
Abstract
Voltage-gated sodium channels play a crucial role in the initiation and propagation of neuronal action potentials. Genistein, an isoflavone phytoestrogen, has long been used as a broad-spectrum inhibitor of protein tyrosine kinases (PTK). In addition, genistein-induced modulation of ion channels has been described previously in the literature. In this study, we investigated the effect of genistein on voltage-gated sodium channels in rat superior cervical ganglia (SCG) neurons. The results show that genistein inhibits Na+ currents in a concentration-dependent manner, with a concentration of half-maximal effect (IC50) at 9.1 ± 0.9 μM. Genistein positively shifted the voltage dependence of activation but did not affect inactivation of the Na+ current. The inactive genistein analog daidzein also inhibited Na+ currents, but was less effective than genistein. The IC50 for daidzein-induced inhibition was 20.7 ± 0.1 μM. Vanadate, an inhibitor of protein tyrosine phosphatases, partially but significantly reversed genistein-induced inhibition of Na+ currents. Other protein tyrosine kinase antagonists such as tyrphostin 23, an erbstatin analog, and PP2 all had small but significant inhibitory effects on Na+ currents. Among all active and inactive tyrosine kinase inhibitors tested, genistein was the most potent inhibitor of Na+ currents. These results suggest that genistein inhibits Na+ currents in rat SCG neurons through two distinct mechanisms: protein tyrosine kinase-independent, and protein tyrosine kinase-dependent mechanisms. Furthermore, the Src kinase family may be involved in the basal phosphorylation of the Na+ channel.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Voltage-gated sodium channels (VGSC) play a critical role in the generation and propagation of action potentials in excitable cells including neurons, muscle cells, and endocrine cells. VGSC are composed of a complex of a α subunit that forms the voltage-sensitive and ion-selective pore, in association with one or more auxiliary β subunits. To date, nine α subunits of the VGSC superfamily, Nav1.1–Nav1.9, have been identified, which are widely distributed in mammalian tissue (see review [10] and [37]). Mutations in VGSC have been associated with a number of neurological diseases including inherited epilepsy and pain conditions and have been implicated in various to psychiatric disorders (see review [21]).
Within the VGSC family, Nav1.1, Nav1.2, Nav1.3, and Nav1.6 are distributed in central nervous system (CNS) neurons and Nav1.7, Nav1.8, and Nav1.9 are selectively distributed in peripheral nervous system (PNS) neurons. Nav1.1, Nav1.2, Nav1.3, Nav1.6, and Nav1.7 are highly tetrodotoxin (TTX) sensitive whereas Nav1.8 and Nav1.9 are TTX resistant (see review [10] and [14]). Thus, TTX-sensitive VGSC currents in SCG neurons are most likely Nav1.7 currents.
Genistein, an isoflavone phytoestrogen, known as a potent and specific inhibitor of protein tyrosine kinases (PTK) [1], has been reported to modulate ion channel function through PTK-dependent and PTK-independent mechanisms. A PTK-independent effect was considered for genistein-induced inhibition of L-type Ca2+ currents in rat and guinea pig ventricular cells [36, 5], K+ currents in guinea pig ventricular cells [32], 22Na+ influx and Na+ currents in rat cerebellar granule cells [24], fast sodium currents in uterine leiomyosarcoma cells [19], and pacemaker (I f) currents in rabbit sinoatrial node cells [3]. On the other hand, a PTK-dependent mechanism was suggested for genistein-induced inhibition of L-type Ca2+ channel activity in pituitary GH3 cells [9], ATP-sensitive K+ channels in rabbit portal vein smooth muscle cells [23], fast sodium currents in rabbit ventricular myocytes [31], slowly delayed-rectifier K+ currents (I k) in mouse Schwann cells [25], and guinea pig ventricular myocytes [22].
To date, there have been no studies investigating the role of genistein on the function of sodium channels from PNS neurons such as the SCG. Furthermore, no information is available regarding the role of tyrosine phosphorylation in the functional modulation of Na+ channels such as Nav1.7. In this paper, we investigate the effect of genistein on Na+ currents in rat SCG neurons and demonstrate that genistein inhibits Na+ current via two distinct mechanisms: a protein tyrosine kinase-independent and protein tyrosine kinase-dependent pathway.
