
Abstract
Mental stress is an important factor contributing to recognized mechanisms underlying cardiovascular events. Among these, stress-related endothelial dysfunction is an early risk factor that predicts future development of severe cardiovascular disorders. Acute mental stress by a variety of tests impairs endothelial function in humans, although the opposite results have been reported by some investigators. Chronic stress always deteriorates endothelial function in humans and experimental animals. Stress hormones, such as glucocorticoids and pro-inflammatory cytokines, and endothelin-1 liberated in response to mental stress participate in endothelial dysfunction possibly via downregulation of endothelial nitric oxide synthase (eNOS) expression, eNOS inactivation, decreased nitric oxide (NO) actions, and increased NO degradation, together with vasoconstriction counteracting against NO-induced vasodilatation. Catecholamines do not directly affect endothelial function but impair its function when blood pressure elevation by the amines is sustained. Endogenous opioids favorably affect endothelial function, which counteract deteriorating effects of other stress hormones and mediators. Inhibition of cortisol and endothelin-1 production, prevention of pro-inflammatory mediator accumulation, hypnotics, mirthful laughter, humor orientation, and lifestyle modification would contribute to the prevention and treatment for stress-related endothelial dysfunction and future serious cardiovascular disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mental stress has been well recognized as a factor in the generation of pathophysiological sequelae in many disease states. Psychosocial stress activates the hypothalamic–pituitary–adrenal axis and the sympathetic nervous system; corticosteroids and catecholamines are liberated into blood flow as major stress hormones. Mental stress is one of the important risk factors contributing to mechanisms underlying endothelial dysfunction, atherosclerosis, and coronary and cerebral artery diseases [3, 100, 123]. Stress-related hormones and mediators affect vascular actions of nitric oxide (NO) generated from the endothelium and regulations of endothelial NO synthase (eNOS) expression, eNOS activation, and NO degradation.
Endothelial NO, first discovered as an endothelium-derived relaxing factor by Furchgott and Zawadzki [37], has emerged as a unique biological messenger. This labile inorganic molecule acts as a physiologically important vasodilator and has anti-hypertensive, anti-platelet, anti-thrombogenic, and anti-atherosclerotic actions. Impairment of endothelial function plays a key role in the pathogenesis of blood supply deficiency to various organs and tissues including the brain [118], heart [120], and eye [119]; blood pressure elevation [24, 116]; and insulin resistance [113].
The present review article summarizes information concerning modulations of endothelial function, evaluated as blood flow regulation by endothelial NO, induced by acute and chronic mental stress in humans and chronic stress in experimental animals, possible mechanisms of action of major stress hormones and mediators on endothelial function, including glucocorticoids, catecholamines, endothelin-1 (ET-1), pro-inflammatory cytokines, and endogenous opioids/opioid peptides; and describes the possibilities for prophylactic and therapeutic measures against stress-related endothelial dysfunction.
Synthesis, degradation, and actions of endothelial nitric oxide
NO is produced when l-arginine is transformed to l-citrulline via catalysis by NOS in the presence of oxygen and cofactors including tetrahydrobiopterin (BH4). Ca2+ is required for the activation of endothelial NOS (eNOS) that is constitutively expressed mainly in endothelial cells [35]. eNOS binds to caveolin-1 in the caveolae, microdomains of the plasma membrane. The eNOS intracellularly migrates in response to increased cytosolic Ca2+ in the presence of calmodulin (Fig. 1) and is activated for NO synthesis. The transmembrane influx of Ca2+ and its mobilization from intracellular storage sites are caused via stimulation of drug receptors located on the endothelial cell membrane by acetylcholine (ACh) and bradykinin (BK) or via mechanical stimuli such as shear stress. On the other hand, shear stress, BK, or insulin induce the phosphorylation of Ser1177/1179 of eNOS through phosphatidylinositol 3-kinase (PI3K) and the downstream serine/threonine protein kinase Akt, resulting in enhanced NO formation [26]. Endothelial NO causes vasodilatation, decreases vascular resistance, increases regional blood flow, and lowers blood pressure; it also inhibits platelet aggregation and adhesion, reduces leukocyte adhesion and migration, and inhibits smooth muscle proliferation, thus, leading to prevention of atherosclerosis. These NO actions are mediated by cyclic guanosine monophosphate (cyclic GMP) from GTP synthesized through soluble guanylyl cyclase.

Information pathways via NO (right panel) and endothelin-1 (ET-1) liberated from endothelial cells to vascular smooth muscle cells. Superoxide generation via NADPH oxidase is also included in the upper right part of the figure. Sites of action of glucocorticoids (GC) and endothelin-1 (ET) released by mental stress are shown in the right panel. R in the square on the endothelial membrane is the drug receptor or mechanoreceptor, pool Ca2+ storage site, CV caveolin-1, CaM calmodulin, BH 4 tetrahydrobiopterin, PI 3 K phosphatidylinositol 3-kinase, Akt serine/threonine protein kinase Akt, eNOS* activated eNOS, L -Arg. L-arginine, L -Citru. L-citrulline, nNOS* activated nNOS, O 2 – superoxide anion, ONOO – peroxynitrite, SOD superoxide dismutase, sGC soluble guanylyl cyclase, cGMP cyclic GMP, ODQ 1H[1,2,4] oxadiazolo[4,3-a]quinoxalin-o1-one, PDE-5 phosphodiesterase-5. Solid line denotes stimulation, dotted line denotes inhibition
The synthesis of NO by eNOS is inhibited by l-arginine analogs, including N G-monomethyl-l-arginine (l-NMMA), N G-nitro-l-arginine (l-NA), and l-NA methylester (l-NAME). Nitro compounds, such as nitroglycerin (GTN) and sodium nitroprusside (SNP), are capable of liberating NO. 1H[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one decreases the synthesis of cyclic GMP by inhibiting guanylyl cyclase activity. Insufficiency of BH4 causes uncoupling of NOS consequentially resulting in superoxide anions being produced instead of NO. Superoxide anions are also generated by NADPH oxidase (Fig. 1) and xanthine oxidase. Superoxide dismutase (SOD), catalase, and dimethyl sulfoxide scavenge free radicals. NO reacts with superoxide anions, generating highly toxic compounds such as peroxynitrite.
Vascular endothelial cells generate not only vasodilator mediators, such as NO, prostacyclin, and endothelium-derived hyperpolarizing factor, but also vasoconstrictor endothelin-1 (ET-1), a 21 amino acid polypeptide synthesized through the process shown in Fig. 1. The potent vasoconstrictor property is mediated via activation of ETA receptors located on smooth muscle cell membranes. ET-1 has been implicated in the pathogenesis of many cardiovascular-related diseases.
Studies on humans subjected to mental stress
Effects of acute stress
Mental stress (color-word conflict test)-induced increases in platelet activation, as measured by the level of β-thromboglobulin, and endothelium activity, as evaluated by factor VIII/von Willebrand factor, were observed in healthy normotensive volunteers and hypertensive patients to a similar extent [78]. Mental arithmetic testing increased forearm blood flow, and l-NMMA infusion inhibited the response [10]. Mental stress provoked by an arithmetic challenge induced endothelium-dependent, flow-mediated vasodilatation in healthy men and women to a similar extent [45]. Coronary segments with stenosis and coronary irregular segments were constricted in response to mental arithmetic stress, whereas smooth segments did not show this response; coronary blood flow increased in smooth vessels but decreased in irregular vessels; the degree of constriction or dilatation during mental stress correlated with the responses to infusions of ACh [138]. In atherosclerosis, unopposed constriction caused by a local failure of endothelium-dependent vasodilatation appears to result in coronary arteries having an abnormal response to mental stress. The vasodilator response to mental stress was blunted in hypertensive patients but not in hypercholesterolemic ones compared with healthy controls; l-NMMA administration reduced mental stress-induced vasodilatation in healthy subjects and hypercholesterolemic patients but not hypertensive patients [9]. Different mechanisms may underlie endothelial dysfunction in these pathological conditions; a susceptibility to vascular damage over repeated exposure to stressful situations may be increased in hypertensive patients. Forearm blood flow increase observed during mental stress with a color word test in humans was blunted by administration of l-NMMA; both atropine and l-NMMA caused a greater reduction in the response, suggesting that forearm vasodilatation during mental stress is mediated by NO liberated possibly through cholinergic stimulation [25]. In response to mental stress (forced arithmetic) in healthy young males, forearm blood flow and net release of endothelium-derived tissue-type plasminogen-activator activity increased [56]. Mental stress by Stroop color word conflict, math, or anger recall tests caused carotid arterial dilatation and an increase in carotid and middle cerebral arterial blood flow in young and old healthy subjects; these responses were attenuated in patients with essential hypertension; there was no difference in the carotid artery response to GTN in healthy subjects and hypertensive patients [79]. Impaired cerebrovascular reactivity to mental stress in hypertension may be associated with cerebral vascular endothelial dysfunction.
In contrast to the findings described so far, recent studies provided evidence suggesting that acute mental stress impairs endothelial function in humans. Systemic vascular resistance responses during a battery of four diverse laboratory stressors were greater for healthy individuals with decreased endothelium-dependent arterial dilatation [103]. Exaggerated systemic vasoconstriction during mental stress may reflect endothelial dysfunction. Brachial artery flow-mediated dilatation in healthy men was reduced at 30 and 90 min after a standardized mental stress test, which however had no effect on the response to GTN, suggesting that a brief episode of mental stress, similar to those encountered in everyday life, may cause transient (up to 4 h) endothelial dysfunction [40]. Mental stress induced by an arithmetic test opposed only methacholine (MCh)-induced, endothelium-dependent forearm vasodilatation in young healthy people, while cold pressor and isometric handgrip tests induced attenuations of endothelium-dependent and endothelium-independent vasodilatation [98]. Mental stress provoked by anger recall and mental arithmetic and trait hostility attenuated flow-mediated brachial arterial dilatation in normal subjects and those with hypercholesterolemia; the behavioral trait of hostility was associated with enhancement of the adverse effect of mental stress on endothelial function [42]. Endothelial function did not change following a 5-min mathematics stressor or the consumption of 75 g of glucose; however, the combination of glucose and stress impaired endothelium-dependent cutaneous vasodilatation in young healthy females [7]. In postmenopausal women with typical angina and normal coronary arteries, mental stress by the anger recall task for 5 min provoked myocardial ischemia associated with endothelial dysfunction [86]. Endothelium-dependent forearm vasodilatation elicited by MCh was impaired by a 5-min mental arithmetic stress test, this effect being blocked by propranolol and neurogenic blockade but not by phentolamine [29]. β-Adrenoceptor activation appears to be involved in the NO-mediated vasodilatation.
