
Abstract
Phosphatidylinositol (4,5)-bisphosphate (PIP2) is an important lipid mediator that has multiple regulatory functions. There is now increasing evidence that the phosphatidylinositol 4-phosphate 5 kinases (PIP5Ks), which synthesize PIP2, are regulated spatially and temporally and that they have isoform-specific functions and regulations. This review will summarize the highlights of recent developments in understanding how the three major PIP5K isoforms regulate the actin cytoskeleton and other important cellular processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Phosphatidylinositol (4,5)-bisphosphate (PIP2) is particularly abundant at the plasma membrane (PM) and is proposed to be a PM organelle marker, which distinguishes the PM from the internal organelles that are more highly enriched in other types of phosphoinositides [35, 111, 140]. As such, PIP2 is the hub for the docking of multicomponent signaling complexes [56] and the maintenance of cytoskeletal-PM adhesion [122]. PIP2 is also a regulator of ion channels [58, 130], endocytic and exocytic membrane trafficking [72, 89], integrin signaling [32], cytokinesis [84], epithelial cell morphogenesis [88], and apoptosis [53, 92]. Some of these roles are mediated through PIP2-dependent modulation of the actin cytoskeleton [59, 120, 149], while others are not [111]. In addition, PIP2 is the immediate precursor to three pivotal second messengers, diacylglycerol (DAG), inositol (1,4,5)-triphosphate (InsP3), and phosphatidylinositol (3,4,5)-triphosphate (PIP3) [38].
PIP2 dynamics
PIP2 is found primarily on the cytoplasmic leaflet of the PM where it accounts for approximately 1% of membrane phospholipids [91]. PIP2 is generated from phosphatidylinositol monophosphates (PIPs) through two distinct pathways: first, by the type I phosphatidylinositol 4-phosphate 5 kinases (PIP5Ks) which phosphorylate phosphatidylinositol 4-phosphate [PI(4)P] on the D-5 position of the inositol ring, and second, by the type II PIP4Ks which phosphorylate PI(5)P at the D-4 position. These two types of PIP kinases are non-redundant and have distinct functions [60]. PIP4Ks exist as dimers, which form a flattened surface that docks on the lipid bilayer [107]. PIP5Ks are likely to have a similar overall organization. Domain-swapping experiments show that the recognition of PI(4)P vs PI(5)P is dictated primarily by the specificity loop within the kinase core; PIP4Ks have a conserved ala that recognizes PI(5)P, while PIP5Ks have a conserved glu that recognizes PI(4)P [75]. As PI4P is much more abundant than PI(5)P [132], PIP5Ks are likely to be the major source of PIP2. This is confirmed by pulse-labeling studies [128]. We will focus on PIP5Ks exclusively in this review.
PIP2 level is determined by a balance between synthesis and dissipation. PIP2 can be decreased in many ways. First, PIP2 is hydrolyzed by phospholipase C (PLC) to generate InsP3 and DAG. This provides an effective mechanism for downshifting the PIP2 signal [109]. Second, PIP2 is converted by the class I phosphoinositide 3 kinases (PI3Ks) to generate PIP3, which is important for signaling, growth regulation, and cell migration [44, 144]. Third, the D-5 phosphate on the PIP2 inositol ring is dephosphorylated by phosphoinositide 5 phosphatases [6], such as synaptojanin [104] and Ocrl [42], which have been implicated in the maintenance of PIP2 homeostasis at the PM and trans Golgi network (TGN), respectively. Fourth, PIP2 that is generated locally may be dissipated by diffusion, but a gradient can be maintained by continuous generation locally or by PIP2 binding to scaffolding molecules at a site of synthesis to immobilize the lipid [27].
There is now overwhelming evidence suggesting that some pools of PIP2 are generated in a spatially and temporally regulated manner and that downregulation of the PIP2 signal is critically important for the cycling of almost all PIP2-dependent processes [18, 28, 119].
New tools for studying PIP2 dynamics
PH-PLCδ-GFP has been widely used to monitor PIP2 dynamics in cells by high-resolution live cell imaging [13]. It strongly labels the PM [12], and its ability to accurately report PIP2 levels at the PM, when overexpressed at low level, has been corroborated in fixed cells using anti-PIP2 antibody [78, 140].
There have been many attempts to change the PIP2 level in cells. A constitutively membrane-targeted yeast inositol polyphosphate 5 phosphatase (Inp54p) is used to assess the role of PIP2 in membrane–cytoskeleton interactions [108]. A cell permeant PIP2-binding peptide derived from gelsolin’s PIP2-binding domain depletes PM PIP2 in 3T3-L1 adipocytes and inhibits glucose transport in an actin-dependent manner [45]. PIP2 is shuttled into intact cells through histone carriers to increase PIP2 globally [140] or selectively on the basal vs apical side of polarized epithelial cells [88].