Materials and methods
Chemicals
The drugs and chemicals used in this experiment were obtained as follows: Genistein, daidzein, erbstatin analog, tyrphostin 23, PP2, Perorthovanadate, 5, 4-[2-hydroxyethyl]-1-piperazineethane-sulfonic acid (HEPES), Dulbecco’s modified Eagle’s medium (DMEM), dimethyl sulfoxide (DMSO), were purchased from Sigma-Aldrich (St Louis, MO, USA). TTX was purchased from Swellxin Science and Technology (Guangzhou, China). Perorthovanadate (Na3VO4) in the bath solution was prepared by a 20-min incubation with the oxidant H2O2 (0.003%) and subsequent quenching of residual H2O2 with 100 U/ml catalase for 5 min [16].
Cell culture
Primary cultures of neurons were prepared from SCG from 3- to 5-week-old Sprague–Dawley rats using a previously described procedure [17]. Briefly, rats were killed by cervical dislocation and ganglia were rapidly removed from the carotid bifurcation and placed in modified D-Hanks’ solution. Ganglia were digested at 37°C with collagenase (1 mg/ml, Worthington) and dispase (5 mg/ml, Sigma-Aldrich) for 30 min, followed by another 30-min digestion with trypsin (2.5 mg/ml, Worthington). They were subsequently resuspended at least three times in DMEM medium plus 10% bovine calf serum (Hy-Clone, USA) to stop the digestion. Ganglia were then dissociated into a suspension of individual cells and planted on poly-d-lysine coated glass coverslips in 24-well tissue culture plates (Costar, USA). Cells were incubated at 37°C with a 5% CO2 and 95% air atmosphere. The medium was changed to neurobasal A medium plus 2% B27 supplement (Invitrogen) after 12 h and neurons were used for recording within 48 h. Within this time frame, no processes developed in these neurons. Procedures for the primary culture of neurons from the hippocampus and cortex were similar to SCG neurons, except these neurons were digested at 37°C with trypsin (1.25 mg/ml, Worthington) for 12 min.
Electrophysiology
Perforated-patch whole-cell recordings were used in this study. Recordings were made at room temperature (20–25°C). Pipettes were pulled from borosilicate glass capillaries and had a resistance of 3–5 MΩ when filled with internal solution. Series resistance compensation has always been used. Normally up to 80–90% compensation can be reached in our condition. Under this condition, the maximum access resistance is about 2 MΩ. Currents were recorded using Axon patch 200B amplifier and pClamp 9.0 software (Axon Instruments, CA), and were filtered at 10 KHz. The voltage protocol used to study Na currents of SCG neurons was as follows: the cells were held at −70 mV and 20 ms depolarizing step to 0 mV was applied every 3 s. The current-clamp technique was used to record the action potentials of SCG neurons. The action potentials were elicited by an approximate twofold threshold depolarizing current of 0.1 nA. The sampling rate was 10 KHz for membrane currents and was 2.5 KHz for membrane potential recordings. Compounds were delivered to the cell through a home-made perfusion pipette which was placed just beside the cell. The bath chamber was continuously superfused with background control solution. For the perforated patch recording, a pipette was first front-filled with the standard internal solution, then backfilled with the same internal solution containing amphotericin B (120 ng/ml). The external solution used to record the Na currents from SCG neurons contained (in mM): NaCl 120, KCl 3, HEPES 5, NaHCO3 23, glucose 11, MgCl2 1.2, CaCl2 2.5, BaCl2 0.2, and CdCl2 0.2 (adjusted to pH 7.4 with NaOH). The internal solution for sodium current recording consisted of (in mM): CsCl 90, KCl 40, HEPES 20, and MgCl2 3 (adjusted to pH 7.3–7.4 with CsOH). The external solution used to record neuronal action potentials contained (in mM): NaCl 120, KCl 3, HEPES 5, NaHCO3 23, glucose 11, MgCl2 1.2, and CaCl2 2.5, (adjusted to pH 7.4 with NaOH). The internal solution for action potential recording contained (in mM): KAC 90, KCl 40, HEPES 20, and MgCl2 3 (adjusted to pH 7.3–7.4 with KOH).
Cell immunofluorescence and confocal imaging
Neurons were seeded on poly-lysine coated coverslips and were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 3% BSA for 30 min before incubation for labeling. The cells were washed three times with 0.01 M phosphate buffered saline (PBS, pH 7.4) before each of the above treatments. Subsequently, neurons were incubated with primary rabbit anti-Nav1.7 Ab or anti-Nav1.1 Ab (1:200, Santa Cruz Biotechnology) in blocking solution overnight at 4°C, followed by three washes with PBS and incubation with goat anti-rabbit IgG-TRITC secondary Ab (1:100, Chemicon) for 30 min at 37°C. Cells were examined on an inverted laser-scanning microscope (SP2, Leica, Germany). TRITC was excited at 564 nm and the emitted fluorescence signal was at 570 nm.
