, and 58-mM sucrose, pH 7.3–7.4, osmolarity 310–320 mOsm]. The cochlear capsule was then opened. The modiolus containing SGNs was isolated and then minced into small pieces and transferred into the dissection medium containing 0.25% EDTA-trypsin (HyClone, Logan, UT) at 37°C for 10 min. Trypsin was then inactivated by adding DMEM/F12 (Mediatech Inc. Manassas VA) containing 10% fetal bovine serum (VWR) to the solution. Following centrifugation at 450 g for 3 min and the removal of supernatant, the pellet was re-suspended and gently triturated. Dissociated SGN tissue was then plated onto poly-D-lysine-coated 35-mm culture dishes and cultured with DMEM/F12 containing 10% fetal bovine serum for 12 to 72 h in a humidified CO2 (5%) incubator (Thermo Forma, Asheville, NC) at 37°C before performing electrophysiological recordings. All animal experiments were conducted following the guidelines of the NIH and approved by the Animal Care and Use Committee at Florida State University.
For immunocytochemical staining, SGNs cultured on glass cover-slips coated with poly-D-lysine were fixed in 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 for 5 min. After incubation in bovine serum albumin (BSA, 10 mg/ml) for 15 min, the cultures were then, respectively, incubated in BSA containing the antibodies against neurofilament 200 (1:400, mouse) and SFKs (1:500, rabbit, Millipore, Billerica, MA) overnight at 4°C. The double labeling with these antibodies was then visualized with fluorescence Alexa-488 conjugated goat anti mouse (1:8,000, Invitrogen, Eugene, OR) for neurofilament 200 and Alexa-568 conjugated donkey anti rabbit (1:1,000, Invitrogen) for SFKs, overnight at 4°C. Fluorescence images of SGNs were observed under an inverted microscope (Axiovert 200 M, Carl Zeiss, USA) equipped with Differential Interference Contrast (DIC) System and a CCD camera (Orca-ER, Hamamatsu, Bridgewater, NJ). Images were recorded and analyzed with AxioVision 4.5 (Carl Zeiss) and NIH-ImageJ (NIH, Bethesda, MD) software, respectively.
Whole-cell patch clamp recordings
The methods used for whole-cell patch clamp recordings in SGNs are similar to that described previously [19]. In brief, SGN cultures were placed in a recording chamber on an inverted microscope (Axiovert S200 TV, Carl Zeiss) equipped with 64 × DIC System. The image was magnified further by 63 × and displayed on a 17" TV monitor. The morphology of SGNs was monitored. None of the bath solutions or experimental manipulations produced significant changes in size or shape of SGNs recorded. Voltage-gated Na+ and K+ currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) in the whole-cell configuration from cultured SGNs bathed with a standard extracellular solution containing (in mM): NaCl (140), KCl (5.4), CaCl2 (1.2), CdCl2 (0.1), HEPES (12), and glucose (16). pH and osmolarity of the solution were adjusted to 7.35 and 310–320 mOsm, respectively. Recording electrodes pulled from borosilicate glass capillaries were filled with intracellular solution composed of (in mM): KCl (130), EGTA (5), HEPES (5), MgCl2 (4), and K-ATP (4). pH and osmolarity of the solution were adjusted to 7.25 and 290–300 mOsm. DC resistances of electrodes were 3–5 MΩ. Series resistance compensation (up to 80–90%) was always used in recordings, and thereby, the maximum access resistance was not greater than 3 MΩ. Na+ in the extracellular solution was replaced with N-methyl-d-glucamine (NMDG), or K+ in both the extracellular and intracellular solutions was replace with Cs+ to block Na+ or K+ currents. Voltage-gated inward or outward currents were evoked every 30 s by cell depolarization from potentials of −90 mV to −20 mV or +50 mV. The peak amplitudes of inward and outward currents were 0.43 ± 0.07 nA (n = 122) and 1.66 ± 0.2 nA (n = 19), respectively. The capacitance of recorded SGNs was 13 ± 6.5 pF (mean ± SEM, n = 194). The density of peak inward and outward currents (D I: the ratio of peak current amplitude versus cell capacitance) was calculated.
Effects produced by bath application of SFK inhibitors or by intracellular delivery of a peptide which may activate SFKs or inhibit Src were investigated. For intracellular delivery of peptides, the peptides were added into the intracellular solution filling recording electrodes. Recorded voltage-gated Na+ currents were compared with that recorded immediately (2.1 ± 0.1 s, which was defined as “0” min in this work) after breakthrough.
To determine the current–voltage relationship and the voltage-dependent activation, SGNs were held at −110 or −90 mV, and Na+ or K+ currents were evoked every 200 ms by depolarization from −110 or −90 mV to +55 mV for 25 ms in 5-mV steps. Data of normalized currents to maximum peak amplitude (I max) were plotted versus testing potentials, and curves were fitted by the Boltzmann function [I/I max = 1/(1+ exp(V-V 1/2)κ v], where V 1/2 is the potential for half-maximal activation and κ v is the slope factor. Na+ conductance was calculated by relation G Na = I Na/(V - V Rev).