Effects of chronic stress
Psychosocial stress adversely affects autonomic and hormonal homeostasis, resulting in metabolic abnormalities, inflammation, and endothelial dysfunction [20]. Adolescents exposed to chronic, negative stressors that worsen over time show increasing magnitude of cardiovascular stress responses [65].
Healthy male college students, who had chronic sleep deprivation for 4 week and were under great stress to pass an examination, showed decreased flow-mediated, endothelium-dependent vasodilation [115]. In young medical students, showing a high incidence of stress, smoking, unhealthy nutritional habits, and sedentary lifestyle, the main factors which determine endothelial function imbalance were stress and smoking [67]. Both objective and subjective socioeconomic status were related to brachial artery flow-mediated dilatation in healthy adults; the MacArthur Scale of Subjective Social Status, which assessed perceived social standing in the local community, positively correlated with flow-mediated dilatation; no other socioeconomic status measures were significant for flow-mediated dilatation [17]. Lower subjective social status in one’s community may be linked to cardiovascular disease via impaired vasodilatation. Mausbach et al. [71] noted that chronic stress of caregiving in the elderly was associated with impaired endothelial function, as evaluated by reactive hyperemia-induced flow-mediated vasodilatation. Stress-induced endothelial dysfunction may be a potential mechanistic link to the increased risk of cardiovascular disease in elderly caregivers. In healthy adults, increases in Profile of Mood States total mood disturbance scores were associated with decreases in endothelial function. The following Profile of Mood States subscales were inversely correlated with flow-mediated brachial arterial dilation: depression/dejection, tension/anxiety, anger/hostility, fatigue/inertia, and confusion/bewilderment [18]. Mood disturbance may contribute to cardiovascular disease via impaired vasodilatation
There was a significant interaction between carotid artery compliance and vital exhaustion, characterized by fatigue and irritability, on intima-media thickness for men but not women; no interaction was found between flow-mediated vasodilatation and vital exhaustion for men and women [13].
Taken together, most of studies performed in the 1990s indicate that acute mental stress induces NO-mediated flow-dependent blood flow in humans, which is hypothesized to play a role in minimizing blood pressure increase associated with catecholamine release. In contrast, stress-induced endothelial dysfunction has been reported from most investigations carried out after 2000 (Table 1). The reason for such a discrepancy in the effect of acute mental stress in humans placed under similar experimental designs (kinds and durations of mental stress, ages, etc.) remains to be determined. Possible individual differences in the neuroendocrine coping mechanisms may affect mood regulation and the state of health [39]. Hyper-responsivity of the sympathetic nervous system to psychological stress is an intrinsic characteristic among some individuals [95] that may be a factor to differentiate their responsiveness to stress to other majorities. According to Gottdiener et al. [42], behavioral hostility is associated with enhancement of the adverse effect of mental stress on endothelial function. As summarized in the later section “Stress hormones and mediators,” there are factors adversely affecting endothelial function, such as glucocorticoids, inflammatory cytokines, ET-1, and sustained increase in blood pressure. There is no evidence that the beneficial effects of endogenous opioids/opioid peptides generated under mental stress on endothelial function overcome stress-related impairment of endothelial function. At present, on the basis of recent studies, in which mechanisms of action of mental stress are analyzed, endothelial dysfunction induced by acute mental stress may be more plausible. Under various kinds of chronic mental stress, endothelial function is always impaired, leading to atherosclerosis and cardiovascular disease.
Studies on experimental animals subjected to mental stress
Cynomolgus monkeys
Intracoronary infusion of ACh caused coronary vasodilatation (+4%) in control monkeys but vasoconstriction (−11%) in those fed a high cholesterol diet in the unstable group (stressful environment); in monkeys consuming the cholesterol-lowering diet, the change in artery diameter was +2% in the stable group and −10% in the unstable one; the arterial response to GTN was similar among all groups [136]. Iliac arteries isolated from atherosclerotic monkeys of the “late stress” group (stable social groups for the first half of the experiment and unstable groups for the second half) had impaired dilatation to ACh and the Ca2+ ionophore A23187, compared with those from unstressed or “early stress” (unstable social groups for the first half of the experiment and stable groups for the second half) monkeys, suggesting that current, but not previous, exposure to chronic stress impairs endothelium-dependent vasodilatation [135]. Chronic social disruption appears to be associated with endothelial dysfunction in atherogenic monkeys consuming a cholesterol-lowering diet. Social disruption was also associated with both sympathetic nervous system arousal and indexes of endothelial dysfunction in male monkeys, and the effects were prevented by treatment with a β-adrenoceptor blocker [114]. Psychosocial stress in monkeys placed under socially unstable conditions for 72 h increased the number of injured (IgG-positive) endothelial cells of the descending thoracic aorta, an effect that had not been demonstrated previously; β1-adrenoceptor blockade by metoprolol and atenolol inhibited the stress effect, suggesting that chronic mental stress induces endothelial injury and that this effect is mediated via β1-adrenoceptor activation [109].
Swine
Chronic NOS inhibition with l-NAME increased the adrenocorticotropic hormone (ACTH) response to handling and treadmill exercise but decreased the ACTH response to restraint in female Yucatan miniature swine, together with a decrease in vascular conductance in the frontal cortex and hypothalamus [54]. NO produced by NOS appears to function in a context-specific manner to inhibit or activate neuroendocrine activity.
Rats and mice
Coronary arteries isolated from borderline hypertensive rats exposed to chronic exposure to air–jet stress for 2 h/day for 10 days responded to ACh with relaxation to a lesser extent than those from rats without stress; SNP-induced relaxations were also attenuated, but the response to isoproterenol was without effect, suggesting that vasodilator effects of NO, irrespective of endogenous or exogenous origin, may be blunted by mental stress [36]. Stress-induced inhibition in coronary arterial relaxation by ACh was seen only in old borderline hypertensive rats [41]. This inhibition may be associated with superoxide anions, vasoconstrictor cyclooxygenase products, and a loss of K+ channel-mediated relaxation. Chronic mild unpredictable stress induced increased responsiveness to phenylephrine in endothelium-intact aortic rings from rats without changes in aortas denuded of the endothelium; rats exposed to chronic stress showed hypertrophy of the intima and media of the aorta and increased serum levels of triglycerides and cholesterols, when compared with control rats [80]. Chronic stress-induced functional and histological changes appear to be mediated by deficiency of NO production and dyslipidemia. Immobilization stress (120 min/day, 14 days) increased systolic blood pressure and decreased ACh-induced ex vivo relaxation of arteries in rats; immobilization increased the plasma levels of angiotensin II and angiotensin-converting enzyme activity, increased the plasma levels of malondialdehyde and expression of gp91(phox), and Rho-associated kinase-1 in arteries, and it decreased the arterial eNOS mRNA and nitrate/nitrite (NOx); ramipril or losartan reversed the functional and biochemical alterations [14]. Chronic stress induces vascular oxidative stress by activating the angiotensin II AT1 receptor-signaling pathway, thereby provoking endothelial dysfunction. In vascular rings isolated from male mice that were exposed to chronic mild stress, endothelium-dependent relaxations in response to MCh were partially attenuated, and the remaining response was abolished by catalase; arterial NO production was decreased and H2O2 production was increased in stressed mice [19].
Cage-switch stress elevated the mean arterial pressure and heart rate in ovariectomized rats, the responses being attenuated by estrogen replacement; treatment with l-NAME reduced the difference in the pressor response to stress; eNOS expression in the mesenteric artery was increased in the estrogen group compared to that in the control group, suggesting that mesenteric overexpression of eNOS via chronic estrogen treatment appears to suppress the enhanced cardiovascular responses to mental stress [77]. In female rats, a scheduled stress for 10 days elevated the serum testosterone level, reduced the reactivity to their male mates, and decreased eNOS and neuronal NOS expressions in vaginal tissue [139]. Chronic stress modifies the sexual behavior of female rats possibly through complex changes in sex hormones, endocrine factors, and NO. Exposure of male and female rats to mild unpredictable stress for 6 weeks induces different oxidative stress and compensatory responses in both sexes probably due to differences in the mechanisms underlying oxidant/antioxidant pathways; the responses to chronic stress in females is accompanied by lower soluble intercellular adhesion molecule type 1 levels than in males, suggesting lower endothelial injury in females [58].
In contrast to the stress-induced endothelial dysfunction reported so far, Púzserová et al. [88] found that the endothelium-dependent relaxation of the aorta and NO synthesis were increased in rats exposed to chronic social stress by crowding, and red wine polyphenols blunted the elevation of NO production and vasorelaxation.
Effects of stress hormones and mediators
Glucocorticoids
Studies on humans
Following activation of the limbic–hypothalamic–pituitary–adrenal or stress axis, glucocorticoids are thought to downregulate the transcription and activity of NOS via a feedback mechanism [64]. Plasma levels of cortisol are elevated under the fire strategies condition in firefighters, and there are positive correlations between cortisol and interleukin-6, ET-1, and thromboxane B2 [129]. Glucocorticoid is one of the important stress hormones responsible for impairment of endothelial function [5, 38].