Statistics
The programs “Origin” (Version 7.0, OriginLab Corporation) and Excel (Microsoft) were used for data analysis. Data were presented as the mean ± SEM. Statistical significance was computed using Student’s t test. The differences were considered significant if p < 0.05.
Results
Genistein inhibits Na+ currents in rat SCG neurons in a concentration-dependent manner
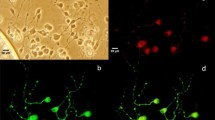
As discussed in the Introduction, the TTX-sensitive Na+ current in SCG neurons most likely comes from Nav1.7. Indeed, immunocytochemistry revealed the presence of Nav1.7 channels in SCG neurons (Fig. 1a). We also tested for the presence of the Nav1.1 channel in SCG neurons (Fig. 1b), which was previously reported to be present at lower levels in DRG neurons [4]. Similarly, we also detected a very weak signal for Nav1.1 in SCG neurons (Fig. 1b). To confirm the specificity of the channel-antibody, hippocampal and cortical neurons were used in control experiments and no immunoreactivity for Nav1.7 was found in these neurons (Fig. 1a). Thus, TTX-sensitive Nav1.7 currents were the dominant Na+ currents in SCG neurons. The maximal Na+ current amplitude is about 5 nA, but most are around 3.5 nA.

Genistein inhibits Na+ currents in rat SCG neurons. a Immunofluorescence of Nav1.7 channel in rat SCG neurons. The lower panel shows confocal images of SCG, hippocampal, and cortical neurons probed with an anti-Nav1.7 antibody and TRITC-conjugated anti-rabbit antibody, respectively. The upper panel shows phase-contrast microscopy of the same cells (scale bars, 10 μm). b Immunofluorescence of Nav1.1 channel in rat SCG neurons (scale bars, 10 μm). c Perforated-patch recording of Na+ currents in rat SCG neurons. Cells were held at −70 mV and Na+ current were elicited by depolarization to 0 mV for 20 ms every 3 s. Bath application of genistein (100 μM) for 5 min induced a bi-phasic inhibition of Na+ current. TTX (0.05 μM) abolished the Na+ current. d Representative traces of Na+ current elicited at 0 mV and taken from the corresponding time shown in c. e Concentration-dependent inhibition of Na+ current by genistein. f Concentration–response relationship for genistein. The data was fitted by the Hill function which produced an IC50 of 9.1 ± 0.9 μM and a coefficient of 1.1 (n = 11). Error bars represent SEM
We then tested the effect of genistein on Na+ currents in SCG neurons. Genistein (100 μM), when applied to the bath, induced an inhibition of peak Na+ current by 73.3% ± 5.4% (p < 0.01, n = 12) (Fig. 1c,d). The Na+ current almost fully recovered when genistein was washed out. Maximal inhibition was obtained within 195.9 ± 17.7 s of genistein application (n = 12) (Fig. 1c), which was much longer than the reported inhibition of sodium currents in CNS neurons by genistein [24]. It is interesting to note that two phases of inhibition were evident for most cases (Fig. 1c): an initial fast inhibition followed by a relatively slow inhibition. These two processes were fitted with a double exponential function and time constants were obtained. The fast time constant was 10.6 ± 1.2 s (n = 6) and the slow time constant was 55.9 ± 2.4 s (n = 6), respectively. Genistein (0.1 ∼ 100 μM) inhibited peak Na+ currents in a concentration-dependent manner (Fig. 1e,f). Genistein could inhibit Na+ currents at concentrations as low as 0.3 μM and reached maximal inhibition at a concentration of 100 μM (Fig. 1e,f). We did not use genistein at concentrations higher than 100 μM due to its poor solubility at these concentrations. The concentration–response curve for genistein was well-described by the Hill equation with a concentration for half-maximum inhibition (IC50) of 9.1 ± 0.9 μM and a coefficient of 1.1 ± 0.2 (n = 11) (Fig. 1f). TTX (0.05 μM) could rapidly and completely inhibit the Na+ current (Fig. 1c,e).