To examine voltage-dependent steady-state inactivation, neurons were depolarized every 500 ms from potential −110 or −90 mV to −20 mV for 200 ms in 5-mV increments. Since the peak current density was mainly recorded at −20 ± 6 mV (n = 48, see Fig. 3), inward currents evoked by a testing pulse to −20 mV (25 ms) immediately following the depolarization were measured. Since no difference was found by using depolarization protocols from −110 mV versus from −90 mV, the data were pooled. Normalized peak currents were plotted versus pre-pulse potentials, and curves were fitted by the Boltzmann function as mentioned above.
The time course of Na+ current recovery was investigated. SGNs were given a 10-ms conditioning potential to −20 mV (to fully inactivate the current) every 200 ms, followed by recovery periods at −90 mV, ranging from 0 to 25 ms with 0.5-ms increment and a test potential to −20 mV. Normalized peak current amplitudes were plotted and fitted by a single-exponential function [y = a(1-exp-bx)] or by the Boltzmann function. The time constant of Na+ current recovery was determined.
Statistical significance between the means before and during application of SFK inhibitors or an SFK activator was determined with Student t test or repeated measures two-way ANOVA. All chemicals and reagents were purchased from Sigma (St. Louis, MO) except those as indicated.
Results
The inhibition of endogenous SFKs significantly reduced voltage-gated Na+, but not K+, currents
Immunofluorescence labeling of SFKs in cultured SGNs showed SFK co-labeling in all observed SGNs labeled with an antibody against neurofilament 200 (Supplementary Fig. 1). Consistent with data reported previously [45], the cell depolarization by changing the holding potential from −90 to +50 mV in whole-cell patches induced fast inward and slow outward currents in SGNs (Fig. 1a). We investigated effects of inhibiting endogenous SFKs on outward currents in SGNs bathed with Na+-free extracellular solution in which Na+ was replaced with NMDG (Fig. 1b) by application of the SFK inhibitors, PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine, 10 μM) [4, 17, 23, 29, 54] and SU6656 (2-oxo-3-(4,5,6,7-tetrahydro-1H-indol-2-ylmethylene)-2,3-dihydro-1H-indole-5-sulfonic acid dimethylamide, 2 μM) [7, 54], respectively.

Effects of SFK inhibitor application on voltage-gated outward currents recorded in cultured SGNs. a, b Examples of recorded whole-cell current traces evoked by altering holding potentials in 10-mV increments from −90 to +50 mV (insertion in a) in SGNs bathed with standard (a) and Na+-free (b) extracellular solution. c, d Examples of recorded whole-cell current traces before (Control) and during PP2 (c) and SU6656 (d) application. e, f Summary data showing relationships (mean ± SEM) between voltage and peak current density (D I) before and during PP2 (e) and SU6656 (f) application. Values in brackets indicate the number of SGNs tested
Figure 1c, d illustrates outward currents before and during PP2 and SU6656 applications. Figure 1e, f shows the relationships between voltage and peak current densities which were normalized to that before PP2 or SU6656 application. We found that no change in outward currents was induced following application of either PP2 or SU6656. Outward currents recorded in SGNs were substantially blocked by replacing K+ with Cs+ in solutions for bathing SGNs and filling recording electrodes (Fig. 2a). This finding suggests that voltage-gated K+ channels expressed in SGNs may not be tonically regulated by endogenous SFKs.

Effects of SFK inhibitor application on voltage-gated Na+ currents recorded in cultured SGNs. a Examples of recorded whole-cell current traces evoked by cell depolarization in 5-mV increments from −90 to +55 mV from an SGN. K+ in extracellular solution bathing SGNs and intracellular solution filling recording electrodes was replaced with Cs+. b Examples of recorded current traces before (Control) and during TTX application. c, d Examples of current traces recorded before (Control), during, and after (Wash) PP2 (c) and SU6656 (d) applications. e Summary data showing peak amplitudes of Na+ currents (mean ± SEM) evoked by cell depolarization from −90 to -20 mV, which were normalized to the mean amplitude (=100%, Control) of the currents before application of vehicle, PP2, PP3, or SU6656 (bar). Values in brackets indicate the number of SGNs tested. *P < 0.05, repeated measures two-way ANOVA in comparison with the effect of vehicle application
Application of tetrodotoxin (TTX, 1 μM) considerably abolished inward currents (Fig. 2b), demonstrating that the currents were predominately mediated by TTX-sensitive voltage-gated Na+ channels. The peak Na+ current density was −35 ± 60 pA/pF (n = 78). Figure 2c, d illustrates recorded whole-cell currents evoked by cell depolarization from −90 to −20 mV before (Control), during, and after application (Wash) of PP2 (10 μM) and SU6656 (2 μM), respectively. Compared with controls, peak Na+ currents were inhibited by 40 ± 10% (mean ± SEM, n = 7) and 35 ± 6% (n = 7) following PP2 and SU6656 applications (Fig. 2e). Apparent recovery of Na+ currents was noted following wash of PP2, but not SU6656 (Fig. 2c, d). PP2 and SU6656 applications significantly inhibited peak current densities (Fig. 3a, g) and Na+ conductance (Table 1), while no significant change in either macroscopic activation or inactivation kinetics was detected (insertions in Fig. 3a, g). Application of vehicle or PP3 (4-amino-7-phenylpyrazol[3,4-d]pyrimidine, an inactive structure analog of PP2 [52], 10 μM) did not induce any change in Na+ current (Figs. 2e and 3d).