Gerra et al. [39] provided evidence concerning the involvement of the hypothalamic–pituitary–adrenal axis and catecholamines in response to acute mental stress in healthy male subjects. Elevated cortisol levels inhibit endothelium-dependent vasodilator responses to ACh but did not affect vasodilatation in response to SNP [131]. Acute mental stress impaired flow-mediated vasodilation and increased cortisol levels; impaired endothelial function was prevented by blocking cortisol production with metyrapone in subjects without coronary heart disease, suggesting a direct or facilitative role for cortisol in mental stress-induced endothelial dysfunction [8]. Liu et al. [63] noted that the natural glucocorticoid cortisol decreased the expression levels of eNOS in human endothelial cells and that a suppressive glucocorticoid response element was present in the eNOS promoter region, suggesting that endogenous 11-β-hydroxysteroid dehydrogenases play important roles in modulating the effect of glucocorticoids on eNOS expression. Forearm blood flow increase in response to intra-arterial infusion of ACh was impaired by exogenous cortisol in healthy men [69]. It was suggested that cortisol-induced reduction in NOx production in healthy male subjects may occur at a point distal to l-arginine availability in the NOS pathway [59]. Forearm blood flow increase after the release of arterial occlusion (reactive hyperemia) was attenuated in patients with various autoimmune disorders treated with glucocorticoid and administration of vitamin C normalized blood flow responses; in human umbilical vein endothelial cells (HUVEC) treated with dexamethasone, production of H2O2 was increased, intracellular amounts of peroxynitrite was increased, and those of NO was decreased, suggesting that glucocorticoid excess causes overproduction of reactive oxygen species (ROS) and thereby perturbs NO availability [50]. The progesterone/glucocorticoid receptor antagonist RU-486 and introduction of glucocorticoid receptor siRNA caused attenuation of the inhibitory effect of medroxyprogesterone acetate on the estrogen-induced eNOS phosphorylation and eNOS activity in HUVEC [82].
Studies on experimental animals
Ingestion of dexamethasone by rats increased blood pressure, decreased plasma NOx, and downregulated the expression of eNOS; dexamethasone administration attenuated the vasodilator response to ACh in mice; incubation of HUVEC or bovine aortic endothelial cells with several glucocorticoids reduced eNOS mRNA and protein expression [128]. Dexamethasone decreased the expression of eNOS in the mouse heart, liver, and kidney [127]. In cultured bovine coronary artery endothelial cells, pretreatment with cortisol decreased eNOS protein levels, ATP-induced intracellular Ca2+ elevation, and NO release, suggesting that cortisol suppresses NO release by downregulating eNOS protein and inhibiting intracellular Ca2+ mobilization [94].
Glucocorticoids regulate NOx production following cytokine exposure in cardiac microvascular endothelial cells by limiting BH4 and l-arginine availability [107]. Endothelium-dependent relaxations in response to Ca2+ ionophore were reduced in aortic rings from dexamethasone-hypertensive rats; dexamethasone incubation abolished l-NA-induced contraction in endothelium-intact aortic rings and inhibited expression of the rate-limiting enzyme for BH4 synthesis, GTP cyclohydrolase, suggesting that inhibition of BH4 synthesis by the glucocorticoid may contribute to impaired endothelial function [57]. Michell and Webb [72] discuss the roles of NO and BH4 in the pathogenesis of glucocorticoid-induced hypertension. Under reduced BH4 levels, eNOS synthesizes superoxide rather than NO in vitro [92] and in vivo [105, 106]. There is increasing evidence for a role of oxidative stress and NO deficiency/endothelial dysfunction in experimental glucocorticoid-induced hypertension; NADPH oxidase, rather than xanthine oxidase and eNOS uncoupling, plays an important role in the pathogenesis in rats [83]. In addition, glucocorticoids decrease NO production by decreasing eNOS gene transcription [28].
Endothelium-dependent relaxation and eNOS mRNA expression were greater, and aortic superoxide production was lower in rats co-treated with dexamethasone and atorvastatin than those treated with dexamethasone alone, suggesting that atorvastatin improves endothelial function and reduces superoxide production [76].
Repeated restraint stress in male rats activated the pituitary adrenal system, as indicated by increases in adrenal weight and plasma corticosterone concentration that were accompanied by a decrease in constitutive NOS activities [93].
In short, glucocorticoids generated by psychological stress impairs endothelial function through downregulation of eNOS, reduced eNOS phosphorylation, and increased production of superoxide anions possibly via NADPH oxidase activation and BH4 deprivation.
Catecholamines
Catecholamines released by stimulation of the sympathetic nervous system, as well as glucocorticoids, are the main stress hormones [38].
Studies on humans
There is evidence that exaggerated systemic vascular resistance responses during mental stress may reflect endothelial dysfunction [103]. Santos et al. [97] provided evidence suggesting that sympathetic activation mediates the blunted endothelium-mediated brachial arterial dilatation during mental stress with the color–word test in heart failure patients. Female patients with apical ballooning syndrome showed abnormal vasoreactive and sympathetic responses to acute mental stress tests, evidenced by impaired endothelium-dependent vasodilatation, excessive vasoconstriction, and catecholamine release [70]. Increased release of catecholamines from the sympathetic nerve and adrenal medulla during mental stress may interfere with the vasodilator action of endothelial NO.
On the other hand, Spieker et al. [110] found that intra-arterial infusion of norepinephrine of similar duration as mental stress did not inhibit flow-mediated vasodilatation, suggesting that sympathetically released amine does not seem to participate in stress-induced endothelial dysfunction. Ghiadoni et al. [40] noted that flow-mediated vasodilation was still impaired 90 min after the stress test; this was in contrast with the rapid return of heart rate and blood pressure to the respective pre-stress value [8], suggesting that factors other than catecholamines may contribute to endothelial function interference. In HUVEC, norepinephrine stimulates eNOS activity via RhoA attenuation, VEGF mRNA synthesis, and mitogenic activity [102]. Epinephrine and norepinephrine through activation of the α1B-receptor subtype stimulate phosphorylation of eNOS and increase DNA synthesis in human epicardial coronary endothelial cells [55]. These findings obtained in vivo and in vitro suggest that catecholamines are not involved in an impairment of endothelial function.
Studies on experimental animals
In rat carotid arteries, α1D-adrenoceptors mediate endothelium-dependent relaxation induced by phenylephrine [21]. Cage-switch stress induced the pressor response and increased norepinephrine level in plasma; treatment with l-NAME attenuated the stress-related responses, suggesting that NO is involved in the norepinephrine surge in response to mental stress [124]. In stroke-prone spontaneously hypertensive rats, treatment with hydralazine lowered blood pressure and increased urinary norepinephrine excretion but did not change expressions of eNOS, neuronal NOS, and inducible NOS in the brain [60]. Increased norepinephrine does not appear to impair endothelial function.
The NO production induced by epinephrine and the β3-adrenoceptor agonist BRL 37344 was associated with the activation of the PI3K/Akt pathway and phosphorylation of eNOS at serine 1,177 in the perfused arterial mesenteric bed of rats; isoproterenol, salbutamol, or BRL 37344 produced NO-dependent reductions of blood pressure in anesthetized rats, suggesting that β1-, β2-, and β3-adrenocptors are coupled to the NO/cyclic GMP pathway, highlighting the role of the endothelium in the vasodilator action elicited by epinephrine and related β-adrenoceptor agonists [32]. Epinephrine-dependent eNOS activation was accompanied by an increase in phosphorylation of eNOS at serine 1,179 in cultured bovine aortic endothelial cells; epinephrine promoted activation of the small G protein Rac 1; all of these responses were blocked by the β3-adrenoceptor blocker SR59230A, suggesting that the small G protein Rac 1 is a key regulator of β3 receptor signaling with potentially important implications for the pathways involved in adrenergic mechanism of eNOS pathways [62].
Taken together, epinephrine and norepinephrine by themselves, potentially liberated in response to mental stress, seem unlikely to be involved in the stress-induced endothelial dysfunction. However, vasodilatation due to endothelial NO may be antagonized physiologically to some extent by vasoconstrictor catecholamines. Repetitive or chronic stress leads to increased blood pressure, possibly mediated via catecholamines that can result in impaired endothelial damage and vasoconstriction.
Endothelin-1
ET-1, an endothelium-derived peptide, is a potent vasoconstrictor that counteracts the vascular actions of endothelial NO. An exaggerated ET-1 release in response to mental stress was noted in healthy subjects [68], firefighters [129], and patients with coronary artery disease [31]. There are sex and ethnicity differences in acute stress-induced ET-1 release [122].
Endothelium-dependent flow-mediated vasodilatation in healthy subjects was reduced by the 3-min mental stress task with high-resolution A-mode ultrasonic echo-tracking device for about 45 min, whereas vasodilatation in response to GTN remained unaffected; BQ-123, a selective endothelin-A (ETA) receptor antagonist, prevented the impairment of endothelium-dependent vasodilatation; intra-arterial infusion of norepinephrine did not inhibit flow-mediated vasodilatation [110]. Mental stress induces prolonged endothelial dysfunction possibly through generation of ET-1 that counteracts vasodilator NO liberated from the endothelium. Baseline ET-1 values were higher in patients with intermittent claudication than in control subjects; at the end of mental arithmetic performed for 10 min, ET-1 levels rose in both groups for 10 min [68]. Exaggerated release of ET-1 in the diseased patients could be a potential pathophysiological mechanism, through which mental stress may trigger acute cardiac events. One of stress neuropeptides, corticotrophin-releasing hormone (CRH), induced an increase in ET-1 release from human endothelial cells but did not affect NO release; the CRH receptor–antagonist astressin abolished the ET-1-releasing effect of CRH [133]. CRH receptor 1 modulates ET-1 release and ET-1 mRNA activation caused by hypoxia in rats [46]. In human microvascular endothelial cells, ET-1 release was increased by exposure to acute (epinephrine and norepinephrine) and chronic stress hormones (dexamethasone, β-endorphin, and CRH), whereas NO release was increased and decreased by short-term stimulation of dexamethasone and norepinephrine, respectively [81].