Genistein inhibits voltage-dependent activation but not inactivation of the Na+ current in SCG neurons
The effects of genistein (50 μM) on the current–voltage relationship and the voltage-dependence of activation and inactivation of the Na+ current were tested (Fig. 2). Neurons were held at −100 mV and Na+ currents were evoked by depolarization for 50 ms from −80 mV to +50 mV in 5-mV steps to obtain the current–voltage relationship curve (I–V curve) (Fig. 2a). Genistein clearly reduced peak Na+ currents. The effect of genistein on the voltage-dependent activation and inactivation of Na+ currents is shown in Fig. 2b. The protocols used to elicit voltage-dependent activation and inactivation of the Na+ currents are shown in the inset of Fig. 2b. Genistein significantly shifted the voltage-dependent activation of Na+ current to the right, but did not affect voltage-dependent inactivation (Fig. 2b). The half-maximal activation potential (V 1/2) was significantly more positive for genistein-treated neurons (−20.6 ± 0.1 mV, n = 9) than in control cells (−32.4 ± 0.2 mV, n = 9). The half-maximal inactivation potentials were −41.3 ± 0.4 mV (n = 9) for genistein and −41.8 ± 0.3 mV (n = 9) for controls, respectively.

Effect of genistein on current–voltage curve, activation, and inactivation of Na+ current. a Current–voltage (I–V) curve of Na+ currents recorded from SCG neurons. The neurons were held at −100 mV and depolarized for 50 ms from −80 to 50 mV in 5-mV steps. Genistein (50 μM) was applied for 5 min. Filled square and open square indicated before and after genistein application, respectively. b Effects of genistein (50 μM) on voltage-dependent activation and inactivation of Na+ currents. Filled square/open triangle, before and open circle/open inverted triangle, after application of genistein for 5 min, respectively. Steady-state inactivation curves were elicited by a 200-ms prepulse from −100 to −10 mV in 5-mV steps, and the activation curves were elicited by a test pulse from −60 to −5 mV in 5-mV steps. Error bars represent SEM
Genistein-induced inhibition of Na+ currents involves two mechanisms: PTK-independent and PTK-dependent mechanisms
It was previously shown that genistein inhibits sodium currents in CNS neurons through its direct interaction with channels and not through tyrosine kinase inhibition [24]. To investigate the underlying mechanisms involved in genistein-induced inhibition of Na+ currents in rat SCG neurons, we tested daidzein, an inactive structural analog of genistein with no activity against protein tyrosine kinases. Daidzein rapidly inhibited Na+ currents in a concentration-dependent manner (Fig. 3a). Maximal inhibition was reached at a concentration of 100 μM, which reduced the Na+ peak current by 28.5% ± 3.1% (n = 10) (Fig. 3a,b). The Hill fitting of the concentration–response data produced an IC50 of 20.7 ± 0.1 μM and a coefficient of 1.2 ± 0.2 (n = 10) (Fig. 3b).

Daidzein inhibits Na+ currents in rat SCG neurons. a Daidzein was applied at various concentrations (10, 30, 100, 300 μM) for the indicated period of time. b Concentration–response relationship of daidzein-induced inhibition on Na+ current. The data was fitted by the Hill function. IC50 is 20.7 ± 0.1 μM and coefficient is 1.2 (n = 10). c Representative time course of Na+ current in the presence of daidzein (100 μM) alone and in the presence of both daidazein and genistein (100 μM). d Summary data for genistein- and daidzein-induced inhibition of Na+ currents (**p < 0.01, n = 7). Error bars represent SEM
To compare the effects of genistein and daidzein, experiments were conducted in the same cell with the maximal inhibitory concentrations of 100 μM for both agents (Fig. 3c,d). Daidzein (100 μM) was first applied for 5 min and then genistein (100 μM) was applied for another 5 min (Fig. 3c) in the continuous presence of daidzein. Daidzein alone and genistein in the presence of daidzein reduced peak Na+ currents by 27.8% ± 3.6% and 57.3% ± 10.1% (p < 0.01, n = 7), respectively (Fig. 3c,d). Thus, at the same maximal concentration, the inhibition produced by daidzein was substantially smaller. Furthermore, daidzein-only-induced inhibition of Na+ currents was faster than genistein-induced inhibition (Fig. 3c). Since daidzein is an inactive structural analog of genistein, these data suggest that part of genistein-induced inhibition is through a PTK-independent mechanism. The data also suggest that genistein utilizes additional mechanisms since genistein is much more potent than daidzein in inhibiting Na+ currents (Fig. 3d).