Effects of SFK inhibitor application on the voltage-dependent activation, steady-state inactivation, and the recovery of Na+ currents. a, d, and g show voltage-peak current density relationships (mean ± SEM) before (Control) and during PP2, PP3, and SU6656 applications, respectively. Insertions in a, d, and g show examples of superimposing the control and treatment current traces elicited by depolarization to −20 mV and normalized to the peak amplitude of each trace before and during the agent application as indicated. b, e, and h show the voltage-dependent activation and steady-state inactivation of Na+ currents (mean ± SEM) recorded before and during application of the agents as indicated. Solid and dash lines represent the fitting curves for data before and during the agent application with the Boltzmann function (see “Methods”), respectively. c, f, and i show the recovery of Na+ currents (mean ± SEM). Except for data during PP2 application shown in c (which was fitted with the Boltzmann function), solid and dash lines represent the fitting curves for data before and during the agent application with a single-exponential function (see “Methods”)
The inhibition of endogenous SFKs significantly shifted voltage-dependent steady-state inactivation and delayed the recovery of Na+ currents from inactivation
Furthermore, we examined effects of SFK inhibitor application on the voltage-dependent activation and steady-state inactivation of Na+ currents. Compared with controls before SFK inhibitor applications, no significant change was found in the voltage-dependent activation of Na+ current (Fig. 3b, h). However, the steady-state inactivation curves of Na+ currents shifted significantly towards the left (Fig. 3b, h). The half-maximal inactivation potential became significantly more negative than that before PP2 or SU6656 application (Table 2). PP2 or SU6656 application also altered the recovery from inactivation. The time constants for recovery from inactivation were significantly increased (Fig. 3c, i, Table 3). PP3 application did not produce such changes in Na+ currents (Fig. 3e, f, Table 3).
The inhibition of endogenous Src significantly reduced voltage-gated Na+ current, shifted the steady-state inactivation, and delayed the recovery of Na+ current from inactivation
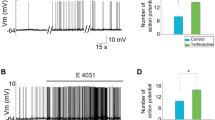
We investigated the effects of delivering a peptide, which has the amino acid sequence of Src 40–58 (Src40–58) and selectively inhibits Src activity [28, 39, 43, 55], into SGNs through recording electrodes. Figure 4a shows examples of voltage-gated Na+ current traces recorded immediately (“0” min; Control) and 5 min after breakthrough with electrodes filled with intracellular solution containing Src40–58 (0.3 mg/ml, left). We found that when compared with those recorded at “0” min after breakthrough, Na+ currents recorded with electrodes filled with the solution containing Src40–58 were significantly decreased (Fig. 4a–c). The peak current amplitude and conductance at 5 min after breakthrough were significantly decreased without significantly altering either macroscopic activation or inactivation kinetics (Fig. 4b, c, Table 1). When compared with those recorded at “0” min, the steady-state inactivation curves of Na+ currents recorded at 5 min after breakthrough significantly shifted towards the left (Fig. 4e), the half-maximal inactivation potential became more negative (Table 2), and the time constants for recovery from inactivation were increased (Fig. 4g, Table 3). However, we also noted that when compared with those recorded in other groups with electrodes containing no Src40–58, the time constants of Na+ current recovery from inactivation recorded with electrodes containing Src40–58 were significantly reduced (Fig. 4g, Table 3).

