
Abstract
Obstructive sleep apnea (OSA) is very common in the general population and is characterized by ineffective inspiratory efforts against a collapsed upper airway during sleep. Collapse occurs mainly at the level of the velopharynx and oropharynx due to a combination of predisposing anatomy and the withdrawal of pharyngeal dilator activity during sleep. Central sleep apnea (CSA) is a manifestation of chemoreflex control instability, leading to periods of inadequate respiratory drive sufficient to trigger breathing, usually alternating with periods of hyperventilation. While both forms of apnea are the result of differing pathophysiology, it has become increasingly clear that OSA and CSA often coexist in the same patient, the existence of one can predispose to the other, and that the two are not as distinct as previously thought. Both OSA and CSA exert a number of acute deleterious effects including intermittent hypoxia, arousals from sleep, and swings in negative intrathoracic pressure, which in turn lead to chronic physiologic consequences such as autonomic dysregulation, endothelial dysfunction, and cardiac remodeling. These underlying pathophysiological mechanisms provide a framework for understanding why OSA and CSA may predispose to cardiovascular diseases like ischemic heart disease and stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obstructive sleep apnea (OSA) is present in approximately 3–7% of the general population [149] and is characterized by repetitive collapse of the upper airway during sleep, leading to ineffectual respiratory efforts and apnea. Apneas are typically followed by large recovery breaths, referred to as hyperpneas. Lesser reductions in ventilation that fall short of outright apnea are termed hypopneas. The severity of sleep apnea is most commonly quantified as the apnea–hypopnea index (AHI), the number of apneas and hypopneas per hour of sleep. While there is some disagreement about the exact thresholds to be used in defining the severity of sleep apnea, in general, AHI > 5 events per hour is considered to be normal, 5–15 mild, 15–30 moderate, and >30 severe [43].
In addition to causing nonrestorative sleep that leads to excessive daytime sleepiness, OSA has been implicated in a host of serious cardiovascular consequences, including hypertension, coronary artery disease, stroke, and congestive heart failure (CHF) [7, 96, 111, 142, 178]. The prevalence of OSA in patients with these conditions is much higher than in the general population, approaching 30–50% [15].
Central sleep apnea (CSA) differs from OSA in that central apneas are associated with absent respiratory effort. Hypercapnic CSA is a result of diminished central drive or respiratory muscle weakness and will not be discussed further in this review. Instead, the focus will be restricted to eucapnic or hypocapnic CSA which results from instability in the chemoreflex control of breathing. Cheyne–Stokes respiration is a form of CSA in which an apneic phase alternates with ventilatory periods (hyperpneic phase) having a crescendo–decrescendo pattern of tidal volume. Although much less common than OSA in the general population, CSA is very common among patients with certain disease states, including CHF and stroke, and can also be frequently observed in normal individuals at altitude and, not uncommonly, in patients with OSA immediately following establishment of upper airway patency with continuous positive airway pressure (CPAP) (sometimes referred to as “complex sleep apnea”). The latter observation, among others, has led to the recognition in recent years that OSA and CSA are not completely distinct pathophysiological entities as has previously been thought. Patients often display both kinds of apnea, and one or the other form may predominate at different times of the night during sleep in a single individual. It is increasingly thought that OSA and CSA are more usefully conceptualized as extremes along a continuum of sleep-disordered breathing (SDB) with considerable overlap between the two apnea types.
Obstructive sleep apnea
OSA, by definition, consists of repetitive pharyngeal airway collapse (obstruction) leading to the absence of ventilation (apnea) during sleep (Fig. 1). Apneas may result in significant oxyhemoglobin desaturation and are generally terminated by central nervous system arousals, which in turn lead to activation of the pharyngeal dilator muscles, relieving the obstruction. Lesser reductions in ventilation that fall short of outright apnea are termed hypopneas.

Obstructive sleep apnea in an obese patient. Note the ineffectual paradoxical motion of the chest and abdomen during apneas, during which there is no nasal airflow. Apneas are terminated with arousals visible on EEG and are accompanied by fluctuations in oxygen saturation. EEG (C3, C4) electroencephalogram, Sum the total effort of the abdomen and chest, SaO 2 oxygen saturation
Owing to the complexities of the functional anatomy of the upper airway, the pathogenesis of OSA is not fully understood, but fundamentally must relate to the balance between the action of dilating upper airway muscles and the inherent tendency for the upper airway to collapse. It is also worthwhile to consider that OSA is a state-dependent phenomenon. If the passive anatomy of the upper airway were solely responsible for its collapse, obstruction would occur during both wakefulness and sleep, instead of during sleep alone. Even within sleep, the various sleep stages are not uniform with respect to their tendency to cause upper airway obstruction. It has long been observed that OSA worsens during rapid eye movement (REM) sleep. Therefore, any theory which purports to explain upper airway obstruction in OSA must account for its state dependency.
Historically, there were two theoretical mechanisms for this state-dependent airway collapse, termed the “active” and “passive” theories. The theory of active collapse was proposed by Weitzman et al., based on the observation of spasmodic closure of the lateral pharyngeal walls at end expiration [213]. However, direct upper airway electromyography has not confirmed the presence of muscle activity with airway collapse, leading to the “active” theory being largely discarded [53]. In contrast, there is a considerable weight of evidence supporting the theory of passive collapse of the upper airway. Stated more explicitly, airway obstruction during OSA is thought to result from the loss of the dilating activity of pharyngeal muscles in an anatomically predisposed upper airway [154].
Anatomic considerations
The human upper airway is a complicated passage that transmits air, liquids, and solids. In other mammals, the respiratory and alimentary canals are more separate, with the larynx being closer to the skull base [100]. Laryngeal descent, while facilitating speech development, predisposes the supraglottic pharyngeal airway to obstruction and requires compensatory neurological and physiological changes [107, 138].
The upper airway consists of four regions, the nasopharynx, velopharynx, oropharynx, and hypopharynx (Fig. 2). The anatomic relationships are defined below:

Anatomical representation of the upper airway showing important structures affecting upper airway patency. The most common point of collapse during obstructive apneas is the retropalatal oropharynx or velopharynx or both. Reproduced with permission from Fogel et al. [47]
-
1.
Nasopharynx: extends from the nasal turbinates to hard palate.
-
2.
Velopharynx: approximates and is posterior to the soft palate. In some classification schemes, this region is considered to be part of the oropharynx, called the “retropalatal oropharynx” to distinguish it from the “glossopalatal oropharynx” inferiorly.
-
3.
Oropharynx: extends from the hard palate to the base of the epiglottis.
-
4.
Hypopharynx: extends from the base of tongue to the larynx.
The velopharynx (or retropalatal oropharynx) has the narrowest cross-sectional area in the upper airway, making it a high risk location for obstruction [171, 173]. It is bounded anteriorly by the soft palate and tongue, posteriorly by the superior, middle, and inferior constrictor muscles and laterally by orophargyngeal muscles (hyoglossus, styloglossus, stylohyoid, stylopharyngeus, palatoglossus, palatopharyngeus, pharyngeal constrictors (superior, middle, and inferior)), lymphoid tissue (lingual and palatine tonsils, located at the base of the tongue, and on either side of the oropharynx, respectively, and adenoids, located in the roof of the nasopharynx), and adipose tissue (parapharyngeal fat pads). Decreases in oropharyngeal cross-sectional area due to crowding or dysfunction of any of these adjacent structures may predispose to obstruction.
Static and dynamic properties of the upper airway
There are many static features that influence upper airway patency, including surface adhesive forces, neck and jaw posture, lung volume and tracheal tug, as well as the effects of gravity. Surface adhesive forces tend to cause the soft palate to remain in opposition to the back of the tongue during mouth closure and may delay separation of these structures following mouth opening [72, 158]. Manipulation of surface adhesive forces through instillation of exogenous surfactant and saline has been found to improve upper airway patency [84, 122]. Neck extension facilitates airway opening, whereas neck flexion promotes airway closure [121, 164]. Increases in lung volume lead to caudal displacement of the thoracic notch, thereby passively opening the pharynx [62, 161, 201, 208]. Finally, the effects of gravity have long been recognized in the tendency of OSA and snoring to worsen when patients sleep supine. When supine, the tongue and soft tissue fall posteriorly, further compromising the upper airway [175, 186].
Dynamic features of the upper airway affecting patency include upstream resistance within the nasal airway and pharynx, the Bernoulli effect, and the dynamic compliance of the airway. Upstream resistance during inspiration develops mainly within the nose and pharynx. Nasal resistance leads to a more negative nasopharyngeal intraluminal pressure, as does resistance within the pharynx itself. These falls in intraluminal pressure predispose the airway to obstruction [8]. The Bernoulli effect refers to the conversion of fluid potential energy to kinetic energy in high resistance regions of the airway. As a result, flow velocity increases when lumen size decreases, leading to a drop in lateral wall pressure that further narrows the airway [8]. Airway compliance dictates the degree of the resultant narrowing with the aforementioned intraluminal pressure variations [71].
Physiology of upper airway and dilator muscles
The state dependency of OSA is illustrated by the routine observation that even patients with the most severe OSA do not suffer airway collapse during wakefulness. The state-dependent cross-sectional area of the pharyngeal airway is determined by muscles regulating the position of the soft palate, tongue, hyoid, and posterior lateral wall. Upper airway muscle function depends both on which muscles are simultaneously active and also their anatomic relationship to each other. For example, the position of the hyoid, as a bone with no bony attachments, is in turn determined by the sternohyoid, thyrohyoid, genioglossus, and geniohyoid [209]. Similarly, mouth opening decreases genioglossus and geniohyoid length and, hence, the force developed by muscle action [87, 88].
Given the multiple and complex effects of various muscle movements on airway caliber, an effective pressure, termed P mus, can simplify and quantify the effect of muscle action. This effective pressure of muscle activation can be defined as the change in transmural pressure required to yield the equivalent change in airway compliance under passive conditions (i.e., with no muscle activation) [8].
The degree of muscle activation and, hence, P mus, is influenced by a number of factors including changes in sleep state and chemical and proprioceptive stimulation. Sleep increases supraglottic airway resistance (from the nares to the region above the glottis) even in healthy persons from 1–2 to 5–10 cmH2O/L/s and to 50 cmH2O in heavy snorers. Supraglottic airway resistance is typically elevated even during wakefulness in patients with OSA and further increases to the point of airway closure with sleep [3, 4, 185, 199, 206, 218]. Chemical and proprioceptive stimuli also play a role in maintaining upper airway patency. Arterial CO2 changes around the CO2 threshold for upper airway motor neuron activity may cause an imbalance of forces leading to airway closure. Responses to an increase in CO2 appear first in the phrenic nerve and later in the upper airway at higher CO2 levels [135, 200, 212]. Similarly, upper airway and intrathoracic propioceptors can increase or decrease motor output to inspiratory muscles [64, 69, 112, 113].
OSA worsens during REM sleep. The muscular atonia accompanying REM further relaxes the upper airway dilators, lowering P mus and favoring collapse, and the decreased chemosensitivity and arousability serve to delay the termination of apneas by arousals, leading to more profound hypoxemia. REM-related muscular atonia also reduces end-tidal lung volume and, hence, oxygen stores, an effect that may be further exacerbated by the presence of obesity [141].