ET-1 depresses NOS activity in cultured rat vascular smooth muscle cells [49] and increased production of ET-1 in the rat kidney causes a decrease in renal blood flow possibly via attenuating NO production [66]. Elevated ET-1 levels reduced eNOS protein expression in human saphenous vein endothelial cells; protein kinase C inhibition downregulated eNOS protein expression, whereas the protein kinase C agonist PMA upregulated its expression, suggesting that high levels of ET-1 impair endothelial NO production via a protein kinase C-mediated inhibition of eNOS expression [90]. Activation of ETA receptors in type 1 diabetic rats plays an important role in impaired eNOS- and neuronal NOS-dependent dilatation of cerebral arterioles [2]. ET-1 increased H2O2 levels in fetal lamb pulmonary arterial smooth muscle cells in an ETA receptor-dependent manner and decreased eNOS promoter activity, this activity being restored in the presence of catalase; high concentrations of H2O2 decreased eNOS promoter activity and protein levels, suggesting that H2O2 generated by ET-1 from smooth muscle may play a role in downregulation of eNOS expression [130].
Taken together, elevated levels of ET-1 in response to mental stress appear to impair endothelial function through eNOS downregulation, reduced eNOS activity, and superoxide generation. The potent vasoconstrictor property of ET-1 via activation of ETA receptors counteracts vasodilator actions of NO. The stress-related CRH release may play a role in increasing the release of ET-1 from endothelial cells.
Pro-inflammatory cytokines
Various psychological stressors induce cytokine production and cytokines involved may vary according to the type of stress [5, 38]. Acute systemic inflammation evoked by Salmonella typhi vaccine induces temporal but profound dysfunction of the arterial endothelium in both resistance and conduit vessels in healthy volunteers [47].
In non-diabetic subjects, levels of C-reactive protein were related to those of interleukin (IL)-6 and tumor necrosis factor (TNF)-α; concentrations of C-reactive protein were related to insulin resistance and to markers of endothelial dysfunction [140]. Chronic inflammatory state may induce insulin resistance and endothelial dysfunction. In the human cutaneous vein, TNF-α and IL-1β but not IL-6 attenuated the dilatation to BK and arachidonic acid; hydrocortisone and aspirin prevented the endothelial dysfunction, suggesting that pro-inflammatory cytokines induce reversible endothelial dysfunction and that cyclooxygenase products may contribute to the response [4]. Exposure of porcine pulmonary artery endothelial cells to the combination of interferon-γ, TNF-α, and IL-1 reduced eNOS activity in association with reductions of eNOS mRNA and protein content; treatment with the protein synthesis inhibitor, cycloheximide, prevented cytokine-induced reduction of eNOS mRNA expression [142]. TNF-α downregulates eNOS mRNA in the isolated rat heart [85]. TNF-α and IL-1β also downregulates eNOS mRNA levels in cultured human coronary artery endothelial cells [101]. These authors also found that opioids (β-endorphin, endomorphin-1, and endomorphin-2) did not change eNOS mRNA expression; however, when applied together with cytokines, eNOS downregulation was enhanced compared to that found with cytokine alone, suggesting that opioids do not affect eNOS expression in normal conditions but might do so upon stress stimuli. TNF-α impairs mitochondrial biogenesis and function in different tissues of obese rodents by downregulating eNOS expression [125].
A variety of cytokines, such as TNF-α, IL-1, IL-6, and interferon-γ, increase oxidative stress, downregulate eNOS activity, and induce endothelial apoptosis [61]. eNOS expression was reduced by TNF-α in both in vitro and in vivo experiments with female mice, whereas IL-10 restored the eNOS expression, suggesting that the anti-inflammatory cytokine IL-10 prevents impairment of endothelium-dependent vasodilatation caused by TNF-α by protecting eNOS expression [141].
Endogenous opioids and opioid peptides
The endogenous opioid system is one of the important contributors to mediate, modulate, and regulate stress responses, including vascular endothelial and blood flow responses. There was a transient but significant increase in β-endorphin levels immediately after jumping in inexperienced tandem parachutists [99]. β-Endorphin levels 5 min before non-emergency surgery increased compared with those 24 h earlier in children, whereas leucine enkephalin levels remained unchanged [16]. Morphine biosynthesis occurs in mammalian neural tissues; morphine releases NO in the limbic system that modulates emotion [143]. Supporting morphine as an endogenous signaling molecule is the presence of the newly cloned μ3 opiate–receptor subtype in vascular, neural, and immune tissues, which is coupled to NO release [87].
Healthy subjects were classified according to their increase in systolic blood pressure after a mental arithmetic test as low and high responders; during the mental test, low responders showed increased levels of β-endorphin, cortisol, catecholamines, and atrial natriuretic factor and decreased levels of ET-1, whereas high responders showed increased levels of methionine enkephalin, dynorphin B, and catecholamines [34]. The individual difference in hemodynamic and endocrine responses to mental stress may depend on a different activation of the endogenous opioid system. β-Endorphin increased human endothelial and monocytic ET-1 release and decreased NO release; these effects were mediated via μ1 opioid-receptor activation, suggesting that β-endorphin plays a role in the pathogenesis of stress-induced endothelial dysfunction [132].
Exposure to morphine of human saphenous endothelium, internal thoracic artery endothelium, or rat microvascular endothelial cells results in NO release via the stimulation of μ3 opiate-receptor subtype [112]. Morphine stimulates both [Ca2+]i and NO production in human arterial endothelial cells [33]. There is evidence suggesting that morphine-induced ocular hypotension and miosis are mediated by μ3 receptors and NO release [6, 27].
The in vivo stimulation of δ-opiate receptor by DSLET, but not the μ-agonist DAMGO, increased the tolerance of the rat heart to oxidative stress through activation of NOS and SOD [91].
In rat hindquarter vascular beds, vasodilator responses to endomorphin 1 and DAMGO, μ-opioid receptor agonists, and ACh were attenuated by treatment with l-NAME but not by sodium meclofenamate and the ATP-sensitive K+-channel blocker U-37883A, suggesting that endomorphin-induced vasodilatation is mediated by the release of NO but not by the release of vasodilator prostaglandins or the opening of ATP-sensitive K+ channels [12]. Endomorphin-1 and endomorphin-2 also relaxed rat aortic ring preparations by an endothelium-dependent mechanism [48]. Endomorphin-1 and endomorphin-2 inhibited contractile responses of rat thoracic aorta rings to phenylephrine and angiotensin II by a naloxone-sensitive mechanism [89]. The release of NO from endothelial cells appears to be involved. Endomorphin analogs caused relaxations of rat aortic rings, which were naloxone-sensitive and endothelium-dependent [30].
Methionine enkephalin elicited pial artery dilatation in the newborn pig that was blocked by the selective eNOS inhibitor N-iminoethyl-l-ornithne [134], and it also stimulated cyclic GMP release into cerebrospinal fluid [1], suggesting that eNOS contributes to opioid-induced pial artery dilatation. Leucine enkephalin, dynorphin, DAMGO, DPDPE, deltorphin, and U50,488H, which are μ-, δ1-, δ2-, and κ-agonists, respectively, also elicited increases in pial artery diameter and cyclic GMP release that were attenuated by l-NA [23].
Nociceptin/orphan FQ, one of the stress-related neuropeptides, regulates neuroendocrine function of the limbic–hypothalamic–pituitary–adrenal axis [22]. Rats exposed to acute social defeat showed elevated nociceptin/orphan FQ and NOP receptor mRNA expression in their limbic regions [43]. Nociceptin induces relaxation in feline renal, mesenteric, carotid, and femoral artery rings with intact endothelium [44]. In porcine coronary artery rings, nociceptin-induced relaxations were reduced by endothelial removal and treatment with l-NA but were unaffected by naloxane [137]. On the other hand, Champion et al. [11] noted that vasodilator responses to nociceptin were not altered by l-NAME in rat hindquarters.
Taken together, endogenous opioids and opioid peptides, such as morphine, endomorphin, methionine enkephalin, and nociceptin, if liberated in response to acute and chronic mental stress, appear to act beneficially on endothelial function and minimize the effects of glucocorticoids, ET-1, and proinflammatory cytokines. Determining the effect of opioid antagonists, selectively acting on specific opioid receptor subtypes, on stress-induced endothelial dysfunction may provide evidence that endogenous opioids/opioid peptides liberated as stress hormones contribute to minimizing or enhancing impaired endothelial function.
Therapeutic and prophylactic measures
Drug therapy
Inhibition of cortisol by oral metyrapone (750 mg ×2), an inhibitor of adrenal 11-hydroxylase, prevented stress-related endothelial dysfunction in healthy volunteers [8]. In patients with autoimmune disorders, glucocorticoid therapy reduced endothelium-dependent forearm blood flow increase, the response being normalized by administration of vitamin C [50]. Overproduction of ROS by excess glucocorticoid appears to perturb NO bioavailability; therefore, antioxidants, such as vitamins C and E, apocynin, BH4, and polyphenols, would efficiently protect and restore endothelial function under exposure to mental stress. Estrogen supplementation suppresses the enhanced cardiovascular responses to mental stress [77]. Major pathways accounting for the ability of estrogens to increase eNOS function consist of rapid signaling by estrogen receptors through the PI3K/Akt pathway, resulting in eNOS phosphorylation and increased eNOS activity and genomically regulated increases in eNOS mRNA and protein [28]. Atorvastatin reduces superoxide and improves endothelial function in dexamethasone-treated rats [76].
Intra-arterial infusion of the selective ETA receptor antagonist BQ-123 prevented the stress-induced impairment of endothelium-dependent vasodilatation in healthy individuals [110]. ET-1 antagonism by bosentan is suggested to provide a novel therapeutic strategy for improving vascular homeostasis [90]. Blockade of vasoconstrictor actions of ET-1 appears to revive the vascular effect of endothelial NO. The stress neuropeptide CRH induced an increase in ET-1 release from human endothelial cells, and the effect was abolished by the CRH-receptor antagonist astressin, whereas NO release was not affected by CRH [133]. CRH-receptor antagonists may prevent CRH-induced disorders, including impairment of endothelial function through ETA-receptor activation by ET-1.