Effects of Src inhibitory peptide (Src40–58) application. a Examples of current traces recorded immediately (solid line) and 5 min (dash line) after breakthrough with electrodes filled with intracellular solution containing the Src inhibitory peptide, Src40–58 (0.3 mg/ml) in SGNs bathed with extracellular solution added without (left) and with PP2 (10 μM, right). b Summary data showing relative changes of peak Na+ currents when compared with that recorded immediately after breakthrough. Src40–58/PP2: Src40–58 was applied into neurons bathed with the extracellular solution containing PP2. *P < 0.05, repeated measures two-way ANOVA in comparison with the effect of Src40–58 applied into neurons treated with PP2. c, d Summary data showing voltage-peak current density (D I) relationships recorded immediately (0 min) and 5 min after breakthrough in neurons treated without (c) and with (d) PP2. Insertions in c and d show superimposition of current traces recorded immediately (0 min) and 5 min after breakthrough. e, f The voltage-dependent activation and steady-state inactivation of Na+ channels recorded immediately (0 min, Control) and 5 min after breakthrough with electrodes filled with intracellular solution containing Src40–58 from neurons treated without (e) and with (f) PP2. g, h The recovery of Na+ currents recorded immediately (0 min) and 5 min after breakthrough from neurons treated without (g) and with (h) PP2
Since voltage-dependent activation curves of Na+ currents were also found to shift towards the left when Src40–58 was applied (Fig. 4e, Table 2), we investigated effects of Src40–58 application when SFKs had been inhibited by application of PP2 (10 μM, see Fig. 4b, d, f, h, g; Tables 1, 2 and 3). Following the intracellular application of Src40–58, no significant change in Na+ currents was found in SGNs bathed with extracellular solution containing PP2, except the shift of the voltage-dependent activation curves (Fig. 4b, d, f, h, g; Tables 1, 2 and 3).
The activation of endogenous SFKs significantly potentiates voltage-gated Na+ currents
To confirm the role of endogenous SFKs in the regulation of voltage-gated Na+ currents in SGNs, we examined effects of intracellular application of a peptide, EPQ(pY)EEIPIA (1 mM), which activates SFKs (SFK activator peptide [28, 38, 55]), on Na+ currents in SGNs. Figure 5a shows examples of voltage-gated Na+ current traces recorded immediately (“0” min) and 5 min after breakthrough with electrodes filled with intracellular solution containing the SFK activator peptide (left) or a control peptide, EPQYEEIPIA (1 mM, right) which produces no change in the enzyme activity of SFKs [28, 38, 55]. We found that when compared with those recorded immediately after breakthrough, the Na+ currents recorded with electrodes filled with the solution containing the SFK activator peptide were significantly increased (Fig. 5a–c). The peak current amplitude and conductance recorded at 5 min after breakthrough were significantly increased without apparent alteration of either macroscopic activation or inactivation kinetics (Fig. 5b, c, Table 1). In contrast, no significant change was noted in the Na+ currents recorded with electrodes filled with intracellular solution containing the control peptide or no peptide (vehicle) (Fig. 5b, d, Table 1). The intracellular application of the Src activator peptide (1 mM) did not induce any significant change in inward currents recorded from SGNs bathed with extracellular solution containing TTX (1 μM) (Supplementary Fig. 2).

Effects of SFK activator application on voltage-gated Na+ currents recorded in cultured SGNs. a Examples of current traces recorded immediately (0 min, solid line) and 5 min (dash line) after breakthrough with electrodes filled with intracellular solution containing the SFK activator peptide, EPQ(pY)EEIPIA (1 mM, left), and the control peptide, EPQYEEIPIA (1 mM, right). b Summary data (mean ± SEM) showing relative changes of peak Na+ currents when compared with that recorded immediately after breakthrough (0 min). Vehicle: recording electrodes were filled with standard intracellular solution. *P < 0.05, repeated measures two-way ANOVA in comparison with the effect of vehicle application. c, d Summary data showing voltage-peak current density (D I) relationships recorded immediately (0 min, Control) and 5 min after breakthrough with electrodes filled with intracellular solution containing the SFK activator peptide and the control peptide. Insertions in c and d show superimposition of the control and treatment current traces. e, f The voltage-dependent activation and steady-state inactivation of Na+ channels recorded immediately (0 min, Control) and 5 min after breakthrough with electrodes filled with intracellular solution containing the SFK activator peptide and the control peptide. g The recovery of Na+ currents recorded from neurons intracellularly applied with the SFK activator peptide immediately (0 min, Control) and 5 min after breakthrough
We then examined effects produced by application of the SFK activator peptide on the voltage-dependent activation and steady-state inactivation of Na+ current. We found that the voltage-dependent activation curve of Na+ currents shifted significantly towards the left (Fig. 5e). Compared with that recorded immediately, 5 min following breakthrough, the half-maximal activation potentials were shifted (Table 2). However, no significant change was noted in either steady-state inactivation or recovery of Na+ currents from inactivation (Tables 2 and 3; Fig. 5g). Intracellular application of the control peptide did not produce any change in either the activation or inactivation of Na+ currents (Fig. 5f, Tables 2 and 3). To confirm that the effects induced by intracellular application of the SFK activator peptide were produced through the activation of SFKs, we examined effects of the SFK activator peptide applied into SGNs treated with PP2 (10 μM). We found that the application of PP2 effectively blocked the increase in voltage-gated Na+ currents (Fig. 6a–c, Table 1) and reduced the left shift of voltage-dependent activation induced by the SFK activator peptide (Fig. 6d).