Model of passive pharyngeal collapse
Given the complex anatomy of the upper airway, it is clear that many physiologic processes enable airway patency including upper airway muscle tone, mechanical forces from the airway wall, tissue masses, and surface adhesive forces. Further, upstream and downstream pressures from the nose and trachea also affect intraluminal airway pressure. In order to simplify the functional anatomy of the upper airway, it is useful to model the airway as a Starling resistor and seek explanations for the role of various factors on upper airway patency by considering their net effects on transmural pressure (P tm), defined as the distending force of the upper airway resulting from the difference between the sum of the dilating forces (P out) and the sum of the collapsing forces (P in).
where P tm is transmural pressure, P out is the sum of dilating forces, and P in is the sum of collapsing forces.
In this simplified view, the upper airway is considered to be a collapsible tube, wherein the cross-sectional area depends on the transmural pressure and the tube compliance. Poiseuille’s law states that flow through a rigid tube is a function of the driving pressure and resistance through the tube.
where V is flow, P 1 is the pressure upstream, P 2 is the pressure downstream, (P 1 − P 2) is the driving pressure, and R is resistance.
Similarly, resistance in a rigid tube varies with tube length, fluid viscosity and, most importantly, the radius of the tube.
where R is resistance, L is the length of tube, η is the fluid viscosity, and r is the radius.
The Starling resistor concept builds on Poiseuille’s law, to describe flow through a tube which is not rigid, but rather is collapsible, with infinite resistance at one transmural pressure, but low resistance at another transmural pressure. This behavior is explainable by the tube shifting from closed to open at a certain intraluminal pressure, termed the critical pressure (P crit). In normal subjects, P crit would be negative, whereas it would be positive in subjects with OSA during sleep. Within this framework, the main treatment for maintaining airway patency in OSA, CPAP, can be viewed as a means to achieving an intraluminal pressure that exceeds P crit.
In a collapsible tube, resistance varies with airway lumen size, airflow and transmural pressure, and the upstream and downstream pressures. The downstream pressure (P downstream) is the negative inspiratory pressure at the trachea and the upstream pressure (P upstream) is the ambient pressure at the nose. As airway size varies, unimpeded flow, flutter, and obstruction can occur, corresponding to normal breathing, snoring, and obstruction as follows:
-
Normal breathing: The airway is like a rigid tube; airway size and resistance are constant. P crit is lower than both the upstream and downstream pressures.
-
Flutter (snoring): In snorers, the upstream (nasal) pressures are higher than P crit, but the downstream (tracheal) pressures are less than P crit. Thus, when the airway is open, it is exposed to the downstream pressure, which leads to airway collapse. However, when the choke point closes, the airway is exposed only to the positive upstream pressures and the airway reopens.
-
Obstruction: The airway is always closed. P crit is higher than both the upstream and downstream pressures.
Anatomical and physiological abnormalities in patients with sleep apnea
Many studies have assessed the anatomic and physiologic basis for OSA. Broadly, predisposing factors can be considered in two categories. It is clear that some patients develop OSA mainly due to congenital predisposing skeletal relationships of the head, neck, and jaw. Alternatively, upper airway soft tissue enlargement, most commonly due to an excess of adipose tissue is often at fault.
Cephalometry, the study of skeletal relationships in the head, has found many anatomical correlates of OSA. Compared to normals, patients with OSA have a decreased mandibular body length (retrognathia), an inferiorly positioned hyoid bone, and retroposition of the mandible [19, 38, 86, 117, 163, 171, 172, 174, 181].
Similarly, upper airway soft tissue enlargement is associated with OSA. CT and MRI examination of patients with and without OSA have revealed increased dimensions of the soft palate, tongue, parapharyngeal fat pads, and lateral pharyngeal walls in patients with OSA [9, 29, 35, 103, 147, 173]. Upper airway soft tissue enlargement in OSA may relate to gender, obesity, muscle injury, edema, and genetic considerations, based on epidemiological and physiological studies. For example, upper airway size and neck size are smaller in women than men [54, 91, 119]. Conversely, airway length, soft palate size, and tongue size are larger in men than women [106, 217]. Body mass index has long been recognized as an excellent predictor of OSA and is associated with increased fat deposition in lateral pharyngeal fat pads [179], thereby reducing airway cross-sectional area [65, 124]. There is also evidence for pathologic airway remodeling in patients with OSA, although whether it is the cause or the result of sleep apnea is unclear. Type II fibers, which are more fatiguable than type I fibers, are more common in the genioglossus muscles of patients with OSA than controls [21, 48, 176]. Edema and fluid shifts have been implicated in OSA pathogenesis. Edema from negative pressure from airway closure and trauma leads to an increase in soft tissue volume [168, 170]. Fluid shifts from dependent areas of the body into the upper airway soft tissues, particularly in patients in edematous states such as CHF and renal failure may also contribute to airway obstruction [49, 152, 153, 195]. There are also genetic factors predisposing patients to OSA. The familial predominance of OSA may be due to the heritability of facial structure [114]. Another uncommon but strong risk factor for OSA is macroglossia, as in patients with trisomy 21 [110].
Central sleep apnea
Commonly, repetitive nonhypercapnic central apneas can occur during non-REM sleep under conditions of high altitude or in association with congestive CHF or stroke [130, 216]. Much less commonly, CSA can occur in some subjects with no apparent cardiac, pulmonary, or neurologic disease. For these individuals, the term “idiopathic” CSA is used [17]. Idiopathic CSA is relatively rare and constitutes <5% of patients referred to sleep clinics [105]. In contrast, although rare in the general population, CSA is very common in the setting of CHF, being present in 30–40% of these patients in the two largest reported series [74, 183].
Repetitive central apneic events wherein tidal volume fluctuates in a crescendo–decrescendo pattern (Fig. 3) are often referred to as Cheyne–Stokes respiration or periodic breathing; the two terms are generally used interchangeably. However, some authors distinguish between the two. If intervals of hyperventilation (hyperpneas) are separated by periods of reduced tidal volume (hypopneas), the term periodic breathing may be used, but if mainly apneas are present, the event is described as Cheyne–Stokes respiration [85, 118]. It has been suggested that three or more successive oscillation periods of tidal volume are necessary for the diagnosis of periodic breathing or Cheyne–Stokes respiration [5], but no specific definition has been generally accepted.

Cheyne–Stokes respiration in a patient with CHF. Note the crescendo–decrescendo of the nasal flow during hyperpneas and the absence of abdomen and chest effort during apneas indicating their central nature. There are fluctuations in oxygen saturation in response to the periodic breathing pattern. EEG (C3, C4) electroencephalogram, Sum the total effort of the abdomen and chest, SaO 2 oxygen saturation
Loop gain and ventilatory control instability
Hypopneas or apneas manifest in patients with CSA due to destabilized ventilatory control. The chemical control of respiration operates in a negative feedback, closed-loop fashion. For a change in metabolic rate, there is a transient change in ventilation that results in a transient change in alveolar gas tension (i.e., PaCO2, PaO2). These changes are sensed by chemoreceptors resulting in a further change to the ventilatory response. The ventilatory output to a given change in PaCO2 or PaO2 (i.e., chemosensitivity) can vary between individuals and with disease status. The concept of “loop gain”, an engineering term to define the sensitivity of a variable system, was first used to describe ventilatory control in the early 1980s [83]. Loop gain has a number of components: with ventilation, these can be considered the ventilatory response to CO2 (i.e., controller gain), the blood gas response to change in ventilation (i.e., plant gain), and the delays in feedback between the two imposed by hemoglobin binding and cardiac output (i.e., feedback gain) [17, 82].
Chemosensitivity (controller gain) and the apnea threshold
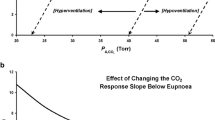
The transition from wakefulness to non-REM sleep is characterized by a decrease in ventilatory motor output, a fall in ventilation, and resultant increases in PaCO2 (~5 mmHg). The sensitivity to small changes in PaCO2 and the speed of the ventilatory response provides stability, allowing these fluctuating PaCO2 levels to stay within a PaCO2 reserve of ~5 mmHg. This reserve is often referred to as the apnea threshold [222]. If PaCO2 falls below the bounds of the apnea threshold, respiratory drive is curtailed and breathing ceases. In the context of loop gain theory, ventilation is destabilized when the ratio between the size of the response to the size of a disturbance is greater than one. This value predicts an overshoot in ventilation to an increase in PaCO2 and progressive instability. A ratio of less than one, however, would predict an appropriate ventilatory response and stability of the system [214]. Individuals with elevated chemosensitivity (i.e., controller gain) will hypoventilate to decreases in PaCO2 and, consequently, hyperventilate to increases in PaCO2 causing ventilatory control to become destabilized [26, 83] (Fig. 4).

Ventilatory control (stability)–loop gain theory: In patients with high chemosensitivity, small increases in PaCO2 are responded to with resultant hyperventilation, until PaCO2 is reduced below the apnea threshold resulting in hypoventilation and possible apnea. During apnea, PaCO2 is increased; thus, the cycle of periodic breathing pattern is repeated
A number of studies have shown that CHF patients with periodic breathing while awake or Cheyne–Stokes respiration during sleep demonstrate higher central [73, 219] and peripheral chemoresponsiveness [145, 189] than among those with OSA or without sleep apnea. Interestingly, Xie et al. [222] showed that in CHF patients with CSA, the apnea thresholds are not different from patients without CSA. However, an increased ventilatory drive during sleep in CHF patients with CSA narrows the proximity of the eupneic PaCO2 to the apnea threshold, predisposing them to the development of apnea and subsequent breathing instability. It still remains unclear, however, whether such increases in central and peripheral chemoresponsiveness predate or is a consequence of CHF [220], though increased peripheral chemoreflexes have been induced in a rabbit model of CHF though a nitric oxide-dependent mechanism [196, 197]. In a study investigating chemosensitivity in patients with CHF, Solin et al. [189] made the following observations: (1) the timing of the peripheral CO2 ventilatory response was consistent with the response time of the carotid chemoreceptor allowing for circulatory delay, (2) the peripheral CO2 ventilatory response was similarly elevated in patients with CHF and CSA and in patients with idiopathic CSA—suggesting that CSA can be evoked solely by raised chemosensitivity independent of underlying cardiac function, and (3) in the CHF patients as a whole, the peripheral CO2 ventilatory response correlated significantly with CSA severity. These findings suggest that in patients with CHF, elevated central CO2 ventilatory response lowers PaCO2 toward the apnea threshold, narrowing the difference between PaCO2 levels and the apnea threshold, predisposing a patient with CHF to develop central apneas and breathing instability.
Circulatory delay (feedback gain)
The circulation time for changes in arterial blood gas tension to travel from the lungs to the chemoreceptors affects the length of the subsequent ventilatory phase and is inversely proportional to cardiac output. Patients with CSA and CHF, compared to patients without CHF, demonstrate a longer ventilatory phase during which tidal volume rises and falls more gradually [56]. Thus, the prolonged circulation period in CHF patients induces a periodic breathing pattern. This was first speculated by Pryor [148] over 60 years ago. However, later studies have not been able to show a predisposition or statistical association between low cardiac output and periodic breathing in patients with CHF [57, 183]. Indeed, among CHF patients with and without CSA, no significant differences in lung to peripheral chemoreceptor circulation time or cardiac output have been observed [56, 187]. Rather, the central control of ventilation appears to be the primary defect leading to instability in patients with CSA rather than their impaired cardiac function (and resulting circulatory delay) [187]. The genesis of abnormal central control of ventilation may lie, at least in part, in the observation that pulmonary vascular pressure is significantly elevated in patients with CSA. Highly significant correlations have been demonstrated between raised pulmonary vascular pressure on the one hand, and hypocapnia and CSA severity on the other [187]. Data from animal models have shed light on this relationship, by demonstrating that the hyperventilation results from elevation of pulmonary interstitial pressure. Indeed, pulmonary edema and the concomitant increase in interstitial pressure stimulate pulmonary J receptors, which lie within the interstitium in close proximity to the pulmonary capillaries [139]. Neural impulses are transmitted via afferent pulmonary vagal C fibers to the ventilatory control center in the medulla [157], whereby stimulation of this vagal afferent system results in brief central apnea followed by tachypnea and hyperventilation [28, 139, 157].