Aromatherapy and acupuncture
Flow-mediated brachial artery dilatation was lower in medical staff after nightshift work (average sleeping time of 3.3 h) than on a regular workday, which was improved after aromatherapy (30 min by inhalation of the essential oil of lavender) [104].
Stress-related endothelial dysfunction, evaluated as impairment of flow-mediated brachial artery dilatation, was not modulated by acupuncture in healthy subjects; no endothelial dysfunction was found in highly hypnotizable subjects, whereas non-susceptible individuals exhibited stress-related reduction in flow-mediated vasodilatation [51]. Hypnotizability appears to be more effective than acupuncture. In contrast, in maternal separation rats, acupuncture reduced anxiety-related behaviors and reduced corticosterone and ACTH levels in plasma, suggesting that acupuncture may be useful as a therapeutic measure in this animal model [84]. Further study is required to determine whether acupuncture is beneficial for minimizing stress-induced untoward effects.
Chronic stress is estimated to increase the risk of cardiovascular events. In patients with coronary artery disease, endothelium-dependent vasodilatation improved with 6 weeks of yoga training [108].
Hypnotic modulation
Awake subjects with a high hypnotic susceptibility did not show any stress-related endothelial dysfunction, while in contrast flow-mediated vasodilatation was impaired by cognitive stress in those with a low hypnotic susceptibility [52, 53].
Lifestyle
To determine the impact of lifestyle factors, Mancaş et al. [67] studied a group of young medical students who showed a high incidence of smoking, unhealthy nutritional habits, stress, and sedentary lifestyle; the odds for endothelial function alteration were significant only in subjects who associated stress and smoking. Chronic stress could lead to an unhealthy lifestyle: smoking, excessive alcohol drinking, overeating, and insomnia, all factors that further impair endothelial function and increase cardiovascular risk [117, 121]. Lifestyle modifications that included consumption of low-fat, high antioxidant, and high fiber diets; doing moderate aerobic exercise; and stress management for 6 months ameliorated oxidative stress and serum lipids in patients with coronary artery disease; in addition, endothelial cell survival was increased, while intracellular ROS production was decreased in vitro as determined by examining the serum of the lifestyle-modified patients [111].
Laughter, hobbies, and joyful music
In contrast to acute and chronic mental stresses, positive emotions, such as mirthful laughter, would beneficially influence endothelial function. Volunteers watching a movie or segment of popular comedies showed a 22% increase in endothelium-dependent flow-mediated brachial artery dilatation, whereas those viewing a movie known to promote mental stress exhibited a 35% reduction of flow-mediated vasodilatation [74].
Mirthful laughter may serve as a useful and important vehicle for minimizing endothelial damage and promoting vascular health [73]. The percent change in coronary blood flow and coronary artery diameter in response to ACh, but not to GTN, was significantly greater in the hobby group, who enjoyed a variety of hobbies, than in the non-hobby group after the average follow-up period of 916 days in patients with almost normal coronary arteries [96]. Enjoying hobbies may improve cardiovascular outcomes via its favorable effects on coronary endothelial function. There was a significant inverse correlation between humor production and antisocial Type A personality traits, suggesting that the propensity to laugh may contribute to coronary vascular protection [15].
In healthy, nonsmoking volunteers, music that evoked joy was associated with increases in flow-mediated brachial artery dilatation, whereas reductions of flow-mediated vasodilatation were observed after listening to music that elicited anxiety [75]. Listening to joyful music may contribute to the promotion of vascular endothelial health. Worries and fear raised during two psychometric tests were reduced by listening to especially composed relaxation music in patients from a coronary sport group [126].
Exposure of human or rat endothelial cells to morphine results in NO release via the μ3-opiate-receptor subtype [112]. Endogenous opioid peptides, such as endomorphin, activate μ-opiate receptors which in turn upregulate eNOS to enhance NO production [12, 48]. Nociceptin, the endogenous ligand of the opioid receptor-like 1 receptor, also induce vasodilatation through endothelial NO [44, 137]. It is intriguing to determine whether endogenous opioids/opioid peptides in concentrations sufficient to act in favor of endothelial function in humans are released by mirthful laughter, hobbies, and joyful music.
Summary and conclusion
In this review, we have discussed mental stress-related impairments of endothelial function mediated by stress hormones and mediators, including glucocorticoids, catecholamines, endothelin-1, pro-inflammatory cytokines, and endogenous opioids/opioid peptides, and their mechanisms of action on humans and experimental animals. Acute mental stress induces either an impairment or an enhancement of endothelial function, a difference that may possibly depend on the environment in which the study is conducted, behavioral hostility in the studied individuals, and the attitudes or personalities of examiners, examinees, or both. However, most of the recent studies (2000 to date) support the idea that acute mental stress negatively affects endothelial function. This untoward action appears to be mediated by elevated glucocorticoids and ET-1 concentrations that result in inhibition of eNOS activity, downregulation of eNOS mRNA and protein, and increased production of ROS via BH4 deficiency and NADPH oxidase activation. However, catecholamines released by acute stress do not directly affect endothelial function, but when blood pressure is repeatedly increased or sustained at a high level via the released catecholamines, endothelial function is impaired. Chronic mental stress always evokes endothelial dysfunction in humans and experimental animals. Together with glucocorticoids, catecholamines, and ET-1, pro-inflammatory cytokines and endogenous opioids are involved in the regulation of endothelial function, cytokines being harmful and opioids/opioid peptides (endogenous morphine, endomorphin, methionine enkephalin, and nociceptin) being beneficial. Stress-related changes in endothelial function are summarized in Fig. 2. There is an evident imbalance in the regulation of endothelial function that is associated with a greater increase in stress hormones and mediators showing negative effects under chronic mental stress. Therapeutic and prophylactic measures to suppress the release or action of deleterious hormones and mediators as well as increasing the release of beneficial opioids/opioid peptides should lessen or counteract stress-related endothelial dysfunction. Lifestyle modifications, such as mirthful laughter, humor orientation, positive socialization, and cessation of smoking and excess drinking, are recommended. In our home country Japan, we have unfortunately suffered (March 2011) an enormous earthquake followed by a huge “tsunami” in the eastern region, and the magnitude of this disaster could never be adequately expressed in words. The suffering residents and their relatives must be exposed to severe and prolonged mental as well as physical stresses. We send our heartfelt prayers to them to raise positive thinking and hope for a better future for those affected by this disaster. The numbers of domestic and foreign humanitarian volunteers are continuing to provide joyful music, comedy shows, plays to evoke mirthful laughter, delicious healthy meals, and medical and psychological consultations that are hopefully beneficial for the good health of those affected by this disaster via reversing endothelial dysfunction and improving cerebral and cardiac circulation.

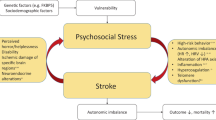
Mechanisms underlying deleterious and beneficial effects of mental stress on endothelial function via adrenocorticotropic hormone (ACTH), glucocorticoids (GC), endothelin-1 (ET-1), pro-inflammatory cytokines (Cyto), opioids/opioid peptides (Opi), catecholamine (CA), and blood pressure elevation (BP↑). Endothelial function was evaluated as blood flow regulation by endothelial NO. Minus sign deleterious action, plus sign beneficial action, plus–minus sign no action
Abbreviations
- ACh:
-
Acetylcholine
- ACTH:
-
Adrenocorticotropic hormone
- BH4 :
-
Tetrahydrobiopterin
- BK:
-
Bradykinin
- Cyclic GMP:
-
Cyclic guanosine monophosphate
- CRH:
-
Corticotropine-releasing hormone
- eNOS:
-
Endothelial nitric oxide synthase
- ET:
-
Endothelin
- GTN:
-
Nitroglycerin
- HUVEC:
-
Human umbilical vein endothelial cell
- IL:
-
Interleukin
- l-NA:
-
N G-nitro-l-arginine
- l-NAME:
-
l-NA methylester
- l-NMMA:
-
N G-monomethyl-l-arginine
- MCh:
-
Methacholine
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- NOx:
-