SFK activator-induced up-regulation of voltage-gated Na+ currents was blocked by SFK inhibitor application. a Examples of current traces recorded immediately (0 min) and 5 min after breakthrough with electrodes filled with intracellular solution containing the SFK activator peptide, EPQ(pY)EEIPIA (1 mM), from neurons bathed with extracellular solution containing PP2 (10 μM). b Summary data showing relative changes of Na+ currents recorded with electrodes filled with intracellular solution containing the SFK activator peptide (1 mM) from neurons bathed with extracellular solution containing PP2. c Voltage-peak current density (D I) relationships recorded with electrodes filled with intracellular solution containing the SFK activator peptide immediately (0 min, Control) and 5 min after breakthrough from neurons treated with PP2. d The voltage-dependent activation and steady-state inactivation of Na+ channels recorded with electrodes filled with intracellular solution containing the SFK activator peptide immediately (0 min, Control) and 5 min after breakthrough from neurons treated with PP2
Discussion
Cation currents mediated by voltage-gated Na+ and K+ channels expressed in SGNs are responsible for generation and propagation of afferent signals along auditory nerves innervating the hair cells of the organ of Corti. According to their anatomical connections, SGNs are classified into two types [42, 47]. We did not test for neuronal type in this study. However, 95% of cells in previous studies were shown to be type I [42, 47]. Thus, almost all of the cells studied here were probably type I.
Previous data recorded from different type of cells, e.g., vascular smooth muscle, HEK-293, tsA-201, renal, Schwann cells, or cervical ganglion neurons (CGNs), have shown that SFKs have different effects on voltage-gated K+ or Na+ currents and that this difference may be due to the different types of channels expressed [1, 2, 5, 27, 33, 36, 37, 46, 48, 51]. Thus, the SFK regulation of voltage-gated K+ and Na+ currents appeared to be “cell-type specific.” Previous studies have indicated that rat SGNs exhibit a unique pattern of voltage-gated Na+ channel subunit expression and that this may be related to the independent embryological origins of SGNs [18, 20]. Since it remains unknown whether SFKs are involved in the regulation of voltage-gated K+ and Na+ channels expressed in SGNs, we conducted the present study.
Our data showed that either PP2 or SU6656 application produced no effect on voltage-gated K+ currents. This appears consistent with the finding that application of the wide-spectrum protein tyrosine kinase inhibitor, genistein, inhibits voltage-gated Na+ currents but does not alter the resting membrane potential in CGNs [27]. Previous studies have shown that SGNs possess all K+ channels responsible for the depolarization-activated K+ currents (for a review see [42]). It has been found that some SFKs may up-regulate [16, 36, 37, 41, 46], while some may down-regulate [3, 12, 16, 24, 26, 49] voltage-gated K+ channels, depending upon the channel structure [21, 33] and intracellular signaling [48]. Thus, more detailed studies dealing with the regulation of voltage-gated K+ channels by SFKs in SGNs are still required.
Our present data showed that voltage-gated Na+ currents recorded in SGNs were inhibited following the bath application of the SFK inhibitor, PP2 or SU6656. Apparent recovery of Na+ currents was noted following wash of PP2, but not SU6656. These findings are similar to data reported previously, which show that the inhibitory effects on N-methyl-d-aspartate receptors can be reversed by removal of PP2 but not SU6656 [32, 54]. Based on findings that (1) voltage-gated Na+ current in SGNs was consistently down-regulated by application of PP2, SU6656, or Src40–58, but not by PP3, (2) the Na+ current was up-regulated by application of the SFK activator, EPQ(pY)EEIPIA, but not EPQYEEIPIA, we conclude that voltage-gated Na+ channels expressed in SGNs are tonically up-regulated by endogenous SFKs under native conditions. The finding that the SFK activator peptide produced no significant effect on the inward currents of neurons treated with TTX implicated that the current mediated by TTX-sensitive Na+ channels was up-regulated by the activation of endogenous SFKs in SGNs.
Our data showed that intracellular application of Src40–58 or bath application of PP2 or SU6656 significantly reduced voltage-gated Na+ currents, shifted the steady-state inactivation curves of Na+ currents towards the left, and delayed the recovery of Na+ current from inactivation. Unexpectedly, however, compared with that in neurons recorded with electrodes containing no Src40–58, there was a significant reduction in the time constant of the recovery of Na+ currents recorded with electrodes filled with a solution containing Src40–58. Moreover, when Src40–58 was applied, voltage-dependent activation curves of Na+ currents shifted towards the left. These findings raised a question: Are the effects produced by intracellular application of Src40–58 through modulation of Src activity? To answer this question, we examined effects of PP2 on changes in Na+ current induced by application of Src40–58. Our hypothesis was that a pharmacological pre-inhibition of SFKs should occlude the effect of Src40–58 if Src played a role in the regulation of Na+ by SFKs.
Src40–58 application along with PP2 produced no significant change in Na+ currents, except the shift of the voltage-dependent activation curves. This finding suggested that Src is involved in the regulation of Na+ current by SFKs. A reduction in the recovery time constants was noted in SGNs treated with Src40–58. PP2 pre-application could block this effect of Src40–58. These data implied that the effect induced by inhibition of just Src could be different from that induced by inhibition of all SFKs. The fact that along with PP2 application the left shift of the activation curves induced by application of Src40–58 was maintained indicated that this effect was probably not produced through Src- or SFK-dependent mechanisms.