The effects of an isolated increased circulation time on respiratory control were investigated by Guyton [55] in a series of classic animal experiments in the 1950s. Guyton was able to induce periodic breathing in dogs by inserting a length of tube between the heart and cerebral circulation to prolong transit time between the lungs and chemoreceptors. However, CSA was only induced when the circulation time was a few minutes in duration. This far exceeds the lung to cerebral circulation time in CHF patients, which in humans is inversely proportional to stroke volume and cardiac output [56].
However, the concept that prolonged circulation time predisposes to CSA has not been completely discarded. Mortara et al. [123], for one, found that a prolonged circulation time was a significant determinant of the presence of CSA in patients with CHF, but a number of other investigators have reported that left ventricular (LV) ejection fraction, cardiac output, and circulation time are not significantly different in CHF patients with and without CSA [57, 130, 187]. Therefore, CSA appears not to be caused by circulatory delay, but rather is proportional to the preceding decrease in PaCO2 [31, 56]. Indeed, the general consensus is that prolonged circulation time is probably not the critical determinant predisposing CHF patients to CSA. Circulatory delay, however, does influence periodic breathing cycle length such that cycle length is proportional to lung carotid body circulation time [56], accounting for the greater length of the periodic breathing cycle in CHF patients than in patients with idiopathic CSA whose cardiac function is normal. A study investigating circulation time and CSA by Solin et al. extended these concepts, by objectively assessing systolic function in subjects with and without CHF [190] and found that the presence of CHF can be inferred in patients with nonhypercapnic CSA by a prolonged circulation time exceeding 15 s.
Sleep transitions and ventilatory control (stability)
The transition from wakefulness to sleep and vice versa may show some instability in breathing as the PaCO2 set point changes. Periodic breathing is commonly seen even in normal individuals during transitional periods between wakefulness and light sleep and is markedly attenuated or disappears during REM sleep. A close relationship between periodic breathing and shift in sleep stages has been reported [41], confirming that stage-specific brain activity has a role in the control of ventilation [184]. Most commonly, arousal from sleep is associated with an abrupt increase in ventilation [67, 78], particularly immediately after awakening. In normal individuals during wakefulness, the PaCO2 approximates 40 mmHg. However, during stable non-REM sleep, the PaCO2 rises by around 5 mmHg. Therefore, during the transition from non-REM sleep to wakefulness, individuals typically briefly hyperventilate to drive down the sleeping PaCO2 to the new waking set point. If the increase in ventilation is associated with ventilatory overshoot and yields a PaCO2 below the apnea threshold [36], a resulting cessation of breathing (apnea) can occur. Similarly, during the transition from wakefulness to non-REM sleep, the waking neural drive to breathe is lost, and the threshold for a ventilatory response to PaCO2 is increased [143]. Sleep onset is therefore very commonly associated with central apneas, resulting from the waking PaCO2 (around 40 mmHg), being too low to stimulate ventilation in association with the higher PaCO2 set point of sleep. Finally, it has been reported that arousals can trigger abrupt increases in ventilation that rise above waking levels independently of PaCO2 [66], and these would further tend to cause instability.
A low eupneic PCO2 has been reported, and a eupneic PCO2 close to the apneic threshold has been suggested as the main mechanism of CSA [102, 130, 203]. Indeed, Xie et al. [222] reported that patients with CHF and CSA show a closer proximity of eupneic PETCO2 to the threshold PETCO2 and a greater hypocapnic ventilatory response below eupnea compared with patients with CHF but without CSA. Furthermore, patients with CSA do not show an increase in their PETCO2 from wakefulness to sleep [203, 222], whereas in normal patients, the hypercapnic effect of sleep persists even in the presence of ventilatory stimulation with progesterone or hypoxia [75]. These findings suggest that in patients with CSA, sleep state is associated with an additional ventilatory drive that offsets the removal of the ventilatory drive associated with wakefulness.
Although periodic breathing can be observed during wakefulness [17, 205], it is much more common in non-REM sleep, where chemical–metabolic factors are the predominant influence on ventilatory control, and less common during REM sleep where behavioral nonmetabolic factors again supervene [17, 130, 144]. Indeed, the arousability to respiratory stimuli is diminished during REM sleep compared with the lighter stages of non-REM sleep. Therefore, during REM sleep, the tendency to increase ventilation above the apnea threshold is lessened, thereby reducing the tendency to blow down PaCO2 and consequently dampening apneic events. Furthermore, in the deeper stages of non-REM sleep where arousability is also decreased, CSA events are also reduced compared to lighter non-REM sleep.
Periodic breathing can also present during exercise in patients with CHF. Indeed, increases in the metabolic responses to incremental exercise can produce an excessive ventilatory response, thus promoting greater ventilatory instability [93].
Interactions between central sleep apnea and obstructive sleep apnea
There is substantial evidence to suggest a mechanistic link between OSA and CSA. Several studies have shown that oscillating ventilatory motor output during periodic breathing is associated with reciprocal changes in upper airway resistance [10, 68]. Indeed, patients who snore without concomitant sleep apnea may be dependent on ventilatory motor output to preserve upper airway patency through the action of the pharyngeal dilators [68, 134], and in these patients, pharyngeal obstruction can occur when ventilatory drive reaches a nadir during induced periodic breathing [68, 134, 211]. Similarly, spontaneous central apnea is often associated with pharyngeal narrowing or occlusion early during the central apneic period in the absence of inspiratory efforts [11]. It has been suggested [224] that the ability of the patient with severe OSA to effectively compensate for airway narrowing and increased mechanical load was a more important determinant of the degree of cycling behavior of airway patency and ventilation than the inherent passive collapsibility of the airway. Therefore, although an inherently collapsible airway may allow for significant airway narrowing and obstruction during sleep, any cycling behavior in airway patency and ventilation is also critically dependent upon chemical control mechanisms.
A number of studies have demonstrated that the concept of ventilatory instability due to increased loop gain holds relevance not only in CSA, but in OSA. In patients with OSA, the CO2 reserve has been reported to be significantly narrower and the controller gain significantly greater compared to controls, whereas the apnea threshold and plant gain are similar. However, following 1 month of CPAP, the CO2 reserve widens and the controller gain is diminished in patients with OSA, both towards normal [165]. The susceptibility to periodic breathing in patients with mild to severe OSA was first assessed in a novel fashion by Younes et al. by artificially increasing controller gain, using proportional assist ventilation [225]. The increase in loop gain at each assist level was quantified from the ratio of assisted tidal volume (VT) to the VT obtained during single-breath reloading tests. The chemical control system has been reported to be more unstable in patients with severe OSA than in patients with milder OSA and was speculated to contribute to the severity of OSA. Finally, the clinical utility of mathematically quantifying loop gain from the cyclic pattern of periodic breathing in heart failure patients has been demonstrated by one group. The calculated estimates of loop gain enabled the quantification of the severity of ventilatory instability underlying periodic breathing. This made possible a priori selection of patients whose periodic breathing is treatable with CPAP therapy; non-responders to CPAP had higher calculated loop gain [166].
In patients with CHF, there is commonly observed both obstructive and central apneas during a single night; in such patients, there is also often a gradual shift from predominantly obstructive apneas at the beginning of the night to predominantly central apneas towards the end [204]. This change occurs in association with a prolongation in circulation time and a downward drift in PaCO2 towards the apnea threshold—suggesting the possibility that repetitive surges in afterload induced by OSA, combined with increased venous return can cause an overnight increase in left ventricular filling pressure that leads to hyperventilation and hypocapnia through stimulation of pulmonary afferents. A cause and effect relationship between central and obstructive apneas is also suggested by the frequent occurrence of mixed apneas, which are characterized by a period of decreased central drive followed by an obstructive breath. Indeed, a high central apnea index with a low obstructive apnea index is encountered in only a small number of patients [34]. Despite similarities and interactions between CSA and OSA, arousals accompanying the two appear to serve different purposes. In OSA, arousals operate as a defense mechanism to terminate apneas and activate pharyngeal muscles to allow the re-opening of the upper airway. However, in CSA, arousals appear to provoke ventilatory overshoot that sustains periodic breathing, as indicated by the strong relationship between the magnitude of arousal, and both the magnitude of ventilation during hyperpnea and subsequent apnea duration [223]. This reflects the tendency of the upper airways to collapse even in healthy subjects when the drive to the inspiratory muscles and especially the upper airway muscles is reduced at the nadir of periodic breathing [218].
Still another manifestation of the linkage between OSA and CSA is the recently described entity “complex sleep apnea”, referring to the unexpected appearance of repetitive central apneas once upper airway patency has been maintained in patients being treated for OSA with CPAP [120]. While the specific mechanisms responsible for complex sleep apnea remain unclear, sleep fragmentation and frequent transitions between sleep and wakefulness due to initial intolerance of CPAP is thought to play a role [22, 37, 92]. Moreover, it may be that patients who display complex sleep apnea may have underlying chemoreflex instability obscured by OSA that only becomes apparent once upper airway patency is established. In support of this concept is the finding that patients with OSA who develop complex sleep apnea following application of CPAP have more frequent central apneas even on their baseline pre-CPAP sleep studies than patients who do not develop complex sleep apnea [76].
Consequences of OSA and CSA
Acute effects
As a consequence of the repetitive apneas characteristic of OSA and CSA, hemodynamic variables and cardiovascular autonomic activity oscillate between the apneic and ventilatory phases. Surges in heart rate (HR) and blood pressure (BP) typically occur 5–7 s after apnea termination in OSA [156, 202], coincident with arousal from sleep, peak ventilation, and the nadir of SaO2. In CSA, these surges occur not at apnea termination, but during hyperpnea [98]. These repetitive surges counteract the usual fall in HR and BP that accompany normal sleep. Three key pathophysiological features of sleep apnea give rise to these abnormal cardiovascular oscillations: hypoxia, arousals from sleep, and generation of negative intrathoracic pressure. In turn, these disturbances give rise to a fourth consequence of sleep apnea: sympathetic activation during sleep.
Hypoxia alternating with normoxia is a hallmark of sleep apnea. Hypoxia can reduce myocardial oxygen delivery, directly depress myocardial contractility and increase LV afterload, and indirectly cause increased pulmonary vasoconstriction and increasing pulmonary arterial pressure [177]. Hypoxia can either increase or decrease HR depending on whether parasympathetic or sympathetic influences predominate [14]. It is well known that apnea-associated hypoxia can lead to episodes of extreme bradycardia and even heart block [51], and severe hypoxia has been reported to acutely trigger ventricular arrhythmias [180]. Moreover, the combination of hypoxia and tachycardia further impairs myocardial contractility [177]. While apnea-associated hypercapnia would tend to favor oxygen unloading to the tissues through a rightward shift of the oxyhemoglobin dissociation curve, hypocapnia, as a result of post-apneic hyperventilation, can further exacerbate matters by inducing coronary artery vasoconstriction [125] and a leftward shift of the dissociation curve, reducing oxygen availability to the tissues. Given the primacy of hypoxia as a candidate mechanism for causing deleterious effects, it is perhaps surprising that the severity of hypoxia is poorly reflected in AHI, the conventional metrics of OSA. However, there is evidence that the frequency of obstructive events associated with at least a 4% drop in oxyhemoglobin saturation is independently associated with cardiovascular disease, whereas events with lesser degrees of desaturation are not [150], raising the possibility that the intermittent pattern of hypoxia is important in disease pathogenesis.