Several lines of evidence have shown that genistein modulates ion channels through a PTK-dependent mechanism (e.g., [9, 23, 25, 31, 22]). To determine whether inhibition of PTK activity is involved in genistein-induced inhibition of Na+ currents, we first used sodium orthovanadate (Na3VO4), a potent inhibitor of protein tyrosine phosphatases (PTP). Vanadate (1 mM) partially rescued Na+ currents from genistein-induced inhibition (Fig. 4a), reversing genistein-induced inhibition of Na+ currents from 57% ± 5.4% to 39.5% ± 4.7% (p < 0.05, n = 9) (Fig. 4b). Pretreatment with vanadate reduced genistein-induced inhibition of Na+ currents by 18.7% ± 3.3% (n = 8) (Fig. 4c,d). Thus, inhibition of PTK activity is also partly responsible for the observed Na+ current inhibition by genistein.

Vanadate partly reduces genistein-induced inhibition of Na+ current. a Vanadate (1 mM) partly reverses the genistein (100 μM)-induced inhibition of Na+ current. b Summary data for a (*p < 0.05, n = 9). c Pretreatment with vanadate (1 mM) reduced genistein (100 μM)-induced inhibition of Na+ current. d Summary data for c (*p < 0.05, n = 8). Error bars represent SEM
We then tested the effects of other PTK inhibitors such as tyrphostin 23, an erbstatin analog, and PP2. Among these three potent inhibitors of tyrosine kinases, the erbstatin analog and PP2 are also regarded as selective inhibitors for the Src kinase family, and tyrphostin 23 is, similar to genistein, a broad-spectrum PTK inhibitor [22]. To get a good comparison between these PTK inhibitors and genistein, genistein was used with each of these three PTK inhibitors in the same cell (Fig. 5a–c). Tyrphostin 23, the erbstatin analog, and PP2 all inhibited Na+ current in SCG neurons. The inhibition was smaller compared to the effect of genistein, but significant. Tyrphostin 23 (100 μM), Erbstatin analog (100 μM), and PP2 (1 μM) inhibited Na+ currents by 14.6% ± 0.5% (p < 0.05, n = 10), 19.3% ± 1.0% (p < 0.01, n = 9), and 17.8% ± 1.9% (p < 0.01, n = 7), respectively, whereas genistein (100 μM) inhibited Na+ currents by 54–58% (Fig. 5a–d). It should be noted that tyrphostin 23-induced inhibition of Na+ current was almost completely reversible, whereas the inhibition produced by the erbstatin analog and PP2 was irreversible. We also noticed that a fast component of inhibition was always present with genistein-induced inhibition, whereas it was absent during inhibition with the other three PTK inhibitors (Fig. 5a–c). Furthermore, the characteristic of the slow component in genistein-induced inhibition was similar to that of inhibition by other PTK inhibitors. It is worth noting that the concentrations of the tyrosine kinase inhibitors used here are much higher than those normally used (10 μM for tyrphostin 23 [23], 10 μM for erbstatin analog [24], and 0.2 μM for PP2 [13]). We used high concentrations of these PTK inhibitors to get a full assessment of the PTK mechanisms involved in genistein-induced inhibition of Na+ current. The combined data suggest that about one-third of the genistein-induced inhibition of Na+ currents occurs through inhibition of tyrosine phosphorylation.

PTK inhibitors inhibit Na+ currents. Tyrphostin 23 (T23, 100 μM) (a), erbstatin analog (EA, 100 μM) (b), and PP2 (1 μM) (c) all inhibited Na+ current to a similar degree, which was significantly less than genistein (GST, 100 μM)-induced inhibition of Na+ current (d) (**p < 0.01, n = 10 for Tyrphostin 23, n = 9 for Erbstatin analog and n = 7 for PP2). Error bars represent SEM
Genistein suppresses excitability in rat SCG neurons
It is well known that VGSCs play a critical role in the generation and propagation of action potentials. Modulation of sodium channel function will inevitably alter the excitability of excitable cells. To confirm that inhibition of Na+ current by genistein reduces neuronal excitability, we performed current-clamp patches to record action potentials elicited by current injection (see the detail in material and methods). Bath application of genistein (100 μM) immediately suppressed the excitability of SCG neurons (Fig. 6a), by reducing the number of action potentials from 8.7 ± 2.1 to 1.3 ± 0.3 (p < 0.01, n = 7) (Fig. 6b). However, genistein did not alter the resting membrane potential (56.1 ± 3.1 and 54.7 ± 4.1 mV before and after application of genistein, respectively) (Fig. 6c). We also quantified the depolarization rate of action potentials before and after the application of genistein (Fig. 6d,e). Genistein significantly reduced the depolarization rate of action potentials from 7.0 ± 1.3 to 4.4 ± 0.9 V/s (p < 0.01, n = 7) (Fig. 6e).