The intracellular application of the SFK activation peptide, EPQ(pY)EEIPIA, shifted the voltage-dependent activation curves towards the left without affecting the steady-state inactivation. The fact that no effect could be found following application of EPQ(pY)EEIPIA in SGNs treated with PP2 confirmed that the activation of SFKs may promote the voltage-dependent activation but not affect the voltage-dependent steady-state inactivation of the channels. Taken together with the effects induced by inhibition of SFKs or just Src, it is very likely that both the voltage-dependent activation and steady-state inactivation of Na+ currents are the subjects of SFK regulation.
Why the inhibition of SFK activity produced different effects on the voltage-dependent activation and steady-state inactivation of Na+ currents than the activation in SGNs and how Src40–58 application caused the shift in steady-state activation are still unclear. It is known that endogenous SFKs are regulated by a variety of mechanisms and therefore may exhibit different levels of enzyme activity coinciding with activation states [8, 9, 22, 23, 28, 40, 56]. Thus, one potential possibility could be that under native conditions, the steady-state inactivation and the recovery of Na+ channels might be controlled by activated SFKs, while less active or inactive SFKs might be involved in the regulation of voltage-dependent activation of Na+ channels. Thus, SFKs might exhibit a bifunctional regulation of Na+, but not K+, channels in SGNs.
It has been found that protein phosphorylation is an important mechanism underlying the regulation of ion channel activity through altering the channel gating and/or expression on the cell surface [10, 11, 13, 15, 16, 30, 31, 43]. Several members in SFKs and types of Na+ channels are expressed in SGNs. To completely understand the mechanisms underlying the regulation of voltage-gated Na+ channels by endogenous SFKs in SGNs, the identification of how every type of Na+ channels is regulated by each member of SFKs and whether SFKs may regulate the gating and surface expression of Na+ channels in SGNs is still required.
Voltage-gated Na+ channels play a critical role in the generation and propagation of action potentials. Encoding sound signals into a neuronal code is central in the process of hearing. Our findings that the inhibition of SFKs may reduce voltage-gated Na+ current amplitude, shift steady-state inactivation, and delay the recovery of Na+ channels and that the augmentation of SFK activity may potentiate Na+ currents and promote the voltage-dependent activation have strongly implied that the excitability and firing behavior (such as discharge frequency and pattern in response to sound stimuli) of SGNs may be critically regulated by SFKs. The observations that inhibition of SFK activity affects recovery from inactivation could be particularly important because hearing (especially detection of the direction of sounds and high pitches) relies on very high firing rates. Thus, characterizing the regulation of voltage-gated Na+ channels by SFKs will be important for the development of therapeutics for treating pathological conditions in the auditory system such as tinnitus and/or hearing loss.
Reference
Ahern CA, Zhang JF, Wookalis MJ, Horn R (2005) Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res 96:991–998
Ahn M, Beacham D, Westenbroek RE, Scheuer T, Catterall WA (2007) Regulation of Na(v)1.2 channels by brain-derived neurotrophic factor, TrkB, and associated Fyn kinase. J Neurosci 27:11533–11542
Alioua A, Mahajan A, Nishimaru K, Zarei MM, Stefani E, Toro L (2002) Coupling of c-Src to large conductance voltage- and Ca2+-activated K+ channels as a new mechanism of agonist-induced vasoconstriction. Proc Natl Acad Sci U S A 99:14560–14565
Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem J 371:199–204
Beacham D, Ahn M, Catterall WA, Scheuer T (2007) Sites and molecular mechanisms of modulation of Na(v)1.2 channels by Fyn tyrosine kinase. J Neurosci 27:11543–11551
Bielefeld EC, Hynes S, Pryznosch D, Liu J, Coleman JK, Henderson D (2005) A comparison of the protective effects of systemic administration of a pro-glutathione drug and a Src-PTK inhibitor against noise-induced hearing loss. Noise Health 7:24–30
Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA (2000) SU6656, a selective Src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol 20:9018–9027
Bradshaw JM (2010) The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal 22:1175–1184
Brown MT, Cooper JA (1996) Regulation, substrates and functions of Src. Biochim Biophys Acta 1287:121–149