However, it is possible that the effects of hypoxia are not entirely harmful: Lavie and Lavie hypothesized that hypoxic “preconditioning” might exert a protective effect against ischemic myocardial injury [90] and one group has described increased coronary vessel collateralization in association with OSA in patients with total coronary artery occlusion [194].
Arousals typically accompany each apneic event in both OSA and CSA. In OSA, arousals are critical to the opening of the upper airway and resumption of ventilation; in CSA, they occur after ventilation has resumed and can contribute to ventilatory control instability [130, 144, 223]. Arousals may contribute to post-apneic surges in HR and BP [66, 133, 205], sympathetic nervous system activation, and catecholamine release [63, 129].
Negative intrathoracic pressure is a result of the patient’s futile inspiratory efforts against a collapsed upper airway and can reach levels as low as −147 cmH2O [198]. Exaggerated negative intrathoracic pressure of a lesser magnitude is also observed during CSA due to pulmonary congestion and reduced lung compliance in conjunction with the increased respiratory efforts accompanying hyperpnea. Negative intrathoracic pressure increases venous return to the right heart, leading to distension of the right ventricle and leftward shift of the interventricular septum during diastole [96], which reduces LV preload. Negative intrathoracic pressure also increases LV transmural pressure by increasing the difference between extracardiac and intracardiac pressures [24, 133, 215]. The consequent increase in systolic wall stress increases afterload. In patients with coexistent cardiac disease, these effects are magnified [16]. This increased mechanical stretch and wall tension of the myocardium may predispose to ventricular hypertrophy, or ventricular arrhythmias through electro-mechanical coupling [95, 162].
Increases in sympathetic nerve activity have been demonstrated during sleep in patients with OSA compared to controls [61, 191], a phenomenon that is attenuated under hyperoxic conditions [94]. Elevations in sympathetic nerve activity during obstructive apneas are largely responsible for the characteristic surges in HR and BP that typically occur shortly following apnea termination [61, 156, 202]. These repetitive surges in BP oppose the usual fall that accompanies normal sleep and may be responsible in many cases for the phenomenon of “non-dipping” of the nocturnal BP profile [146].
Hypoxia is an obvious candidate for the sympatho-excitation that accompanies OSA. Through stimulation of the peripheral chemoreceptors, sympathetic vasomotor outflow is increased [192]. Therefore, despite hypoxia causing vasodilation through local autoregulation, peripheral vasoconstriction actually occurs in most vascular beds [104]. The effect of hypoxia on HR is similarly two-pronged. In the absence of the normal ventilatory response to hypoxia, peripheral chemoreceptor stimulation leads to vagally mediated bradycardia [13, 116]. This response may be relevant to bradycardic episodes during apneas. In contrast, in the presence of the normal ventilatory response to hypoxia, tachycardia is observed, owing to lung inflation reflexes that inhibit cardiac vagal efferents, permitting cardiac sympathetic activity to remain unopposed [115].
While hypoxic stimulation of the peripheral chemoreceptors leads to sympatho-excitation, the act of respiration is itself sympatho-inhibitory. The muscle sympathetic nerve activity (MSNA) response to hypoxia is markedly potentiated by the absence of breathing [192]. Respiration, while diminishing the sympathetic response to hypoxia, does not eliminate it entirely. Therefore, hypoxia is sympatho-excitatory whether breathing is present or absent, with the magnitude of the effect being greater during apnea. Since patients with CHF and CSA display an increased ventilatory response to peripheral chemoreceptor stimulation [145, 189], it is possible that they might also have an exaggerated sympathetic response to hypoxia. However, the chemoreflex response to hypoxia cannot be entirely responsible for the acute autonomic effects of sleep apnea, since elimination of hypoxia only modestly dampens the HR and BP oscillations that accompany OSA and CSA [52, 97].
The MSNA response to hypoxia and the degree of its inhibition by respiration may be further modified by the level of PaCO2. Hypercapnia itself causes increased ventilation, tachycardia, increased cardiac output, and BP. Sympathetic vasoconstrictor activity is increased, which is opposed by the direct vasodilatory action of CO2 [155]. Indeed, hypercapnia is a more potent stimulus for sympatho-excitation than hypoxia, in the sense that for equivalent increases in minute ventilation, greater MSNA is observed with hypercapnia than hypoxia, and the sympatho-inhibitory effect of respiration is less effective [192]. This observation suggests that hypoxia and hypercapnia do not cause sympatho-excitation solely through generation of central respiratory drive, but must also be able to influence the vasomotor centers independently. Combined hypoxia and hypercapnia have a synergistic effect on both sympathetic activity and ventilation and result in a more marked rise in BP.
Chemoreceptor reflexes may be of particular importance in the setting of CHF. Patients with CHF and CSA are known to have increased chemoreceptor sensitivity [73, 189, 219], and an increased sympathetic response to CO2 has also been observed [128]. Thus, the hyperpneic phase of CSA, which is a manifestation of intense chemostimulation by CO2, might be expected to exert considerable sympatho-excitatory effects.
Chronic effects
In recent years, it has become increasingly appreciated that sleep apnea represents not only an acute physiologic insult, but also exerts chronic effects that may promote a number of disease states, particularly cardiovascular disease. While a full discussion of the chronic effects of sleep apnea is beyond the scope of this paper, we will briefly describe some of the most well-accepted pathophysiology, including endothelial dysfunction and atherosclerosis, cardiac remodeling, and neurohormonal dysfunction.
The intermittent hypoxia accompanying sleep apnea may be viewed as essentially a form of ischemia–reperfusion type injury that may have a direct effect on the myocardium or vasculature, an effect exacerbated by hypercapnia and acidosis [42]. Ischemia–reperfusion injury is known to initiate oxidative stress via the production of reactive oxygen species and is recognized to play an important role in the genesis of endothelial dysfunction, via inactivation of nitric oxide [33]. There is increasing evidence that such vascular wall inflammation plays a key role in the pathogenesis of vascular disease and that endothelial dysfunction is a precursor of the atherosclerotic process [160].
The downstream effects of sleep apnea including hypoxia, sympathetic activation, systemic inflammation, production of reactive oxygen species, endothelial dysfunction, and negative intrathoracic pressures provide the basis for a cascade of events that could initiate atherogenesis [221]. In the case of the carotid arteries, which are adjacent to the upper airway, there is also some suggestion that acoustic vibration from snoring may also predispose to atherosclerosis [27]. Evidence suggests that OSA is an independent predictor of endothelial dysfunction [81] and that an imbalance occurs between the vasoconstrictive and vasorelaxant factors with improvement following treatment with CPAP therapy [70, 169]. More recently, direct evidence of endothelial dysfunction and inflammation has been demonstrated in vivo in the vascular endothelium of patients with OSA [77] along with increased carotid intima-media thickness [101]. The role of hypoxia is further supported by Savransky and colleagues who demonstrated induction of atherosclerosis in mice exposed to intermittent hypoxia [167]. Treatment of OSA with CPAP has been found to reverse early atherosclerotic lesions in humans, supporting a causal relationship [39]. OSA may also contribute to atherosclerosis indirectly by causing systolic hypertension, insulin resistance, and impaired lipid metabolism [99, 151].
Cardiac remodeling may be a long-term consequence of OSA. Animal models of chronic intermittent hypoxia induce LV hypertrophy and global LV dysfunction independent of elevations in blood pressure [44, 45]. In a canine model of chronic OSA, acute sleep-related obstructive events were associated with increased LV afterload and decreased fractional shortening, which chronically lead to sustained decreases in LV systolic performance [140]. In these animal models, the LV dysfunction is attributable to cardiomyocyte hypertrophy, apoptosis, and altered gene profile expression [23, 25, 44, 45]. Furthermore, oxidative stress and cytokines are implicated in the pathophysiology of intermittent hypoxia-induced LV remodeling [12, 58].
The interpretation of human studies of cardiac remodeling in the presence of OSA has been obscured by such confounding factors as the presence of cardiac medications, hypertension, or other diseases that could affect LV function, incomplete or varying methodology in the assessment of echocardiographic parameters [6, 50, 131, 137], and lack of an adequately matched control group [40, 50, 182]. However, both hemodynamic load and neurohormonal activation are known mechanisms that predispose to cardiac remodeling [30, 136], and these are present in SDB. In general, the majority of the studies have favored a higher prevalence of LV hypertrophy in OSA patients, especially in those with higher AHI [60, 132]. In nonobese children with OSA, there was an 11-fold increased risk for LV hypertrophy and 83% had eccentric hypertrophy. Those with OSA were also more likely to have right ventricular dysfunction [2]. In contrast, two studies did not find any relationship between LV hypertrophy and OSA, although differences in the calculation of the LV mass may account for some of these differences [32, 131].
Studies examining systolic and diastolic dysfunction in OSA in the absence of other underlying cardiac disease and the effect of CPAP therapy are also conflicting. Most of these studies are uncontrolled and small cross-sectional analyses. One prospective cohort study of 169 patients found systolic dysfunction in 7.7% of OSA patients as assessed by radionuclide imaging. Ischemic cardiac disease was unlikely as there were no segmental LV wall motion abnormalities. However, 69% of these individuals were obese and 54% had hypertension. Nonetheless, normalization of dysfunction following therapy with CPAP was seen in all of the patients who had follow-up imaging [89]. Improvements in early systolic dysfunction manifesting as a reduced cardiac output during exercise in subjects with OSA has also been described following treatment with CPAP [1].
The evidence for OSA contributing to diastolic dysfunction is more robust. Diastolic dysfunction has been associated with moderate to severe OSA in a number of studies [6, 40, 50, 137]. In two studies, OSA independent of obesity was associated with increased left atrial (LA) size as well as impaired LV diastolic function [137, 159]. The authors propose that chronic diastolic dysfunction may cause increased LA size and predispose to atrial fibrillation [79]. The most frequent abnormality observed in these studies is impaired isovolumic relaxation time and mitral deceleration time with a tendency to a higher LV mass, posterior wall, and interventricular septal thickness in OSA subjects. That diastolic dysfunction is associated independently with OSA is suggested by the reversal of some of these changes on application of CPAP [6, 40, 137, 182].
OSA and CSA are also associated with chronic abnormalities of cardiovascular autonomic regulation. It is important to recognize that these abnormalities are not limited to sleep, but carry over into the daytime. Patients with OSA have higher daytime MSNA compared to controls matched for age, sex and body mass index [20, 127]. Treatment of OSA either by tracheostomy [46] or CPAP leads to a reduction in overnight urinary catecholamine levels and daytime MSNA. However, lowering of MSNA with CPAP may take several months to occur, suggesting that the sympatho-excitatory effects of OSA are chronic in nature, and not immediately or easily reversible [59, 126, 210].
While it remains unclear whether OSA can predispose to the development of LV systolic dysfunction in the absence of other cardiac disease, there is evidence that OSA and CSA have particular importance in the setting of established CHF. Patients with the combination of CHF and OSA have higher daytime MSNA than controls matched for age and ejection fraction [193], a finding that portends higher mortality. There is intriguing evidence that the CHF state may actually alter the sympathetic response to obstructive apneas: patients with CHF have a higher MSNA response to simulated obstructive apneas (Mueller maneuvers) than to simple breath holds, whereas healthy controls have similar MSNA responses to both maneuvers [18]. If so, OSA and CHF may act synergistically to undermine normal autonomic regulation. Treatment of OSA for 1 month with CPAP has been reported in a randomized controlled trial to improve LV ejection fraction [80] and to reduce both daytime MSNA and BP, compared to an untreated group [207].