Genistein suppresses the excitability in rat SCG neurons. a Current clamp was used to record the action potentials of rat SCG neurons. Cells were held at 0 pA and injected with 100 pA current for 2 s to elicit action potentials. The left and right panels show the results before and after the application of genistein (100 μM), respectively. b Summary data show the effects of genistein (100 μM) on the spike number of action potentials (**p < 0.01, n = 7). c Summary data show the effect of genistein (100 μM) on the resting membrane potential (n = 7). d Superimposed action potentials in the control (solid line) and in the presence of genistein (dashed line) (100 μM). e Summary data for the depolarization rate before and after genistein (100 μM) application. Error bars represent SEM
Discussion
Genistein is an isoflavone phytoestrogenic molecule with three hydroxyl substituents [34]. Interest in genistein has been raised due to its reported anticancer and cardioprotective properties [2, 38]. Genistein is a frequently used agent in cell signaling studies including studies of ion channel modulation, although some of genistein’s effects on ion channels have been attributed to PTK-independent mechanisms. In the present study, we demonstrated that genistein inhibits voltage-sensitive sodium currents in rat SCG neurons through both PTK-independent and PTK-dependent mechanisms.
Several lines of evidence support that genistein inhibits Na+ currents through two distinct mechanisms. First, the time course of genistein-induced inhibition has two phases, an initial fast phase and a subsequent slow phase, which can be described by a double exponential function with time constants of 10.6 ± 1.2 and 55.9 ± 2.4 s, respectively. These time course characteristics are consistent with a PTK-independent fast effect and a slow process involving alteration of phosphorylation. Second, daidzein, an inactive structural analog of genistein, as well as three other PTK inhibitors tyrphostin 23, an erbstatin analog, and PP2, all inhibited Na+ currents but to a less degree than genistein. In a quantitative estimation, two-thirds of genistein-induced inhibition may come from a PTK-independent effect (based on ∼30% inhibition by daidzein) and one-third may come from the inhibition of PTK activity (based on ∼20% inhibition by tyrphostin 23, erbstatin analog and PP2). Finally, vanadate, presumably through an increase of phosphorylation by the inhibition of phosphatases, reversed or prevented genistein-induced inhibition. Again, ∼20% reduction in inhibition by genistein in the presence of vanadate is consistent with our above quantitative estimation. However, it is possible that the two distinct mechanisms have a synergistic effect since the summed effect of daidzein and other modulators of tyrosine phosphorylation (∼50%) are somewhat smaller than the effect of genistein (∼55–70%).
Sodium channels in cultured CNS neurons are the targets for cAMP-dependent protein kinase (PKA) and protein kinase C (PKC) [20]. Many physiological signals such as dopamine and acetylcholine have been shown to regulate sodium currents in CNS neurons through phosphorylation [7, 8, 29, 33]. Sodium currents also have been shown to be modulated by tyrosine phosphorylation or dephosphorylation. For example, tyrosine phosphorylation induced by Src kinase reduces sodium currents and causes a negative shift in channel inactivation in PC12 cells in the presence of nerve growth factor or other growth factors [15]. Tyrosine dephosphorylation of sodium channels produced by receptor protein tyrosine phosphatase β, which is associated with sodium channels, increases the whole-cell sodium current, slows sodium channel inactivation, and positively shifts its voltage dependence in tsA-201 cells and rat brain cells [26]. However, it is not clear if basal tyrosine phosphorylation of sodium channels plays an important role in the modulation of sodium channel function in neurons. In the present study, our results indicate that basal phosphorylation in resting conditions plays an important role in the function of voltage-gated sodium channels in rat SCG neurons, since all modulators of tyrosine phosphorylation affect channel function.