Cantrell AR, Catterall WA (2001) Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci 2:397–407
Catterall WA (1991) Structure and function of voltage-gated sodium and calcium channels. Curr Opin Neurobiol 1:5–13
Cayabyab FS, Khanna R, Jones OT, Schlichter LC (2000) Suppression of the rat microglia Kv1.3 current by Src-family tyrosine kinases and oxygen/glucose deprivation. Eur J Neurosci 12:1949–1960
Chahine M, Ziane R, Vijayaragavan K, Okamura Y (2005) Regulation of Na v channels in sensory neurons. Trends Pharmacol Sci 26:496–502
Cohen P (2002) Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov 1:309–315
d’Alcantara P, Schiffmann SN, Swillens S (1999) Effect of protein kinase A-induced phosphorylation on the gating mechanism of the brain Na+ channel: model fitting to whole-cell current traces. Biophys J 77:204–216
Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, Wilson E (2001) Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol 281:H1835–H1862
Fang XQ, Xu J, Feng S, Groveman BR, Lin SX, Yu XM (2011) The NMDA receptor NR1 subunit is critically involved in the regulation of NMDA receptor activity by C-terminal Src kinase (Csk). Neurochem Res 36:319–326
Fekete DM, Wu DK (2002) Revisiting cell fate specification in the inner ear. Curr Opin Neurobiol 12:35–42
Feng S, Wei J, Gao M, Tang X, Su J (2010) Inhibition on voltage-gated sodium channels in rat spiral ganglion neurons by salicylate. J Guangxi Med Univ 1:1–4
Fryatt AG, Vial C, Mulheran M, Gunthorpe MJ, Grubb BD (2009) Voltage-gated sodium channel expression in rat spiral ganglion neurons. Mol Cell Neurosci 42:399–407
Gamper N, Stockand JD, Shapiro MS (2003) Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J Neurosci 23:84–95
Groveman BR, Feng S, Fang XQ, Pflueger M, Lin SX, Bienkiewicz EA, Yu XM (2011) The regulation of NMDA receptors by Src kinase. FEBS J 279:20–28
Groveman BR, Xue S, Marin V, Xu J, Ali MK, Bienkiewicz EA, Yu XM (2011) Roles of the SH2 and SH3 domains in the regulation of neuronal Src kinase functions. FEBS J 278:643–653
Gu RM, Wei Y, Falck JR, Krishna UM, Wang WH (2001) Effects of protein tyrosine kinase and protein tyrosine phosphatase on apical K(+) channels in the TAL. Am J Physiol Cell Physiol 281:C1188–C1195
Harris KC, Hu B, Hangauer D, Henderson D (2005) Prevention of noise-induced hearing loss with Src-PTK inhibitors. Hear Res 208:14–25
Holmes TC, Fadool DA, Ren R, Levitan IB (1996) Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science 274:2089–2091
Jia Z, Jia Y, Liu B, Zhao Z, Jia Q, Liang H, Zhang H (2008) Genistein inhibits voltage-gated sodium currents in SCG neurons through protein tyrosine kinase-dependent and kinase-independent mechanisms. Pflugers Arch 456:857–866
Kalia LV, Gingrich JR, Salter MW (2004) Src in synaptic transmission and plasticity. Oncogene 23:8007–8016
Karni R, Mizrachi S, Reiss-Sklan E, Gazit A, Livnah O, Levitzki A (2003) The pp 60c-Src inhibitor PP1 is non-competitive against ATP. FEBS Lett 537:47–52
Kotecha SA, MacDonald JF (2003) Signaling molecules and receptor transduction cascades that regulate NMDA receptor-mediated synaptic transmission. Int Rev Neurobiol 54:51–106
Lau CG, Zukin RS (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci 8:413–426
Lei G, Xue S, Chery N, Liu Q, Xu J, Kwan CL, Fu Y, Lu YM, Liu M, Harder KH, Yu XM (2002) Gain control of N-methyl-d-aspartate receptor activity by receptor-like protein tyrosine phosphatase alpha. EMBO J 21:2977–2989
Li Y, Langlais P, Gamper N, Liu F, Shapiro MS (2004) Dual phosphorylations underlie modulation of unitary KCNQ K(+) channels by Src tyrosine kinase. J Biol Chem 279:45399–45407
Lin X (1997) Action potentials and underlying voltage-dependent currents studied in cultured spiral ganglion neurons of the postnatal gerbil. Hear Res 108:157–179
Lin X, Chen S, Tee D (1998) Effects of quinine on the excitability and voltage-dependent currents of isolated spiral ganglion neurons in culture. J Neurophysiol 79:2503–2512
Ling S, Sheng JZ, Braun AP (2004) The calcium-dependent activity of large-conductance, calcium-activated K+ channels is enhanced by Pyk2- and Hck-induced tyrosine phosphorylation. Am J Physiol Cell Physiol 287:C698–C706
Ling S, Woronuk G, Sy L, Lev S, Braun AP (2000) Enhanced activity of a large conductance, calcium-sensitive K+ channel in the presence of Src tyrosine kinase. J Biol Chem 275:30683–30689
Liu X, Brodeur SR, Gish G, Songyang Z, Cantley LC, Laudano AP, Pawson T (1993) Regulation of c-Src tyrosine kinase activity by the Src SH2 domain. Oncogene 8:1119–1126