Nitrate/nitrite
- PI3K:
-
Phosphatidylinositol 3-kinase
- ROS:
-
Reactive oxygen species
- SNP:
-
Sodium nitroprusside
- SOD:
-
Superoxide dismutase
- TNF:
-
Tumor necrosis factor
- VEGF:
-
Vascular endothelial growth factor
References
Armstead WM (1999) Stimulus duration modulates the interaction between opioids and nitric oxide in hypoxic pial artery dilation. Brain Res 825:68–74
Arrick DM, Mayhan WG (2010) Inhibition of endothelin-1 receptors improves impaired nitric oxide synthase-dependent dilation of cerebral arterioles in type-1 diabetic rats. Microcirculation 17:439–446
Bairey Merz CN, Dwyer J, Nordstrom CK, Walton KG, Saloerno JW, Schneider RH (2002) Psychosocial stress and cardiovascular disease: pathophysiological links. Behav Med 27:141–147
Bhagat K, Vallance P (1997) Inflammatory cytokines impair endothelium-dependent dilatation in human veins in vivo. Circulation 96:3042–3047
Black PH, Garbutt LD (2002) Stress, inflammation and cardiovascular disease. J Psychosom Res 52:1–23
Bonfiglio V, Bucolo C, Camillieri G, Drago F (2006) Possible involvement of nitric oxide in morphine-induced miosis and reduction of intraocular pressure in rabbits. Eur J Pharmacol 534:227–232
Brion MJ, Drummond PD (2005) The effect of glucose and mental stress on cutaneous microvascular endothelial function. Psychophysiology 42:282–289
Broadley AJ, Korszun A, Abdelaal E, Moskvina V, Jones CJ, Nash GB (2005) Inhibition of cortisol production with metyrapone prevents mental stress-induced endothelial dysfunction and baroreflex impairment. J Am Coll Cardiol 46:344–350
Cardillo C, Kilcoyne CM, Cannon RO 3rd, Panza JA (1998) Impairment of the nitric oxide-mediated vasodilator response to mental stress in hypertensive but not in hypercholesterolemic patients. J Am Coll Cardiol 32:1207–1213
Cardillo C, Kilcoyne CM, Quyyumi AA, Cannon RO 3rd, Panza JA (1997) Role of nitric oxide in the vasodilator response to mental stress in normal subjects. Am J Cardiol 80:1070–1074
Champion HC, Bivalacqua TJ, Friedman DE, Zadina JE, Kastin AJ, Kadowitz PJ (1998) Nitric oxide release mediates vasodilator responses to endomorphin 1 but not nociceptin/OFQ in the hindquarters vascular bed of the rat. Peptides 19:1595–1602
Champion HC, Bivalacqua TJ, Zadina JE, Kastin AJ, Kadowitz PJ (1999) Vasodilator responses to the endomorphin peptides, but not nociceptin/OFQ, are mediated by nitric oxide release. Ann NY Acad Sci 897:165–172
Chumaeva N, Hintsanen M, Juonala M, Raitakari OT, Keltikangas-Järvinen L (2010) Sex differences in the combined effect of chronic stress with impaired vascular endothelium functioning and the development of early atherosclerosis: Cardiovascular Risk in Young Finns study. BMC Cardiovasc Disord 10:34
Chung IM, Kim YM, Yoo MH, Shin MK, Kim CK, Suh SH (2010) Immobilization stress induces endothelial dysfunction by oxidative stress via the activation of angiotensin II/its type I receptor pathway. Atherosclerosis 213:109–114
Clark A, Seidler A, Miller M (2001) Inverse association between sense of humor and coronary heart disease. Int J Cardiol 80:87–88
Constantopoulos A, Papadaki-Papandreou U, Papaconstantinou E (1995) Increased β-endorphin but not Leu-enkephalin in plasma due to preoperative stress. Experientia 51:16–18
Cooper DC, Milic MS, Mills PJ, Bardwell WA, Ziegler MG, Dimsdale JE (2010) Endothelial function: the impact of objective and subjective socioeconomic status on flow-mediated dilation. Ann Behav Med 39:222–231
Cooper DC, Milic MS, Tafur JR, Mills PJ, Bardwell WA, Ziegler MG, Dimsdale JE (2010) Adverse impact of mood on flow-mediated dilation. Psychosom Med 72:122–127
d’ Audiffret AC, Frisbee SJ, Stapleton PA, Goodwill AG, Isingrini E, Frisbee JC (2010) Depressive behavior and vascular dysfunction: a link between clinical depression and vascular disease? J Appl Physiol 108:1041–1051
Das S, O’Keefe JH (2006) Behavioral cardiology: recognizing and addressing the profound impact of psychosocial stress on cardiovascular health. Curr Atheroscler Rep 8:111–118
de Andrade CR, Fukada SY, Olivon VC, de Godoy MA, Haddad R, Eberlin MN, Cunha FQ, de Souza HP, Laurindo FR, de Oliveira AM (2006) α1D-Adrenoceptor-induced relaxation on rat carotid artery is impaired during the endothelial dysfunction evoked in the early stages of hyperhomocysteinemia. Eur J Pharmacol 543:83–91
Devine DP, Watson SJ, Akil H (2001) Nociceptin/orphanin FQ regulates neuroendocrine function of the limbic–hypothalamic–pituitary–adrenal axis. Neuroscience 102:541–553
Devine JO, Armstead WM (1995) The role of nitric oxide in opioid-induced pial artery vasodilation. Brain Res 675:257–263
Dharmashankar K, Widlansky ME (2010) Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep 12:448–455
Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ (1994) Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol 480:361–368
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature (Lond) 399:601–605
Dortch-Carnes J, Russell KR (2006) Morphine-induced reduction of intraocular pressure and pupil diameter: role of nitric oxide. Pharmacology 77:17–24
Duckles SP, Miller VM (2010) Hormonal modulation of endothelial NO production. Pflügers Arch 459:841–851
Eriksson M, Johansson K, Sarabi M, Lind L (2007) Mental stress impairs endothelial vasodilatory function by β-adrenergic mechanism. Endothelium 14:151–156
Feng Y, Zhao QY, Chen Q, Wang R (2005) Vasorelaxant responses to endomorphin 1 [psi] and endomorpohin 2 [psi], analogues of endomorphins, in rat aorta rings. Pharmazie 60:85–855
Fernandez AB, Soufer R, Collins D, Soufer A, Ranjbaran H, Burg MM (2010) Tendency to angry rumination predicts stress–provoked endothelin-1 increase in patients with coronary artery disease. Psychosom Med 72:348–353
Figueroa XF, Poblete I, Fernández R, Pedemonte C, Cortés V, Huidobro-Toro JP (2009) NO production and eNOS phosphorylation induced by epinephrine through the activation of β-adrenoceptors. Am J Physiol 297:H134–H143
Fimiani C, Mattocks D, Cavani F, Salzet M, Deutsch DG, Pryor S, Bilfinger TV, Stefano GB (1999) Morphine and anandamide stimulate intracellular calcium transients in human arterial endothelial cells: coupling to nitric oxide release. Cell Signal 11:189–193
Fontana F, Bernardi P, Pich EM, Boschi S, Iasio R, Spampinato S, Grossi G (1997) Opioid peptide modulation of circulatory and endocrine response to mental stress in humans. Peptides 18:169–175
Förstermann U, Pollock JS, Schmidt HH, Heller M, Murad F (1991) Calmodulin-dependent endothelium–derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci USA 88:1788–1792
Fuchs LC, Landas SK, Johnson AK (1997) Behavioral stress alters coronary vascular reactivity in borderline hypertensive rats. J Hypertens 15:301–307
Furchgott RF, Zawadzki JV (1980) The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature (Lond) 288:373–637
García-Bueno B, Caso JR, Leza JC (2008) Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci Biobehav Rev 32:1136–1151
Gerra G, Zaimovic A, Mascetti GG, Gardini S, Zambelli U, Timpano M, Raggi MA, Brambilla F (2001) Neuroendocrine responses to experimentally-induced psychological stress in healthy humans. Psychoneuroendocrinology 26:91–107
Ghiadoni L, Donald AE, Cropley M, Muller MJ, Oakley G, O’Cornnor G, Betteridge J, Klein N, Steptoe A, Deanfield JE (2000) Mental stress induces transient endothelial dysfunction in humans. Circulation 102:2473–2478
Giulumian AD, Clark SG, Fuchs LC (1999) Effect of behavioral stress on coronary artery relaxation altered with aging in BHR. Am J Physiol 276:R435–R440
Gottdiener JS, Kop WJ, Hausner E, McCeney MK, Herrington D, Krantz DS (2003) Effects of mental stress on flow-mediated brachial arterial dilation and influence of behavioral factors and hypercholesterolemia in subjects without cardiovascular disease. Am J Cardiol 92:687–691
Green MK, Devine DP (2009) Nociceptin/orphan FQ and NOP receptor gene regulation after acute or repeated social defeat stress. Neuropeptides 43:507–514
Gumusel B, Hao AQ, Hyman A, Chang JK, Kapusta DR, Lippton H (1997) Nociceptin: an endogenous agonist for central opioid like 1 (ORL1) receptors possesses systemic vasorelaxant properties. Life Sci 60:PL141–PL145
Harris CW, Edwards JL, Baruch A, Riley WA, Pusser BE, Rejeski WJ, Herrington DM (2000) Effects of mental stress on brachial artery flow-mediated vasodilation in healthy normal individuals. Am Heart J 139:405–411
He JJ, Chen XQ, Wang L, Xu JF, Du JZ (2008) Corticotropin–releasing hormone receptor 1 coexists with endothelin-1 and modulates its mRNA expression and release in rat paraventricular nucleus during hypoxia. Neuroscience 152:1006–1014
Hingorani AD, Cross J, Kharbanda RK, Mellen MJ, Bhagat K, Taylor M, Donald AE, Palacios M, Griffin GE, Deanfielod JE, MacAllister RJ, Vallance P (2000) Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation 102:994–999
Hugghins SY, Champion HC, Cheng G, Kadowitz PJ, Jeter JRV Jr (2000) Vasorelaxant responses to endomorphins, nociceptin, albuterol, and adrenomedullin in isolated rat aorta. Life Sci 67:471–478
Ikeda U, Yamamoto K, Maeda Y, Shimpo M, Kanbe T, Shimada K (1997) Endothelin-1 inhibits nitric oxide synthesis in vascular smooth muscle cells. Hypertension 29:65–69