Surprisingly, given the consistency of the studies using MSNA, urinary norepinephrine has not been found to be elevated in CHF patients with OSA compared to those without [188], and although norepinephrine spillover rates are exceedingly high in patients with CHF and CSA, this appears to be a consequence of CHF severity and not the severity of CSA [109]. Nonetheless, treatment of OSA with CPAP for 3 months in the setting of heart failure significantly reduced urinary norepinephrine compared to untreated controls [108].
Conclusion
OSA is very common in the general population and is characterized by ineffective inspiratory efforts against a collapsed upper airway during sleep. It is a state-dependent phenomenon. Although collapse occurs mainly in patients who have narrowing at the level of the velopharynx and oropharynx, predisposing anatomy is not sufficient to cause obstruction without the withdrawal of pharyngeal dilator activity during sleep. CSA is a manifestation of ventilatory instability due to some combination of increased chemoreflex loop gain, circulatory feedback delay, or sleep–wake transitions altering the state-specific control of breathing. While both OSA and CSA arise from different pathophysiology, it has become increasingly clear that both apnea types often coexist in the same patient, that one can predispose to the other, and that the two are not as distinct as previously thought. Withdrawal of ventilatory drive as what occurs during central apnea can cause collapse of the upper airway and obstruction, whereas the ventilatory overshoot that accompanies the termination of obstructive apneas can predispose to hypocapnia and central apneas.
Both OSA and CSA exert a number of acute potentially deleterious effects including intermittent hypoxia, arousals from sleep, and swings in negative intrathoracic pressure, which in turn can lead to chronic physiologic consequences such as autonomic dysregulation, endothelial dysfunction, and cardiac remodeling. Only through understanding of the relevant pathophysiological mechanisms through which OSA and CSA arise and lead to cardiovascular disease will there be sufficient knowledge to develop novel and better approaches to management.
References
Alonso-Fernandez A, Garcia-Rio F, Arias MA, Mediano O, Pino JM, Martinez I, Villamor J (2006) Obstructive sleep apnoea–hypoapnoea syndrome reversibly depresses cardiac response to exercise. Eur Heart J 27:207–215
Amin RS, Kimball TR, Bean JA, Jeffries JL, Willging JP, Cotton RT, Witt SA, Glascock BJ, Daniels SR (2002) Left ventricular hypertrophy and abnormal ventricular geometry in children and adolescents with obstructive sleep apnea. Am J Respir Crit Care Med 165:1395–1399
Anch AM, Remmers JE, Sauerland EK, Degroot WJ (1981) Oropharyngeal patency during walking and sleep in the Pickwickian syndrome: electromyographic activity of the tensor veli palatini. Electromyogr Clin Neurophysiol 21:317–330
Anch AM, Salamy JG, McCoy GF, Somerset JS (1982) Behaviorally signalled awakenings in relationship to duration of alpha activity. Psychophysiology 19:528–530
Andreas S, Hagenah G, Moller C, Werner GS, Kreuzer H (1996) Cheyne–Stokes respiration and prognosis in congestive heart failure. Am J Cardiol 78:1260–1264
Arias MA, Garcia-Rio F, Alonso-Fernandez A, Mediano O, Martinez I, Villamor J (2005) Obstructive sleep apnea syndrome affects left ventricular diastolic function: effects of nasal continuous positive airway pressure in men. Circulation 112:375–383
Arzt M, Young T, Finn L, Skatrud JB, Bradley TD (2005) Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med 172:1447–1451
Ayappa I, Rapoport DM (2003) The upper airway in sleep: physiology of the pharynx. Sleep Med Rev 7:9–33
Bacon WH, Turlot JC, Krieger J, Stierle JL (1990) Cephalometric evaluation of pharyngeal obstructive factors in patients with sleep apneas syndrome. Angle Orthod 60:115–122
Badr MS, Kawak A, Skatrud JB, Morrell MJ, Zahn BR, Babcock MA (1997) Effect of induced hypocapnic hypopnea on upper airway patency in humans during NREM sleep. Respir Physiol 110:33–45
Badr MS, Toiber F, Skatrud JB, Dempsey J (1995) Pharyngeal narrowing/occlusion during central sleep apnea. J Appl Physiol 78:1806–1815
Barth W, Deten A, Bauer M, Reinohs M, Leicht M, Zimmer HG (2000) Differential remodeling of the left and right heart after norepinephrine treatment in rats: studies on cytokines and collagen. J Mol Cell Cardiol 32:273–284
Bernthal T, Green W Jr, Revzin AM (1951) Role of the carotid body chemoreceptors in hypoxic cardiac acceleration. Proc Soc Exp Biol Med 143:361–372
Bonsignore MR, Romano S, Marrone O, Chiodi M, Bonsignore G (1997) Different heart rate patterns in obstructive apneas during NREM sleep. Sleep 20:1167–1174
Bradley TD, Floras JS (2009) Obstructive sleep apnoea and its cardiovascular consequences. Lancet 373:82–93
Bradley TD, Hall MJ, Ando S, Floras JS (2001) Hemodynamic effects of simulated obstructive apneas in humans with and without heart failure. Chest 119:1827–1835
Bradley TD, Phillipson EA (1992) Central sleep apnea. Clin Chest Med 13:493–505
Bradley TD, Tkacova R, Hall MJ, Ando S, Floras JS (2003) Augmented sympathetic neural response to simulated obstructive apnoea in human heart failure. Clin Sci (Lond) 104:231–238
Caballero P, Alvarez-Sala R, Garcia-Rio F, Prados C, Hernan MA, Villamor J, Alvarez-Sala JL (1998) CT in the evaluation of the upper airway in healthy subjects and in patients with obstructive sleep apnea syndrome. Chest 113:111–116
Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG (1993) Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest 103:1763–1768
Carrera M, Barbe F, Sauleda J, Tomas M, Gomez C, Agusti AG (1999) Patients with obstructive sleep apnea exhibit genioglossus dysfunction that is normalized after treatment with continuous positive airway pressure. Am J Respir Crit Care Med 159:1960–1966
Cassel W, Canisius S, Becker HF, Leistner S, Ploch T, Jerrentrup A, Vogelmeier C, Koehler U, Heitmann J (2011) A prospective polysomnographic study on the evolution of complex sleep apnoea. Eur Respir J 38:329–337
Chen L, Einbinder E, Zhang Q, Hasday J, Balke CW, Scharf SM (2005) Oxidative stress and left ventricular function with chronic intermittent hypoxia in rats. Am J Respir Crit Care Med 172:915–920
Chen L, Scharf SM (1997) Comparative hemodynamic effects of periodic obstructive and simulated central apneas in sedated pigs. J Appl Physiol 83:485–494
Chen L, Zhang J, Gan TX, Chen-Izu Y, Hasday JD, Karmazyn M, Balke CW, Scharf SM (2008) Left ventricular dysfunction and associated cellular injury in rats exposed to chronic intermittent hypoxia. J Appl Physiol 104:218–223
Cherniack NS, Longobardo GS (1973) Cheyne–Stokes breathing. An instability in physiologic control. N Engl J Med 288:952–957
Cho JG, Witting PK, Verma M, Wu BJ, Shanu A, Kairaitis K, Amis TC, Wheatley JR (2011) Tissue vibration induces carotid artery endothelial dysfunction: a mechanism linking snoring and carotid atherosclerosis? Sleep 34:751–757
Churchill ED, Cope O (1929) The rapid shallow breathing resulting from pulmonary congestion and edema. J Exp Med 49:531–537
Ciscar MA, Juan G, Martinez V, Ramon M, Lloret T, Minguez J, Armengot M, Marin J, Basterra J (2001) Magnetic resonance imaging of the pharynx in OSA patients and healthy subjects. Eur Respir J 17:79–86
Cohn JN, Ferrari R, Sharpe N (2000) Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. On behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 35:569–582
Datta AK, Shea SA, Horner RL, Guz A (1991) The influence of induced hypocapnia and sleep on the endogenous respiratory rhythm in humans [published erratum appears in J Physiol (Lond) 1991 Dec;444:778]. J Physiol 440:17–33
Davies RJ, Crosby J, Prothero A, Stradling JR (1994) Ambulatory blood pressure and left ventricular hypertrophy in subjects with untreated obstructive sleep apnoea and snoring, compared with matched control subjects, and their response to treatment. Clin Sci (Colch) 86:417–424
De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, Shin WS, Liao JK (1995) Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest 96:60–68
DeBacker WA, Verbraecken J, Willemen M, Wittesaele W, DeCock W, Van deHeyning P (1995) Central apnea index decreases after prolonged treatment with acetazolamide. Am J Respir Crit Care Med 151:87–91
deBerry-Borowiecki B, Kukwa A, Blanks RH (1988) Cephalometric analysis for diagnosis and treatment of obstructive sleep apnea. Laryngoscope 98:226–234
Dempsey JA (2005) Crossing the apnoeic threshold: causes and consequences. Exp Physiol 90:13–24
Dernaika T, Tawk M, Nazir S, Younis W, Kinasewitz GT (2007) The significance and outcome of continuous positive airway pressure-related central sleep apnea during split-night sleep studies. Chest 132:81–87
Do KL, Ferreyra H, Healy JF, Davidson TM (2000) Does tongue size differ between patients with and without sleep-disordered breathing? Laryngoscope 110:1552–1555
Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi GF (2007) Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 176:706–712
Dursunoglu N, Dursunoglu D, Ozkurt S, Kuru O, Gur S, Kiter G, Evyapan F (2007) Effects of CPAP on left ventricular structure and myocardial performance index in male patients with obstructive sleep apnoea. Sleep Med 8:51–59
Eldridge FL, Millhorn DE, Kiley JP, Waldrop TG (1985) Stimulation by central command of locomotion, respiration and circulation during exercise. Respir Physiol 59:313–337
Enson Y, Giuntini C, Lewis ML, Morris TQ, Ferrer MI, Harvey RM (1964) The influence of hydrogen ion concentration and hypoxia on the pulmonary circulation. J Clin Invest 43:1146–1162
Epstein LJ, Kristo D, Strollo PJ Jr, Friedman N, Malhotra A, Patil SP, Ramar K, Rogers R, Schwab RJ, Weaver EM, Weinstein MD (2009) Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 5:263–276
Fletcher EC, Bao G (1996) The rat as a model of chronic recurrent episodic hypoxia and effect upon systemic blood pressure. Sleep 19:S210–S212
Fletcher EC, Lesske J, Behm R, Miller CCd, Stauss H, Unger T (1992) Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol 72:1978–1984
Fletcher EC, Miller J, Schaaf JW, Fletcher JG (1987) Urinary catecholamines before and after tracheostomy in patients with obstructive sleep apnea and hypertension. Sleep 10:35–44
Fogel RB, Malhotra A, White DP (2004) Sleep. 2: pathophysiology of obstructive sleep apnoea/hypopnoea syndrome. Thorax 59:159–163