Liu XJ, Gingrich JR, Vargas-Caballero M, Dong YN, Sengar A, Beggs S, Wang SH, Ding HK, Frankland PW, Salter MW (2008) Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nat Med 14:1325–1332
Marin V, Groveman BR, Qiao H, Xu J, Ali MK, Fang XQ, Lin SX, Rizkallah R, Hurt MH, Bienkiewicz EA, Yu XM (2010) Characterization of neuronal Src kinase purified from a bacterial expression system. Protein Expr Purif 74:289–297
Repp H, Birringer J, Koschinski A, Dreyer F (2001) Activation of a Ca2+-dependent K+ current in mouse fibroblasts by sphingosine-1-phosphate involves the protein tyrosine kinase c-Src. Naunyn Schmiedebergs Arch Pharmacol 363:295–301
Rusznak Z, Szucs G (2009) Spiral ganglion neurones: an overview of morphology, firing behaviour, ionic channels and function. Pflugers Arch 457:1303–1325
Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5:317–328
Salter MW, Pitcher GM (2011) Dysregulated Src upregulation of NMDA receptor activity: a common link in chronic pain and schizophrenia. FEBS J 279:2–11
Santos-Sacchi J (1993) Voltage-dependent ionic conductances of type I spiral ganglion cells from the guinea pig inner ear. J Neurosci 13:3599–3611
Sobko A, Peretz A, Attali B (1998) Constitutive activation of delayed-rectifier potassium channels by a Src family tyrosine kinase in Schwann cells. EMBO J 17:4723–4734
Spoendlin H (1988) Neural anatomy of the ear. In: Janh A, Santos-Sacchi J (eds) Physiology of the ear. Raven, New York, pp 201–219
Strauss O, Rosenthal R, Dey D, Beninde J, Wollmann G, Thieme H, Wiederholt M (2002) Effects of protein kinase C on delayed rectifier K+ channel regulation by tyrosine kinase in rat retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 43:1645–1654
Szabo I, Gulbins E, Apfel H, Zhang X, Barth P, Busch AE, Schlottmann K, Pongs O, Lang F (1996) Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J Biol Chem 271:20465–20469
Tang X, Gao M, Feng S, Su J (2010) Gamma-aminobutyric acid A receptor and N-methyl-d-aspartate receptor subunit expression in rat spiral ganglion neurons. Neural Regen Res 5:1–3
Tong Q, Stockand JD (2005) Receptor tyrosine kinases mediate epithelial Na(+) channel inhibition by epidermal growth factor. Am J Physiol Renal Physiol 288:F150–F161
Traxler P, Bold G, Frei J, Lang M, Lydon N, Mett H, Buchdunger E, Meyer T, Mueller M, Furet P (1997) Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines. J Med Chem 40:3601–3616
Trepanier CH, Jackson MF, MacDonald JF (2011) Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J 279:12–19
Xu J, Weerapura M, Ali MK, Jackson MF, Li H, Lei G, Xue S, Kwan CL, Manolson MF, Yang K, MacDonald JF, Yu XM (2008) Control of excitatory synaptic transmission by C-terminal Src kinase. J Biol Chem 283:17503–17514
Yu XM, Askalan R, Keil GJI, Salter MW (1997) NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275:674–678
Yu XM, Groveman BR (2011) Src family kinases in the nervous system. FEBS J 279:1–1
Acknowledgements
SF is supported by the State Scholarship Fund of China (2009845013). This work was supported by a grant from NIH (5R01 NS053567-04) to XMY. We would like to thank Dr. M. W. Salter for kindly supplying us Src inhibitory peptide (Src40–58), SFK activator peptide (EPQ(pY)EEIPIA), and control peptide (EPQYEEIPIA). We would also like to thank Drs. M. W. Salter, M. Hildebrand, and G. Pitcher for the very constructive discussions.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM doc
(DOC 27.0 KB)
Supplementary Fig. 1
Expression of SFKs in SGNs. N-200: an antibody staining for neurofilament 200; SFKs: an antibody staining for SFKs. Merge: merged image showing co-labeling of neurofilament 200 and SFKs (TIFF 716 KB)
Supplementary Fig. 2
Effects of intracellular application of the SFK activator peptide EPQ(pY)EEIPIA (1 mM) on TTX-resistant inward current. A: example of current traces recorded immediately (0’) and 5 min after breakthrough with electrodes filled with intracellular solution containing the SFK activator peptide, EPQ(pY)EEIPIA (1 mM) from a SGN bathed with extracellular solution containing TTX (1 μM). B: Summary data showing relative changes in TTX-resistant currents recorded from SGNs applied intracellularly with the SFK activator peptide. Values in brackets indicate number of SGNs tested (TIFF 9.10 MB)
Rights and permissions
About this article
Cite this article
Feng, S., Pflueger, M., Lin, SX. et al. Regulation of voltage-gated sodium current by endogenous Src family kinases in cochlear spiral ganglion neurons in culture. Pflugers Arch - Eur J Physiol 463, 571–584 (2012). https://doi.org/10.1007/s00424-012-1072-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-012-1072-4