Previous work has shown that genistein directly inhibits 22Na+ influx through neurotoxin-activated sodium channels in brain neurons and Na+ currents in cerebellar granule cells [24] and inhibits fast sodium currents in uterine leiomyosarcoma cells [19] in a PTK-independent manner. However, it was also reported that genistein inhibits fast sodium currents in rabbit ventricular myocytes through a PTK-dependent mechanism [31]. Our data in the present study suggest that Na+ currents in rat SCG neurons are inhibited by genistein through PTK-dependent as well as PTK-independent mechanisms. Two explanations can be provided for this difference. The first difference may arise from the different Na+ channel isoforms involved in the studies. PNS neurons including DRG [18, 27] and SCG [6, 30] neurons predominantly express Nav1.7 channels whereas cerebellar granule cells predominantly express Nav1.2 and Nav1.6 channels [28]. Sodium currents recorded in muscle cells are most likely from Nav1.4 and Nav1.5 channels [14, 37]. The second possibility is differences in cellular environments where Na+ channels are modulated. In other words, the basal phosphorylation process in different cells could be quiet different.
In the present study, we utilized rat SCG neurons as a platform for recording Na+ currents mainly because they highly express Nav1.7 channels [6, 30] (Fig. 1a). The Nav1.7 channel has been suggested to play an important role in pain pathways. For example, a missense mutation of the Nav1.7 coding gene, SCN9A, leads to primary erythermalgia [12, 35], an inheritable human pain condition characterized by intermittent pain, redness, heat, and swelling in the extremities [11]. To our knowledge, our data is the first to suggest that Nav1.7 channels are targeted for modulation through phosphorylation. It would be interesting to know whether Nav1.7 channel phosphorylation plays a role in physiological and pathophysiological conditions.
Sodium current is a major determinant for neuronal excitability [14]. Modulation of neuronal sodium channels will inevitably alter excitability. Thus, it is not surprising that genistein rapidly suppressed neuronal excitability and the depolarization rate of action potentials in the present study. However, the resting membrane potential was not altered, indicating that potassium currents which contribute mostly to the resting membrane potential are not affected by genistein.
Genistein has been widely used as a blocker of tyrosine kinases and has been used to study signaling mechanisms. The present study and many other reports have suggested its involvement in PTK-independent mechanisms on ion channel functions. However, we emphasize that caution is warranted when a mechanism underlying ion channel modulation is established based on the effect of genistein. Both a PTK-independent effect and an effect through tyrosine phosphorylation must be considered carefully.
References
Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y (1987) Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem 262:5592–5595
Altavilla D, Crisafulli A, Marini H, Esposito M, D, Anna R, Corrado F, Bitto A, Squadrito F (2004) Cardiovascular effects of the phytoestrogen genistein. Curr Med Chem Cardiovasc Hematol Agents 2:179–186
Altomare C, Tognati A, Bescond J, Ferroni A, Baruscotti M (2006) Direct inhibition of the pacemaker (If) current in rabbit sinoatrial node cells by genistein. Br J Pharmacol 147:36–44
Beckh S (1990) Differential expression of sodium channel mRNAs in rat peripheral nervous system and innervated tissues. FEBS Lett 262:317–322
Belevych AE, Warrier S, Harvey RD (2002) Genistein inhibits cardiac L-type Ca2+ channel activity by a tyrosine kinase-independent mechanism. Mol Pharmacol 62:554–565
Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG (1996) Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Mol Brain Res 43:117–131
Cantrell AR, Ma JY, Scheuer T, Catterall WA (1996) Muscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons. Neuron 16:1019–1025
Cantrell AR, Scheuer T, Catterall WA (1997) Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel α subunit. J Neurosci 17:7330–7338
Cataldi M, Taglialatela M, Guerriero S, Amoroso S, Lombardi G, di Renzo G, Annunziato L (1996) Protein-tyrosine kinases activate while protein-tyrosine phosphatases inhibit L-type calcium channel activity in pituitary GH3 cells. J Biol Chem 271:9441–9446
Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26:13–25
Drenth JP, Michiels JJ (1990) Three types of erythromelalgia. BMJ 301:454–455
Drenth JP, Morsche RH, Guillet G, Taieb A, Kirby RL, Jansen JB (2005) SCN9A mutations define primary erythermalgia as a neuropathic disorder of voltage gated sodium channels. J Invest Dermatol 124:1333–1338