Iuchi T, Akaike M, Mitsui T, Ohshima Y, Shintani Y, Azuma H, Matsumoto T (2003) Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ Res 92:81–87
Jambrik Z, Chunzeng L, Santarcangelo EL, Sebastiani L, Ghelarducci B, Picano E (2004) Traditional acupuncture does not modulate the endothelial dysfunction induced by mental stress. Int J Cardiovasc Imaging 20:357–362
Jambrik Z, Sabastiani L, Picano E, Ghelarducci B, Santarcangelo EL (2005) Hypnotic modulation of flow-mediated endothelial response to mental stress. Int J Psychophysiol 55:221–227
Jambrik Z, Santarcangelo EL, Ghelarducci B, Sabastiani L (2004) Does hypnotizability modulate the stress-related endothelial dysfunction? Brain Res Bull 63:213–216
Jankord R, McAllister RM, Ganjam VK, Laughlin MH (2009) Chronic inhibition of nitric oxide synthase augments the ACTH response to exercise. Am J Physiol 296:R728–R734
Jensen BC, Swigart PM, Montgomery MD, Simpson PC (2010) Functional α1B- adrenergic receptors on human epicardial coronary artery endothelial cells. Naunyn-Schmiedebergs Arch Pharmacol 382:475–482
Jern C, Selin L, Jern S (1994) In vivo release of tissue–type plasminogen activator across the human forearm during mental stress. Thromb Haemost 72:285–291
Johns DG, Dorrance AM, Tramontini NL, Webb RC (2001) Glucocorticoids inhibit tetrahydrobiopterin-dependent endothelial function. Exp Biol Med (Maywood) 226:27–31
Kamper EF, Chatzigeorgiou A, Tsimpoukidi O, Kamper M, Dalla C, Pitychoutis PM, Papadopoulou-Daifoti Z (2009) Sex differences in oxidant/antioxidant balance under a chronic mild stress regime. Physiol Behav 98:215–222
Kelly JJ, Williamson P, Martin A, Witworth JA (2001) Effects of oral l-arginine on plasma nitrate and blood pressure in cortisol-treated humans. J Hypertens 19:263–268
Kimura Y, Hirooka Y, Sagara Y, Sunagawa K (2007) Long–acting calcium channel blocker, azelnidipine, increases endothelial nitric oxide synthase in the brain and inhibits sympathetic nerve activity. Clin Exp Hypertens 29:13–21
Kofler S, Nickel T, Weis M (2005) Role of cytokines in cardiovascular disease: a focus on endothelial responses to inflammation. Clin Sci (Lond) 108:205–213
Kou R, Michel T (2007) Epinephrine regulation of the endothelial nitric-oxide synthase: roles of RAC1 and β3-adrenergic receptors in endothelial NO signaling. J Biol Chem 282:32719–32729
Liu Y, Mloadinov D, Pietrusz JL, Usa K, Liang M (2009) Glucocorticoid response elements and 11 β-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression. Cardiovasc Res 81:140–147
López-Figueroa MO, Day HE, Akil H, Watson SJ (1998) Nitric oxide in the stress axis. Histol Histopath 13:1243–1252
Low CA, Salomon K, Matthews KA (2009) Chronic life stress, cardiovascular reactivity, and subclinical cardiovascular disease in adolescents. Psychosom Med 71:927–931
Maeda S, Miyauchi T, Iemitsu M, Tanabe T, Goto K, Yamaguchi I, Matsuda M (2004) Endothelin receptor antagonist reverses decreased NO system in the kidney in vivo during exercise. Am J Physiol 286:E609–E614
Mancaş S, Mihalaş G, Gaitã D, Drăgan S, Duda-Seiman DM, Sarău CA, Noveanu L, Petcov B, Mancaş G, Ionescu V, Păcurar M (2008) Environmental factors and cardiovascular risk in young individuals. Rom J Intern Med 46:69–75
Mangiafico RA, Malatino LS, Attiná T, Messina R, Fiore CE (2002) Exaggerated endothelin release in response to acute mental stress in patients with intermittent claudication. Angiology 53:383–390
Mangos GJ, Walker BR, Kelly JJ, Lawson JA, Webb DJ, Whitworth JA (2000) Cortisol inhibits cholinergic vasodilation in the human forearm. Am J Hypertens 13:1155–1160
Martin EA, Prasad A, Rihal CS, Lerman LO, Lerman A (2010) Endothelial function and vascular response to mental stress are impaired in patients with apical ballooning syndrome. J Am Coll Cardiol 56:1840–1846
Mausbach BT, Roepke SK, Ziegler MG, Milic M, von Känel R, Dimsdale JE, Mills PJ, Patterson TL, Allison MA, Ancoli-Israel S, Grant I (2010) Association between chronic caregiving stress and impaired endothelial function in the elderly. J Am Coll Cardiol 55:2599–2606
Michell BM, Webb RC (2002) Impaired vasodilation and nitric oxide synthase activity in glucocorticoid-induced hypertension. Biol Res Nurs 4:16–21
Miller M, Fry WF (2009) The effect of mirthful laughter on the human cardiovascular system. Med Hypotheses 73:636–639
Miller M, Mangano C, Park Y, Goel R, Plotnick GD, Vogel RA (2006) Impact of cinematic viewing on endothelial function. Heart 92:261–262
Miller M, Mangano CC, Kop W, Vogel RA (2010) Divergent effects of joyful and anxiety–provoking music on endothelial vasoreactivity. Psychosom Med 72:354–356
Mondo CK, Yang WS, Zhang N, Huang TG (2006) Anti-oxidant effects of atorvastatin in dexamethasone-induced hypertension in the rat. Clin Exp Pharmacol Physiol 33:1029–1034
Morimoto K, Kurahashi Y, Shintani-Ishida K, Kawamura N, Miyashita M, Uji M, Tan N, Yoshida K (2004) Estrogen replacement suppresses stress-induced cardiovascular responses in ovariectomized rats. Am J Physiol 287:H1950–H1956
Musumeci V, Baroni S, Cardillo C, Zapoacosta B, Zuppi C, Tutinelli F, Folli G (1987) Cardiovascular reactivity, plasma markers of endothelial and platelet activity and plasma rennin activity after mental stress in normals and hypertensives. J Hypertens 5:S1–S4
Naqvi TZ, Hyuhn HK (2009) Cerebrovascular mental stress reactivity is impaired in hypertension. Cardiovasc Ultrasound 7:32
Neves VJ, Moura MJ, Tamascia ML, Ferreira R, Silva NS, Costa R, Montemor PL, Narvaes EA, Bernardes CF, Novaes PD, Marcondes FK (2009) Proatherosclerotic effects of chronic stress in male rats: altered phenylephrine sensitivity and nitric oxide synthase activity of aorta and circulating lipids. Stress 12:320–327
Nickel T, Deutschumann A, Hanssen H, Summo C, Wilbert-Lampen U (2009) Modification of endothelial biology by acute and chronic stress hormones. Microvasc Res 78:364–369
Oishi A, Takahashi K, Ohmichi M, Mochizuki Y, Inaba N, Kurachi H (2011) Role of glucocorticoid receptor in the inhibitory effect of medroxyprogesterone acetate on the estrogen-induced endothelial nitric oxide synthase phosphorylation in human umbilical vein endothelial cells. Fertil Steril 95:1168–1170
Ong SL, Zhang Y, Whitworth JA (2008) Reactive oxygen species and glucocorticoid-induced hypertension. Clin Exp Pharmacol Physiol 35:477–482
Park HJ, Park HJ, Chae Y, Kim JW, Lee H, Chrng JH (2011) Effect of acupuncture on hypothalamic–pituitary–adrenal system in maternal separation rats. Cell Mol Neurobiol. doi:10.1007/s10571-011-9718-x
Paz Y, Frolkis I, Pevni D, Shapira I, Yuhas Y, Iaina A, Wollman Y, Chemichovski T, Nesher N, Locker C, Mohr R, Uretzky G (2003) Effect of tumor necrosis factor-α on endothelial and inducible nitric oxide synthase messenger ribonucleic acid expression and nitric oxide synthesis in ischemic and nonischemic isolated rat heart. Am J Coll Cardiol 42:1299–1305
Peix A, Tápaga A, Asen L, Ponce F, Infante O, Valiente J, Tornés F, Cabrera LO, Guerrero I, Garcia EJ, Carrillo R, Garcia-Barreto D (2006) Mental stress-induced myocardial ischemia in women with angina and normal coronary angiograms. J Nucl Cardiol 13:507–513
Pryor SC, Zhu W, Cadet P, Bianchi E, Guarna M, Stefano GB (2005) Endogenous morphine: opening new doors for the treatment of pain and addiction. Expert Opin Biol Ther 5:893–906
Púzserová A, Csizmadiová Z, Andriantsitohaina R, Bernátová I (2006) Vascular effects of red wine polyphenols in chronic stress–exposed Wistar–Kyoto rats. Physiol Res 55(suppl 1):S39–S47
Qi YM, Yang DJ, Duan X, Yang F, Li SR, Shen JM, Wang R (2002) Endomorphins inhibit contractile responses of rat thoracic aorta rings induced by phenylephrine and angiotensin II in vitro. Acta Pharmacol Sin 23:40–44
Ramzy D, Rao V, Tumiati LC, Xu N, Sheshgiri R, Miriuka S, Delgado DH, Ross HJ (2006) Elevated endothelin-1 levels impair nitric oxide homeostasis through a PKC-dependent pathway. Circulation 114:1319–1326
Rebrova TY, Maslow LN, Lishmanov AY, Tam SV (2001) Stimulation of μ and δ-opiate receptors and tolerance of isolated heart to oxidative stress: the role of NO–synthase. Biochemistry (Mosc) 66:422–428
Reif A, Fröhlich LG, Kotsonis P, Frey A, Bömmelo HM, Wink DA, Pfeiderer W, Schmidt HH (1999) Tetrahydrobiopterin inhibits monomerization and is consumed during catalysis in neuronal NO synthase. J Biol Chem 274:24921–24929
Rettori V, Mohn C, Scorticati C, Vissio P, Cella M, Farina M, Franchi A, McCann SM (2003) Effects of neurogenic stress and ethanol on nitric oxide synthase and cyclooxygenase activities in rat adrenals. Ann NY Acad Sci 992:86–98
Rogers KM, Bonar CA, Estrella JL, Yang S (2002) Inhibitory effect of glucocorticoid on coronary endothelial function. Am J Physiol 283:H1922–H1928
Rozanski A, Blumenthal JA, Kaplan J (1999) Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation 99:2192–2217
Saihara K, Hamasaki S, Ishida S, Kataoka T, Yoshikawa A, Orihara M, Ogawa M, Oketani N, Fukudome T, Atsuchi N, Shinsato T, Okui H, Kubozono T, Ichiki H, Kuwahata S, Mizoguchi E, Fujita S, Takumi T, Ninomiya T, Tomita K, Tei C (2010) Enjoying hobbies is related to desirable cardiovascular effects. Hear Vessel 25:113–120