Friberg D, Ansved T, Borg K, Carlsson-Nordlander B, Larsson H, Svanborg E (1998) Histological indications of a progressive snorers disease in an upper airway muscle. Am J Respir Crit Care Med 157:586–593
Friedman O, Bradley TD, Chan CT, Parkes R, Logan AG (2010) Relationship between overnight rostral fluid shift and obstructive sleep apnea in drug-resistant hypertension. Hypertension 56:1077–1082
Fung JW, Li TS, Choy DK, Yip GW, Ko FW, Sanderson JE, Hui DS (2002) Severe obstructive sleep apnea is associated with left ventricular diastolic dysfunction. Chest 121:422–429
Guilleminault C, Connolly SJ, Winkle RA (1983) Cardiac arrhythmia and conduction disturbances during sleep in 400 patients with sleep apnea syndrome. Am J Cardiol 52:490–494
Guilleminault C, Connolly S, Winkle R, Melvin K, Tilkian A (1984) Cyclical variation of the heart rate in sleep apnoea syndrome. Mechanisms, and usefulness of 24 h electrocardiography as a screening technique. Lancet 1:126–131
Guilleminault C, Hill MH, Simmons FB, Powell N, Riley R, Stoohs R (1997) Passive constriction of the upper airway during central apneas: fiberoptic and EMG investigations. Respir Physiol 108:11–22
Guilleminault C, Partinen M, Hollman K, Powell N, Stoohs R (1995) Familial aggregates in obstructive sleep apnea syndrome. Chest 107:1545–1551
Guyton AC (1956) Basic oscillating mechanism of Cheyne–Stokes breathing. Am J Physiol 187:395–398
Hall MJ, Xie A, Rutherford R, Ando S, Floras JS, Bradley TD (1996) Cycle length of periodic breathing in patients with and without heart failure. Am J Respir Crit Care Med 154:376–381
Hanly PJ, Zuberi-Khokhar NS (1996) Increased mortality associated with Cheyne–Stokes respiration in patients with congestive heart failure. Am J Respir Crit Care Med 153:272–276
Hayashi T, Yamashita C, Matsumoto C, Kwak CJ, Fujii K, Hirata T, Miyamura M, Mori T, Ukimura A, Okada Y, Matsumura Y, Kitaura Y (2008) Role of gp91phox-containing NADPH oxidase in left ventricular remodeling induced by intermittent hypoxic stress. Am J Physiol Heart Circ Physiol 294:H2197–H2203
Hedner J, Darpo B, Ejnell H, Carlson J, Caidahl K (1995) Reduction in sympathetic activity after long-term CPAP treatment in sleep apnoea: cardiovascular implications. Eur Respir J 8:222–229
Hedner J, Ejnell H, Caidahl K (1990) Left ventricular hypertrophy independent of hypertension in patients with obstructive sleep apnoea. J Hypertens 8:941–946
Hedner J, Ejnell H, Sellgren J, Hedner T, Wallin G (1988) Is high and fluctuating muscle nerve sympathetic activity in the sleep apnoea syndrome of pathogenetic importance for the development of hypertension? J Hypertens Suppl 6:S529–S531
Hoffstein V, Zamel N, Phillipson EA (1984) Lung volume dependence of pharyngeal cross-sectional area in patients with obstructive sleep apnea. Am Rev Respir Dis 130:175–178
Horner RL, Brooks D, Kozar LF, Tse S, Phillipson EA (1995) Immediate effects of arousal from sleep on cardiac autonomic outflow in the absence of breathing in dogs. J Appl Physiol 79:151–162
Horner RL, Innes JA, Holden HB, Guz A (1991) Afferent pathway(s) for pharyngeal dilator reflex to negative pressure in man: a study using upper airway anaesthesia. J Physiol 436:31–44
Horner RL, Mohiaddin RH, Lowell DG, Shea SA, Burman ED, Longmore DB, Guz A (1989) Sites and sizes of fat deposits around the pharynx in obese patients with obstructive sleep apnoea and weight matched controls. Eur Respir J 2:613–622
Horner RL, Rivera MP, Kozar LF, Phillipson EA (2001) The ventilatory response to arousal from sleep is not fully explained by differences in CO(2) levels between sleep and wakefulness. J Physiol 534:881–890
Horner RL, Sanford LD, Pack AI, Morrison AR (1997) Activation of a distinct arousal state immediately after spontaneous awakening from sleep. Brain Res 778:127–134
Hudgel DW, Chapman KR, Faulks C, Hendricks C (1987) Changes in inspiratory muscle electrical activity and upper airway resistance during periodic breathing induced by hypoxia during sleep. Am Rev Respir Dis 135:899–906
Hwang JC, St John WM, Bartlett D Jr (1983) Respiratory-related hypoglossal nerve activity: influence of anesthetics. J Appl Physiol 55:785–792
Ip MS, Lam B, Chan LY, Zheng L, Tsang KW, Fung PC, Lam WK (2000) Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am J Respir Crit Care Med 162:2166–2171
Isono S, Feroah TR, Hajduk EA, Brant R, Whitelaw WA, Remmers JE (1997) Interaction of cross-sectional area, driving pressure, and airflow of passive velopharynx. J Appl Physiol 83:851–859
Issa FG, Sullivan CE (1984) Upper airway closing pressures in snorers. J Appl Physiol 57:528–535
Javaheri S (1999) A mechanism of central sleep apnea in patients with heart failure [see comments]. N Engl J Med 341:949–954
Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L, Roselle GA (1998) Sleep apnea in 81 ambulatory male patients with stable heart failure. Types and their prevalences, consequences, and presentations [see comments]. Circulation 97:2154–2159
Javaheri S, Parker TJ, Wexler L, Liming JD, Lindower P, Roselle GA (1996) Effect of theophylline on sleep-disordered breathing in heart failure. N Engl J Med 335:562–567
Javaheri S, Smith J, Chung E (2009) The prevalence and natural history of complex sleep apnea. J Clin Sleep Med 5:205–211
Jelic S, Padeletti M, Kawut SM, Higgins C, Canfield SM, Onat D, Colombo PC, Basner RC, Factor P, LeJemtel TH (2008) Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation 117:2270–2278
Jordan AS, McEvoy RD, Edwards JK, Schory K, Yang CK, Catcheside PG, Fogel RB, Malhotra A, White DP (2004) The influence of gender and upper airway resistance on the ventilatory response to arousal in obstructive sleep apnoea in humans. J Physiol 558:993–1004
Kanagala R, Murali NS, Friedman PA, Ammash NM, Gersh BJ, Ballman KV, Shamsuzzaman AS, Somers VK (2003) Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 107:2589–2594
Kaneko Y, Floras JS, Usui K, Plante J, Tkacova R, Kubo T, Ando S, Bradley TD (2003) Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med 348:1233–1241
Kato M, Roberts-Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, Somers VK (2000) Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 102:2607–2610
Khoo MC, Gottschalk A, Pack AI (1991) Sleep-induced periodic breathing and apnea: a theoretical study. J Appl Physiol 70:2014–2024
Khoo MC, Kronauer RE, Strohl KP, Slutsky AS (1982) Factors inducing periodic breathing in humans: a general model. J Appl Physiol 53:644–659
Kirkness JP, Christenson HK, Garlick SR, Parikh R, Kairaitis K, Wheatley JR, Amis TC (2003) Decreased surface tension of upper airway mucosal lining liquid increases upper airway patency in anaesthetised rabbits. J Physiol 547:603–611
Kohnlein T, Klante T, Elliott MW, Welte T (2001) Heart failure and central respiratory dysregulation. Cheyne–Stokes respiration during sleep in advanced left heart failure. Pneumologie 55:13–20
Kuna ST, Bedi DG, Ryckman C (1988) Effect of nasal airway positive pressure on upper airway size and configuration. Am Rev Respir Dis 138:969–975
Kuna ST, Brennick MJ (2002) Effects of pharyngeal muscle activation on airway pressure–area relationships. Am J Respir Crit Care Med 166:972–977
Kuna ST, Vanoye CR (1999) Mechanical effects of pharyngeal constrictor activation on pharyngeal airway function. J Appl Physiol 86:411–417
Laaban JP, Pascal-Sebaoun S, Bloch E, Orvoen-Frija E, Oppert JM, Huchon G (2002) Left ventricular systolic dysfunction in patients with obstructive sleep apnea syndrome. Chest 122:1133–1138
Lavie L, Lavie P (2006) Ischemic preconditioning as a possible explanation for the age decline relative mortality in sleep apnea. Med Hypotheses 66:1069–1073
Legato MJ (1997) Gender-specific aspects of obesity. Int J Fertil Womens Med 42:184–197
Lehman S, Antic NA, Thompson C, Catcheside PG, Mercer J, McEvoy RD (2007) Central sleep apnea on commencement of continuous positive airway pressure in patients with a primary diagnosis of obstructive sleep apnea–hypopnea. J Clin Sleep Med 3:462–466
Leite JJ, Mansur AJ, de Freitas HF, Chizola PR, Bocchi EA, Terra-Filho M, Neder JA, Lorenzi-Filho G (2003) Periodic breathing during incremental exercise predicts mortality in patients with chronic heart failure evaluated for cardiac transplantation. J Am Coll Cardiol 41:2175–2181
Leuenberger U, Jacob E, Sweer L, Waravdekar N, Zwillich C, Sinoway L (1995) Surges of muscle sympathetic nerve activity during obstructive apnea are linked to hypoxemia. J Appl Physiol 79:581–588
Leung RS (2009) Sleep-disordered breathing: autonomic mechanisms and arrhythmias. Prog Cardiovasc Dis 51:324–338
Leung RS, Bradley TD (2001) Sleep apnea and cardiovascular disease. Am J Respir Crit Care Med 164:2147–2165
Leung RS, Floras JS, Lorenzi-Filho G, Rankin F, Picton P, Bradley TD (2003) Influence of Cheyne–Stokes respiration on cardiovascular oscillations in heart failure. Am J Respir Crit Care Med 167:1534–1539
Leung RS, Lorenzi-Filho G, Floras JS, Bradley TD (2000) Entrainment of blood pressure and heart rate by Cheyne–Stokes respiration in patients with congestive heart failure. Am J Respir Crit Care Med 161:A865
Libby P (2000) Changing concepts of atherogenesis. J Intern Med 247:349–358
Lieberman DE, McCarthy RC (1999) The ontogeny of cranial base angulation in humans and chimpanzees and its implications for reconstructing pharyngeal dimensions. J Hum Evol 36:487–517
Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M (2007) Prediction of clinical cardiovascular events with carotid intima-media thickness: a systematic review and meta-analysis. Circulation 115:459–467
Lorenzi-Filho G, Rankin F, Bies I, Douglas Bradley T (1999) Effects of inhaled carbon dioxide and oxygen on Cheyne–Stokes respiration in patients with heart failure. Am J Respir Crit Care Med 159:1490–1498
Lowe AA, Fleetham JA, Adachi S, Ryan CF (1995) Cephalometric and computed tomographic predictors of obstructive sleep apnea severity. Am J Orthod Dentofacial Orthoped 107:589–595
Lugliani R, Whipp BJ, Wasserman K (1973) A role for the carotid body in cardiovascular control in man. Chest 63:744–750
Malhotra A, Berry RB, White DP (2004) Central sleep apnea. In: Carney PR, Berry RB, Geyer JD (eds) Clinical sleep disorders. Lippincott, Williams and Wilkins, Philadelphia, pp 331–346
Malhotra A, Huang Y, Fogel RB, Pillar G, Edwards JK, Kikinis R, Loring SH, White DP (2002) The male predisposition to pharyngeal collapse: importance of airway length. Am J Respir Crit Care Med 166:1388–1395
Malhotra A, Pillar G, Fogel RB, Edwards JK, Ayas N, Akahoshi T, Hess D, White DP (2002) Pharyngeal pressure and flow effects on genioglossus activation in normal subjects. Am J Respir Crit Care Med 165:71–77