Gamper N, Stockand JD, Shapiro MS (2003) Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J Neurosci 23:84–95
Goldin AL, Barchi RL, Caldwell JH et al (2000) Nomenclature of voltage-gated sodium channels. Neuron 28:365–368
Hilborn MD, Vaillancourt RR, Rane SG (1998) Growth factor receptor tyrosine kinases acutely regulate neuronal sodium channels through the src signaling pathway. J Neurosci 18:590–600
Holmes TC, Fadool DA, Levitan IB (1996) Tyrosine phosphorylation of the Kv1.3 potassium channel. J Neurosci 16(5):1581–1590
Jia Q, Jia Z, Zhao Z, Liu B, Liang H, Zhang H (2007) Activation of epidermal growth factor receptor inhibits KCNQ2/3 current through two distinct pathways: membrane PtdIns(4,5)P2 hydrolysis and channel phosphorylation. J Neurosci 27(10):2503–2512
Klugbauer N, Lacinova L, Flockerzi V, Hofmann F (1995) Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J 14:1084–1090
Kusaka M, Sperelakis N (1996) Genistein inhibition of fast Na+ current in uterine leiomyosarcoma cells is independent of tyrosine kinase inhibition. Biochim Biophys Acta 1278(1):1–4
Li M, Weat JW, Numann R, Murphy BJ, Scheuer T, Catterall WA (1993) Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science 61:1439–1442
Meisler MH, Kearney JA (2005) Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 115:2010–2017
Missan S, Linsdell P, McDonald TF (2006) Tyrosine kinase and phosphatase regulation of slow delayed-rectifier K+ current in guinea-pig ventricular myocytes. J Physiol 573(2):469–482
Ogata R, Kitamura K, Ito Y, Nakano H (1997) Inhibitory effects of genistein on ATP-sensitive K+ channels in rabbit portal vein smooth muscle. Br J Pharmacol 122:1395–1404
Paillart C, Carlier E, Guedin D, Dargent B, Couraud F (1997) Direct block of voltage-sensitive sodium channels by genistein, a tyrosine kinase inhibitor. J Pharmacol Exp Ther 280:521–526
Peretz A, Sobko A, Attali B (1999) Tyrosine kinases modulate K+ channel gating in mouse Schwann cells. J Physiol 519:373–384
Ratcliff CF, Qu Y, McCormick KA, Tibbs VC, Dixon JE, Scheuer T, Catterall WA (2000) A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase beta. Nat Neurosci 3:437–444
Sangameswaran L, Fish LM, Koch BD, Rabert DK, Delgado SG, Ilnicka M, Jakeman LB, Novakovic S, Wong K, Sze P, Tzoumaka E, Stewart GR, Herman RC, Chan H, Eglen RM, Hunter JC (1997) A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem 272:14805–14809
Schaller KL, Caldwell JH (2003) Expression and distribution of voltage-gated sodium channels in the cerebellum. Cerebellum 2(1):2–9
Smith RD, Goldin AL (1997) Phosphorylation at a single site in the rat brain sodium channel is necessary and sufficient for current reduction by protein kinase A. J Neurosci 17:6086–6093
Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silossantiago I, Halegoua S, Mandel G (1997) Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A 94:1527–1532
Wang Y, Wagner MB, Kumar R, Cheng J, Joyner RW (2003) Inhibition of fast sodium current in rabbit ventricular myocytes by protein tyrosine kinase inhibitors. Pflugers Arch 446(4):485–491
Washizuka T, Horie M, Obayashi K, Sasayama S (1998) Genistein inhibits slow component delayedrectifier K currents via a tyrosine kinase-independent pathway. J Mol Cell Cardiol 30:2577–2590
West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA (1991) A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science 254:866–868
Whaley WL, Rummel JD, Kastrapeli N (2006) Interactions of genistein and related isoflavones with lipid micelles. Langmuir 22(17):7175–7184
Yang Y et al (2004) Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 41:171–174
Yokoshiki H, Sumii K, Sperelakis N (1996) Inhibition of L-type calcium current in rat ventricular cells by the tyrosine kinase inhibitor, genistein, and its inactive analog, daidzein. J Mol Cell Cardiol 28:807–814
Yu FH, Catterall WA (2003) Overview of the voltage-gated sodium channel family. Genome Biology 4:207–213
Zielonka J, Gebicki J, Grynkiwicz G (2003) Radical scavenging properties of genistein. Free Radic Biol Med 35:958–965
Acknowledgements
This work was supported by a National Natural Science Foundation of China Grant 30730031 to HZ. HZ is a beneficiary of the National Science Fund for Distinguished Young Scholars of China (no. 30325038).
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhanfeng Jia and Yueqin Jia contributed equally to this work.
Rights and permissions
About this article
Cite this article
Jia, Z., Jia, Y., Liu, B. et al. Genistein inhibits voltage-gated sodium currents in SCG neurons through protein tyrosine kinase-dependent and kinase-independent mechanisms. Pflugers Arch - Eur J Physiol 456, 857–866 (2008). https://doi.org/10.1007/s00424-008-0444-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0444-2