Santos AC, Alves MJ, Rondon MU, Barrette AC, Middlekauff HR, Negrão CE (2005) Sympathetic activation restrains endothelium-mediated muscle vasodilatation in heart failure patients. Am J Physiol 289:H593–HH599
Sarabi M, Lind L (2001) Mental stress opposes endothelium-dependent vasodilatation in young healthy individuals. Vasc Med 6:3–7
Schedlowski M, Flüge T, Richter S, Tewes U, Schmidt RE, Wagner TO (1995) β-Endorphin, but not substance-P, is increased by acute stress in humans. Psychoneuroendocrinology 20:103–110
Schwartz BG, Mayeda GS, Burstein S, Economides C, Kloner RA (2010) When and why do heart attacks occur? Cardiovascular triggers and their potential role. Hosp Pract (Minneap) 38:144–152
Seidel M, Billert H, Kurpisz M (2006) Regulation of eNOS expression in HCAEC cell line treated with opioids and proinflammatory cytokines. Kardiol Pol 64:153–158
Seya Y, Fukuda T, Isobe K, Kawakami Y, Takekoshi K (2006) Effect of norepinephrine on RhoA, MAP kinase, proliferation and VEGF expression in human umbilical vein endothelial cells. Eur J Pharmacol 553:54–60
Sherwood A, Johnson K, Blumenthal JA, Hinderliter AL (1999) Endothelial function and hemodynamic responses during mental stress. Psychosom Med 61:365–370
Shimada K, Fukuda S, Maeda K, Kawasaki T, Kono Y, Jissho S, Taguchi H, Yoshiyama M, Yoshikawa J (2011) Aromatherapy alleviates endothelial dysfunction of medical staff after night–shift work: preliminary observations. Hypertens Res 34:264–267
Shinozaki K, Kashiwagi A, Nishio Y, Okamura T, Yoshida Y, Masada M, Toda N, Kikkawa R (1999) Abnormal biopterin metabolism is a major cause of impaired endothelium-dependent relaxation through nitric oxide/O −2 imbalance in insulin–resistant rat aorta. Diabetes 48:2437–2445
Shinozaki K, Nishio Y, Okamura T, Yoshida Y, Maegawa H, Kojima H, Masada M, Toda N, Kikkawa R, Kashiwagi A (2000) Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aorta of insulin–resistant rats. Circ Res 87:566–573
Simmons WW, Ungureanu-Longrois D, Smith GK, Smith TW, Kelly RA (1996) Glucocorticoids regulate inducible nitric oxide synthase by inhibiting tetrahydrobiopterin synthesis and l-arginine. J Biol Chem 271:23928–23937
Sivasankaran S, Pollart-Quintner S, Sachdeva R, Pugeda J, Hoq SM, Zarich SW (2006) The effect of a six–week program of yoga and meditation on brachial artery reactivity: do psychosocial interventions affect vascular tone? Clin Cardiol 29:393–398
Skantze HB, Kaplan J, Pettersson K, Manuck S, Blomqvist N, Kyes R, Williams K, Bondjers G (1998) Psychosocial stress causes endothelial injury in cynomolgus monkeys via β1-adrenoceptor activation. Atherosclerosis 136:153–161
Spieker LE, Hürlimann D, Ruschitzka F, Enseleit F, Shaw S, Hayoz D, Deanfield JE, Lüscher TF, Noll G (2002) Mental stress induces prolonged endothelial dysfunction via endothelin-A receptors. Circulation 105:2817–2820
Srimahachota S, Wunsuwan R, Siritanitikorn A, Boonla C, Chaiwongkarjohn S, Tosukhowong P (2010) Effects of lifestyle modification on oxidized LDL, reactive oxygen species production and endothelial cell viability in patients with coronary artery disease. Clin Biochem 43:858–862
Stefano GB, Hartman A, Bilfinger TV, Magazine HI, Liu Y, Casares F, Goligorsky MS (1995) Presence of the μ3 opiate receptor in endothelial cells. Coupling to nitric oxide production and vasodilation. J Biol Chem 270:30290–30293
Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD (1996) Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest 97:2601–2610
Strawn WB, Bondjers G, Kaplan JR, Schwenke DC, Hansson GK, Shively CA, Clarkson TB (1991) Endothelial dysfunction in response to psychosocial stress in monkeys. Circ Res 68:1270–1279
Takase B, Akima T, Uehata A, Ohsuzu F, Kurita A (2004) Effect of chronicstress and sleep deprivation on both flow-mediated dilation in the brachial artery and the intracellular magnesium level in humans. Clin Cardiol 27:223–227
Tang EH, Vanhoutte PM (2010) Endothelial dysfunction: a strategic target in the treatment of hypertension? Pflügers Arch 459:995–1004
Toda N, Ayajiki K (2010) Vascular actions of nitric oxide as affected by exposure to alcohol. Alcohol Alcohol 45:347–355
Toda N, Ayajiki K, Okamura T (2009) Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev 61:62–97
Toda N, Nakanishi-Toda M (2007) Nitric oxide: ocular blood flow, glaucoma, and diabetic retinopathy. Ret Eye Res 26:205–238
Toda N, Tanabe S, Nakanishi S (2011) Nitric oxide-mediated coronary flow regulation in patients with coronary artery disease: recent advances. Int J Angiol 20:121–134
Toda N, Toda H (2010) Nitric oxide-mediated blood flow regulation as affected by smoking and nicotine. Eur J Pharmacol 649:1–13
Treiber FA, Kapuku GK, Davis H, Pollock JS, Pollock DM (2002) Plasma endothelin-1 release during acute stress: role of ethnicity and sex. Psychosom Med 64:707–713
Tsatsoulis A, Fountoulakis S (2006) The protective role of exercise on stress system dysregulation and comorbidities. Ann NY Acad Sci 1083:196–213
Uji M, Yoshida K, Shitani-Ishida K, Morimoto K (2007) Sex difference in norepinephrine surge in response to psychological stress through nitric oxide in rats. Life Sci 80:860–866
Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O (2006) TNF-α downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest 116:2791–2798
Vollert JO, Störk T, RoseM MM (2003) Music as adjuvant therapy for coronary heart disease. Therapeutic music lowers anxiety, stress and β-endorphin concentrations in patients from a coronary sport group. Dtsch Med Wochenschr 128:2712–2716
Wallerath T, Gödecke A, Molojavyi A, Li H, Schrader J, Förstermann U (2004) Dexamethasone lacks effect on blood pressure in mice with a disrupted endothelial NO synthase gene. Nitric Oxide 10:36–41
Wallerath T, Whited K, Schäfer SC, Schwarz PM, Wohlfart P, Kleinert H, Lehr HA, LemmerB FU (1999) Down–regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc Nat Acad Sci USA 96:13357–13362
Webb HE, Garten RS, McMinn DR, Beckman JL, Kamimori GH, Acevedo EO (2011) Stress hormones and vascular function in firefighters during concurrent challenges. Biol Psychol 87:152–160
Wedgwood S, Black SM (2005) Endothelin-1 decreases endothelial NOS expression and activity through ETA receptor-mediated generation of hydrogen peroxide. Am J Physiol 288:L480–L487
Whitworth JA, Mangos GJ, Kelly JJ (2000) Cushing, cortisol, and cardiovascular disease. Hypertension 36:912–916
Wilbert-Lampen U, Trapp A, Barth S, Plasse A, Leistner D (2007) Effects of β-endorphin on endothelial/monocytic endothelin-1 and nitric oxide release mediated by μ1-opioid receptors: a potential link between stress and endothelial dysfunction? Endothelium 14:65–71
Wilbert-Lampen U, Trapp A, Modrzik M, Fiedler B, Straube F, Plasse A (2006) Effects of corticotrophin–releasing hormone (CRH) on endothelin-1 and NO release, mediated by CRH receptor subtype R2: a potential link between stress and endothelial dysfunction? J Psychosom Res 61:453–460
Wilderman MJ, Armstead WM (1998) Role of endothelial nitric oxide synthase in hypoxia-induced pial artery dilation. J Cereb Blood Flow Metab 18:531–538
Williams JK, Kaplan JR, Manuck SB (1993) Effects of psychosocial stress on endothelium-mediated dilation of atherosclerotic arteries in cynomologus monkeys. J Clin Invest 92:1819–1823
Williams JK, Vita JA, Manuck SB, Selwyn AP, Kaplan JR (1991) Psychosocial factors impair vascular responses of coronary arteries. Circulation 84:2146–2153
Xu PH, Chang M, Cheng LX, Cheng Q, Yan X, Wang R (2004) The relaxant effect of nociceptin on porcine coronary arterial ring segments. Can J Physiol Pharmacol 82:993–999
Yeung AC, Vekshtein VI, Krantz DS, Vita JA, Ryan TJ Jr, Ganz P, Selwyn AP (1991) The effect of atherosclerosis on the vasomotor response to coronary arteries to mental stress. N Engl J Med 325:1551–1556
Yoon H, Chung WS, Park YY, Cho IH (2005) Effects of stress on female rat sexual function. Int J Impot Res 17:33–38
Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW (1999) C–reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 19:972–978
Zemse SM, Chiao CW, Hilgers RH, Webb RC (2010) Interleukin-10 inhibits the in vivo and in vitro adverse effects of TNF-α on the endothelium of murine aorta. Am J Physiol 299:H1160–H1167
Zhang J, Patel JM, Li YD, Block ER (1997) Proinflammatory cytokines downregualte gene expression and activity of constitutive nitric oxide synthase in porcine pulmonary artery endothelial cells. Res Commun Mol Path Pharmacol 96:71–87
Zhu W, Ma Y, Bell A, Esch T, Guarna M, Bilofinger E, Bianchi E, Stefano GB (2004) Presence of morphine in rat amygdale: evidence for the μ3 opiate receptor subtype via nitric oxide release in limbic structures. Med Sci Monit 10:BR433–BR439
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Toda, N., Nakanishi-Toda, M. How mental stress affects endothelial function. Pflugers Arch - Eur J Physiol 462, 779–794 (2011). https://doi.org/10.1007/s00424-011-1022-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-011-1022-6