Mansfield DR, Gollogly NC, Kaye DM, Richardson M, Bergin P, Naughton MT (2004) Controlled trial of continuous positive airway pressure in obstructive sleep apnea and heart failure. Am J Respir Crit Care Med 169:361–366
Mansfield D, Kaye DM, Brunner La Rocca H, Solin P, Esler MD, Naughton MT (2003) Raised sympathetic nerve activity in heart failure and central sleep apnea is due to heart failure severity. Circulation 107:1396–1400
Marcus CL, Keens TG, Bautista DB, von Pechmann WS, Ward SL (1991) Obstructive sleep apnea in children with Down syndrome. Pediatrics 88:132–139
Marin JM, Carrizo SJ, Vicente E, Agusti AG (2005) Long-term cardiovascular outcomes in men with obstructive sleep apnoea–hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365:1046–1053
Mathew OP (1984) Upper airway negative-pressure effects on respiratory activity of upper airway muscles. J Appl Physiol 56:500–505
Mathew OP, Abu-Osba YK, Thach BT (1982) Influence of upper airway pressure changes on genioglossus muscle respiratory activity. J Appl Physiol 52:438–444
Mathur R, Douglas NJ (1995) Family studies in patients with the sleep apnea–hypopnea syndrome. Ann Intern Med 122:174–178
MdB D, Scott MJ (1958) The effects of stimulation of the carotid body chemoreceptors on heart rate in the dog. J Physiol 144:148–166
MdB D, Scott MJ (1963) The cardiovascular responses to stimulation of the carotid chemoreceptors in the dog. J Physiol 165:179–197
Miles PG, Vig PS, Weyant RJ, Forrest TD, Rockette HE Jr (1996) Craniofacial structure and obstructive sleep apnea syndrome—a qualitative analysis and meta-analysis of the literature. Am J Orthod Dentofacial Orthoped 109:163–172
Millar TW, Hanly P, Kryger MH (1992) Short technical note: quantification of periodic breathing: preliminary studies. Sleep 15:364–370
Millman RP, Carlisle CC, McGarvey ST, Eveloff SE, Levinson PD (1995) Body fat distribution and sleep apnea severity in women. Chest 107:362–366
Morgenthaler TI, Kagramanov V, Hanak V, Decker PA (2006) Complex sleep apnea syndrome: is it a unique clinical syndrome? Sleep 29:1203–1209
Morikawa S, Safar P, Decarlo J (1961) Influence of the headjaw position upon upper airway patency. Anesthesiology 22:265–270
Morrell MJ, Arabi Y, Zahn BR, Meyer KC, Skatrud JB, Badr MS (2002) Effect of surfactant on pharyngeal mechanics in sleeping humans: implications for sleep apnoea. Eur Respir J 20:451–457
Mortara A, Sleight P, Pinna GD, Maestri R, Capomolla S, Febo O, La Rovere MT, Cobelli F (1999) Association between hemodynamic impairment and Cheyne–Stokes respiration and periodic breathing in chronic stable congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 84:900–904
Mortimore IL, Marshall I, Wraith PK, Sellar RJ, Douglas NJ (1998) Neck and total body fat deposition in nonobese and obese patients with sleep apnea compared with that in control subjects. Am J Respir Crit Care Med 157:280–283
Nakao K, Ohgushi M, Yoshimura M, Morooka K, Okumura K, Ogawa H, Kugiyama K, Oike Y, Fujimoto K, Yasue H (1997) Hyperventilation as a specific test for diagnosis of coronary artery spasm. Am J Cardiol 80:545–549
Narkiewicz K, Kato M, Phillips BG, Pesek CA, Davison DE, Somers VK (1999) Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation 100:2332–2335
Narkiewicz K, Montano N, Cogliati C, van de Borne PJ, Dyken ME, Somers VK (1998) Altered cardiovascular variability in obstructive sleep apnea. Circulation 98:1071–1077
Narkiewicz K, Pesek CA, van de Borne PJ, Kato M, Somers VK (1999) Enhanced sympathetic and ventilatory responses to central chemoreflex activation in heart failure. Circulation 100:262–267
Naughton MT, Benard DC, Liu PP, Rutherford R, Rankin F, Bradley TD (1995) Effects of nasal CPAP on sympathetic activity in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med 152:473–479
Naughton M, Benard D, Tam A, Rutherford R, Bradley TD (1993) Role of hyperventilation in the pathogenesis of central sleep apneas in patients with congestive heart failure [see comments]. Am Rev Respir Dis 148:330–338
Niroumand M, Kuperstein R, Sasson Z, Hanly PJ (2001) Impact of obstructive sleep apnea on left ventricular mass and diastolic function. Am J Respir Crit Care Med 163:1632–1636
Noda A, Okada T, Yasuma F, Nakashima N, Yokota M (1995) Cardiac hypertrophy in obstructive sleep apnea syndrome. Chest 107:1538–1544
O’Donnell CP, Ayuse T, King ED, Schwartz AR, Smith PL, Robotham JL (1996) Airway obstruction during sleep increases blood pressure without arousal. J Appl Physiol 80:773–781
Onal E, Burrows DL, Hart RH, Lopata M (1986) Induction of periodic breathing during sleep causes upper airway obstruction in humans. J Appl Physiol 61:1438–1443
Onal E, Lopata M, O’Connor TD (1981) Diaphragmatic and genioglossal electromyogram responses to CO2 rebreathing in humans. J Appl Physiol 50:1052–1055
Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA (2006) Controversies in ventricular remodelling. Lancet 367:356–367
Otto ME, Belohlavek M, Romero-Corral A, Gami AS, Gilman G, Svatikova A, Amin RS, Lopez-Jimenez F, Khandheria BK, Somers VK (2007) Comparison of cardiac structural and functional changes in obese otherwise healthy adults with versus without obstructive sleep apnea. Am J Cardiol 99:1298–1302
Pae EK, Lowe AA, Fleetham JA (1997) A role of pharyngeal length in obstructive sleep apnea patients. Am J Orthod Dentofacial Orthoped 111:12–17
Paintal AS (1969) Mechanism of stimulation of type J pulmonary receptors. J Physiol 203:511–532
Parker JD, Brooks D, Kozar LF, Render-Teixeira CL, Horner RL, Douglas Bradley T, Phillipson EA (1999) Acute and chronic effects of airway obstruction on canine left ventricular performance. Am J Respir Crit Care Med 160:1888–1896
Peppard PE, Ward NR, Morrell MJ (2009) The impact of obesity on oxygen desaturation during sleep-disordered breathing. Am J Respir Crit Care Med 180:788–793
Peppard PE, Young T, Palta M, Skatrud J (2000) Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 342:1378–1384
Phillipson EA (1978) Control of breathing during sleep. Am Rev Respir Dis 118:909–939
Phillipson EA, Bowes G (1986) Control of breathing during sleep. In: Cherniack NS, Widdicombe JG (eds) Handbook of physiology: vol. 2, control of breathing. Williams and Wilkins, Bethesda, pp 649–689
Ponikowski P, Anker SD, Chua TP, Francis D, Banasiak W, Poole-Wilson PA, Coats AJ, Piepoli M (1999) Oscillatory breathing patterns during wakefulness in patients with chronic heart failure: clinical implications and role of augmented peripheral chemosensitivity. Circulation 100:2418–2424
Portaluppi F, Provini F, Cortelli P, Plazzi G, Bertozzi N, Manfredini R, Fersini C, Lugaresi E (1997) Undiagnosed sleep-disordered breathing among male nondippers with essential hypertension. J Hypertens 15:1227–1233
Pracharktam N, Hans MG, Strohl KP, Redline S (1994) Upright and supine cephalometric evaluation of obstructive sleep apnea syndrome and snoring subjects. Angle Orthod 64:63–73
Pryor WW (1951) Cheyne–Stokes respiration in patients with cardiac enlargement and prolonged circulation time. Circulation 4:233–238
Punjabi NM (2008) The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc 5:136–143
Punjabi NM, Newman AB, Young TB, Resnick HE, Sanders MH (2008) Sleep-disordered breathing and cardiovascular disease: an outcome-based definition of hypopneas. Am J Respir Crit Care Med 177:1150–1155
Punjabi NM, Polotsky VY (2005) Disorders of glucose metabolism in sleep apnea. J Appl Physiol 99:1998–2007
Redolfi S, Arnulf I, Pottier M, Bradley TD, Similowski T (2011) Effects of venous compression of the legs on overnight rostral fluid shift and obstructive sleep apnea. Respir Physiol Neurobiol 175:390–393
Redolfi S, Yumino D, Ruttanaumpawan P, Yau B, Su MC, Lam J, Bradley TD (2009) Relationship between overnight rostral fluid shift and obstructive sleep apnea in nonobese men. Am J Respir Crit Care Med 179:241–246
Remmers JE, deGroot WJ, Sauerland EK, Anch AM (1978) Pathogenesis of upper airway occlusion during sleep. J Appl Physiol 44:931–938
Richardson DW, Wasserman AJ, Patterson JL Jr (1961) General and regional circulatory responses to change in blood pH and carbon dioxide tension. J Clin Invest 40:31–43
Ringler J, Basner RC, Shannon R, Schwartzstein R, Manning H, Weinberger SE, Weiss JW (1990) Hypoxemia alone does not explain blood pressure elevations after obstructive apneas. J Appl Physiol 69:2143–2148
Roberts AM, Bhattacharya J, Schultz HD, Coleridge HM, Coleridge JC (1986) Stimulation of pulmonary vagal afferent C-fibers by lung edema in dogs. Circ Res 58:512–522
Roberts JL, Reed WR, Mathew OP, Menon AA, Thach BT (1985) Assessment of pharyngeal airway stability in normal and micrognathic infants. J Appl Physiol 58:290–299
Romero-Corral A, Somers VK, Pellikka PA, Olson EJ, Bailey KR, Korinek J, Orban M, Sierra-Johnson J, Kato M, Amin RS, Lopez-Jimenez F (2007) Decreased right and left ventricular myocardial performance in obstructive sleep apnea. Chest 132:1863–1870
Ross R (1999) Atherosclerosis—an inflammatory disease. N Engl J Med 340:115–126
Rowley JA, Permutt S, Willey S, Smith PL, Schwartz AR (1996) Effect of tracheal and tongue displacement on upper airway airflow dynamics. J Appl Physiol 80:2171–2178
Ryan CM, Juvet S, Leung R, Bradley TD (2008) Timing of nocturnal ventricular ectopy in heart failure patients with sleep apnea. Chest 133(4):934–940
Ryan CF, Lowe AA, Li D, Fleetham JA (1991) Three-dimensional upper airway computed tomography in obstructive sleep apnea. A prospective study in patients treated by uvulopalatopharyngoplasty. Am Rev Respir Dis 144:428–432
Safar P, Escarraga LA, Chang F (1959) Upper airway obstruction in the unconscious patient. J Appl Physiol 14:760–764
Salloum A, Rowley JA, Mateika JH, Chowdhuri S, Omran Q, Badr MS (2010) Increased propensity for central apnea in patients with obstructive sleep apnea: effect of nasal continuous positive airway pressure. Am J Respir Crit Care Med 181:189–193
Sands SA, Edwards BA, Kee K, Turton A, Skuza EM, Roebuck T, O’Driscoll DM, Hamilton GS, Naughton MT, Berger PJ (2011) Loop gain as a means to predict a positive airway pressure suppression of Cheyne–Stokes respiration in heart failure patients. Am J Respir Crit Care Med http://ajrccm.atsjournals.org/cgi/content/abstract/201103-0577OCv1
Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, Polotsky VY (2007) Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med 175:1290–1297
Schotland HM, Insko EK, Schwab RJ (1999) Quantitative magnetic resonance imaging demonstrates alterations of the lingual musculature in obstructive sleep apnea. Sleep 22:605–613
Schulz R, Schmidt D, Blum A, Lopes-Ribeiro X, Lucke C, Mayer K, Olschewski H, Seeger W, Grimminger F (2000) Decreased plasma levels of nitric oxide derivatives in obstructive sleep apnoea: response to CPAP therapy. Thorax 55:1046–1051
Schwab RJ (1998) Upper airway imaging. Clin Chest Med 19:33–54
Schwab RJ, Gefter WB, Hoffman EA, Gupta KB, Pack AI (1993) Dynamic upper airway imaging during awake respiration in normal subjects and patients with sleep disordered breathing. Am Rev Respir Dis 148:1385–1400
Schwab RJ, Gefter WB, Pack AI, Hoffman EA (1993) Dynamic imaging of the upper airway during respiration in normal subjects. J Appl Physiol 74:1504–1514
Schwab RJ, Gupta KB, Gefter WB, Metzger LJ, Hoffman EA, Pack AI (1995) Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med 152:1673–1689
Schwab RJ, Pasirstein M, Pierson R, Mackley A, Hachadoorian R, Arens R, Maislin G, Pack AI (2003) Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am J Respir Crit Care Med 168:522–530
Schwartz AR, Smith PL, Wise RA, Gold AR, Permutt S (1988) Induction of upper airway occlusion in sleeping individuals with subatmospheric nasal pressure. J Appl Physiol 64:535–542
Series F, Cote C, Simoneau JA, Gelinas Y, St Pierre S, Leclerc J, Ferland R, Marc I (1995) Physiologic, metabolic, and muscle fiber type characteristics of musculus uvulae in sleep apnea hypopnea syndrome and in snorers. J Clin Invest 95:20–25
Serizawa T, Vogel WM, Apstein CS, Grossman W (1981) Comparison of acute alterations in left ventricular relaxation and diastolic chamber stiffness induced by hypoxia and ischemia. Role of myocardial oxygen supply–demand imbalance. J Clin Invest 68:91–102
Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Javier Nieto F, O’Connor GT, Boland LL, Schwartz JE, Samet JM (2001) Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med 163:19–25
Shelton KE, Gay SB, Hollowell DE, Woodson H, Suratt PM (1993) Mandible enclosure of upper airway and weight in obstructive sleep apnea. Am Rev Respir Dis 148:195–200
Shepard JW Jr, Garrison MW, Grither DA, Dolan GF (1985) Relationship of ventricular ectopy to oxyhemoglobin desaturation in patients with obstructive sleep apnea. Chest 88:335–340
Shepard JW Jr, Thawley SE (1989) Evaluation of the upper airway by computerized tomography in patients undergoing uvulopalatopharyngoplasty for obstructive sleep apnea. Am Rev Respir Dis 140:711–716
Shivalkar B, Van de Heyning C, Kerremans M, Rinkevich D, Verbraecken J, De Backer W, Vrints C (2006) Obstructive sleep apnea syndrome: more insights on structural and functional cardiac alterations, and the effects of treatment with continuous positive airway pressure. J Am Coll Cardiol 47:1433–1439
Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD (1999) Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure [see comments]. Am J Respir Crit Care Med 160:1101–1106
Skatrud JB, Dempsey JA (1983) Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol 55:813–822
Skatrud JB, Dempsey JA (1985) Airway resistance and respiratory muscle function in snorers during NREM sleep. J Appl Physiol 59:328–335
Smith PL, Wise RA, Gold AR, Schwartz AR, Permutt S (1988) Upper airway pressure–flow relationships in obstructive sleep apnea. J Appl Physiol 64:789–795
Solin P, Bergin P, Richardson M, Kaye DM, Walters EH, Naughton MT (1999) Influence of pulmonary capillary wedge pressure on central apnea in heart failure. Circulation 99:1574–1579
Solin P, Kaye DM, Little PJ, Bergin P, Richardson M, Naughton MT (2003) Impact of sleep apnea on sympathetic nervous system activity in heart failure. Chest 123:1119–1126
Solin P, Roebuck T, Johns DP, Haydn Walters E, Naughton MT (2000) Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med 162:2194–2200
Solin P, Roebuck T, Swieca J, Walters EH, Naughton MT (1998) Effects of cardiac dysfunction on non-hypercapnic central sleep apnea. Chest 113:104–110
Somers VK, Dyken ME, Clary MP, Abboud FM (1995) Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 96:1897–1904
Somers VK, Mark AL, Zavala DC, Abboud FM (1989) Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. J Appl Physiol 67:2101–2106
Spaak J, Egri ZJ, Kubo T, Yu E, Ando S, Kaneko Y, Usui K, Bradley TD, Floras JS (2005) Muscle sympathetic nerve activity during wakefulness in heart failure patients with and without sleep apnea. Hypertension 46:1327–1332
Steiner S, Schueller PO, Schulze V, Strauer BE (2010) Occurrence of coronary collateral vessels in patients with sleep apnea and total coronary occlusion. Chest 137:516–520
Su MC, Chiu KL, Ruttanaumpawan P, Shiota S, Yumino D, Redolfi S, Haight JS, Yau B, Lam J, Bradley TD (2009) Difference in upper airway collapsibility during wakefulness between men and women in response to lower-body positive pressure. Clin Sci (Lond) 116:713–720
Sun SY, Wang W, Zucker IH, Schultz HD (1999) Enhanced peripheral chemoreflex function in conscious rabbits with pacing-induced heart failure. J Appl Physiol 86:1264–1272
Sun SY, Wang W, Zucker IH, Schultz HD (1999) Enhanced activity of carotid body chemoreceptors in rabbits with heart failure: role of nitric oxide. J Appl Physiol 86:1273–1282
Suzuki M, Ogawa H, Okabe S, Horiuchi A, Okubo M, Ikeda K, Hida W, Kobayashi T (2005) Digital recording and analysis of esophageal pressure for patients with obstructive sleep apnea–hypopnea syndrome. Sleep Breath 9:64–72
Tangel DJ, Mezzanotte WS, White DP (1991) Influence of sleep on tensor palatini EMG and upper airway resistance in normal men. J Appl Physiol 70:2574–2581
Thach BT, Schefft GL, Pickens DL, Menon AP (1989) Influence of upper airway negative pressure reflex on response to airway occlusion in sleeping infants. J Appl Physiol 67:749–755
Thut DC, Schwartz AR, Roach D, Wise RA, Permutt S, Smith PL (1993) Tracheal and neck position influence upper airway airflow dynamics by altering airway length. J Appl Physiol 75:2084–2090
Tilkian AG, Guilleminault C, Schroeder JS, Lehrman KL, Simmons FB, Dement WC (1976) Hemodynamics in sleep-induced apnea. Studies during wakefulness and sleep. Ann Intern Med 85:714–719
Tkacova R, Hall MJ, Liu PP, Fitzgerald FS, Bradley TD (1997) Left ventricular volume in patients with heart failure and Cheyne–Stokes respiration during sleep. Am J Respir Crit Care Med 156:1549–1555
Tkacova R, Niroumand M, Lorenzi-Filho G, Bradley TD (2001) Overnight shift from obstructive to central apneas in patients with heart failure: role of PCO(2) and circulatory delay. Circulation 103:238–243
Trinder J, Merson R, Rosenberg JI, Fitzgerald F, Kleiman J, Douglas Bradley T (2000) Pathophysiological interactions of ventilation, arousals, and blood pressure oscillations during Cheyne–Stokes respiration in patients with heart failure. Am J Respir Crit Care Med 162:808–813
Trudo FJ, Gefter WB, Welch KC, Gupta KB, Maislin G, Schwab RJ (1998) State-related changes in upper airway caliber and surrounding soft-tissue structures in normal subjects. Am J Respir Crit Care Med 158:1259–1270
Usui K, Bradley TD, Spaak J, Ryan CM, Kubo T, Kaneko Y, Floras JS (2005) Inhibition of awake sympathetic nerve activity of heart failure patients with obstructive sleep apnea by nocturnal continuous positive airway pressure. J Am Coll Cardiol 45:2008–2011
Van de Graaff WB (1988) Thoracic influence on upper airway patency. J Appl Physiol 65:2124–2131
Van de Graaff WB, Gottfried SB, Mitra J, van Lunteren E, Cherniack NS, Strohl KP (1984) Respiratory function of hyoid muscles and hyoid arch. J Appl Physiol 57:197–204
Waradekar NV, Sinoway LI, Zwillich CW, Leuenberger UA (1996) Influence of treatment on muscle sympathetic nerve activity in sleep apnea. Am J Respir Crit Care Med 153:1333–1338
Warner G, Skatrud JB, Dempsey JA (1987) Effect of hypoxia-induced periodic breathing on upper airway obstruction during sleep. J Appl Physiol 62:2201–2211
Weiner D, Mitra J, Salamone J, Cherniack NS (1982) Effect of chemical stimuli on nerves supplying upper airway muscles. J Appl Physiol 52:530–536
Weitzman AT, Borowiecki BB, Shprintzen R, Rakoff S (1978) The hypersomnia-sleep apnea syndrome: site and mechanism of upper airway obstruction. In: Guilleminault DW (ed) Sleep apnea syndromes. Kroc Foundation Series. Alan R Liss, New York
White DP (2005) Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med 172:1363–1370
White SG, Fletcher EC, Miller CC 3rd (1995) Acute systemic blood pressure elevation in obstructive and nonobstructive breath hold in primates. J Appl Physiol 79:324–330
White DP, Gleeson K, Pickett CK, Rannels AM, Cymerman A, Weil JV (1987) Altitude acclimatization: influence on periodic breathing and chemoresponsiveness during sleep. J Appl Physiol 63:401–412
Whittle AT, Marshall I, Mortimore IL, Wraith PK, Sellar RJ, Douglas NJ (1999) Neck soft tissue and fat distribution: comparison between normal men and women by magnetic resonance imaging. Thorax 54:323–328
Wiegand L, Zwillich CW, White DP (1989) Collapsibility of the human upper airway during normal sleep. J Appl Physiol 66:1800–1808
Wilcox I, McNamara SG, Dodd MJ, Sullivan CE (1998) Ventilatory control in patients with sleep apnoea and left ventricular dysfunction: comparison of obstructive and central sleep apnoea [see comments]. Eur Respir J 11:7–13
Wilcox I, McNamara SG, Sullivan CE (2000) Central sleep apnea and heart failure [letter]. N Engl J Med 342:293, discussion 293–4
Wolk R, Kara T, Somers VK (2003) Sleep-disordered breathing and cardiovascular disease. Circulation 108:9–12
Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA (2002) Apnea–hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med 165:1245–1250
Xie A, Wong B, Phillipson EA, Slutsky AS, Bradley TD (1994) Interaction of hyperventilation and arousal in the pathogenesis of idiopathic central sleep apnea. Am J Respir Crit Care Med 150:489–495
Younes M (2003) Contributions of upper airway mechanics and control mechanisms to severity of obstructive apnea. Am J Respir Crit Care Med 168:645–658
Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W (2001) Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med 163:1181–1190
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published as part of the Special Issue on Sleep.
Rights and permissions
About this article
Cite this article
Leung, R.S.T., Comondore, V.R., Ryan, C.M. et al. Mechanisms of sleep-disordered breathing: causes and consequences. Pflugers Arch - Eur J Physiol 463, 213–230 (2012). https://doi.org/10.1007/s00424-011-1055-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-011-1055-x