Recently, the arsenal for manipulating PIP2 has become even more sophisticated. 5 phosphatase and PIP5K are targeted to the PM using the rapamycin-inducible FKBP and FRB dimerization [56, 131, 137]. In one study, the cytosolic form of Inp54p is fused to CFP-tagged FKBP (CFP-FKBP-Inp54p) and cotransfected with plasmids encoding FRB attached to the membrane targeting domain of Lyn11 (Lyn11-FRB) and YFP-PH-PLCδ [56]. Rapamycin induces translocation of the normally cytosolic CFP-FKBP-Inp54p to the PM and a reciprocal dissociation of YFP-PH-PLCδ from the PM. PIP2 depletion by PM targeted Inp54p blocks the KCNQ K+ channels, while rapamycin targeting of PIP5Kγ increases K+ current. Significantly, targeting of an activator of PI3K, which increases PIP3 but not PIP2, has no effect on the K+ channels. As PIP2 level is changed acutely in the absence of the cascade of Ca2+, DAG, or InsP3 signals that normally accompany PIP2 signaling, these results show conclusively that PIP2, and not the second messengers generated from PIP2, is the direct regulator of the K+ channels.
PIP2 microdomains
It has been reported that approximately half of the cell’s PIP2 is synthesized preferentially in cholesterol/sphingolipid enriched caveolar light membrane fractions (rafts) [48, 96, 105] and that these PIP2-enriched microdomains exhibit locally regulated PIP2 turnover and restricted diffusion-mediated exchanges with their environment [48]. There are also reports that PIP2 is enriched in noncaveolar microdomains that are the staging platforms for choreographing signaling and cytoskeletal dynamics. The existence of PIP2 microdomains is confirmed by immunofluorescence staining of PM sheets prepared from PC12 cells [3, 94]. The PM PIP2 microdomains are heterogenous; some contain conventional raft markers, while others are enriched for syntaxin, which is involved in Ca2+-mediated exocytosis and mostly excluded from the low-density raft fraction [3]. Other PIP2 clustering proteins have also been identified. These include MARCKS, which sequesters PIP2 under basal conditions and is induced by agonist signaling to release PIP2 for interaction with other PIP2 targets [91]. In the case of phagocytosis, PIP2 accumulates and remains in the phagocytic cup for minutes without diffusing away [27]. This accumulation is not dependent on rafts or the actin cytoskeleton, raising the possibilities that lipids are held in place by the restriction caused by extreme membrane curvature and by binding to proteins within the phagocytic cup.
Raft isolation by floatation on density gradients shows that PIP5K is not enriched in rafts in PC12 cells or platelets [3, 148], while the same PIP5K is recruited to lipid rafts during B cell activation [114]. The yeast PIP5K, Mss4p, is also not present in rafts, although its association with membrane is sphingolipid-dependent [70]. Recently, the existence of rafts per se has been intensely debated [121] because there are suggestions that detergent extraction per se induces artifactual clustering, and optical measurements give mixed results, with only some data supporting the existence of a less mobile lipid population [23].
The questions of whether PIP2 exists in heterogeneous microdomains, and whether these domains are formed by PIP2 in cholesterol-rich rafts by interaction with proteins, or by a combination of both, need to be answered. They hold the key to understanding how PIP2 is regulated spatially and temporally and how the PIP2 pools generated by the PIP5K isoforms are functionally, and possibly physically, segregated.
PIP2 regulates multiple actin-binding proteins
Cytoskeletal proteins were among the first shown to be regulated by PIP2 [59, 149], and many of these proteins regulate actin dynamics at the cell cortex [25, 57, 129]. Some bind PIP2 through well-defined PIP2 recognition modules [80]. For example, ezrin, a member of the ERM family, which links actin filaments to the PM, has a FERM domain which binds PIP2, inducing the relief of the autoinhibited state [103]. However, the majority of actin regulatory proteins bind PIP2 using less obviously structured motifs that contain clusters of basic/aromatic amino acids [149]. Some examples are: WASP family proteins, which promote actin assembly by activating the nucleating Arp2/3 complex; gelsolin family proteins, which sever and cap actin filaments to promote dynamic actin reorganization; vinculin, which regulates focal adhesion (FA) turnover; capping protein, which caps the (+) end of actin filaments; and cofilin, which severs actin filaments to accelerate their in vivo treadmilling. In most cases, PIP2’s charged inositol headgroup and hydrophobic acyl chain are both required for binding [50, 67, 77]. The length of the acyl chain is also critical; di-C16 and di-C8 PIP2 bind cofilin, while di-C4 PIP2 does not [50].
Recently, the three-dimensional structure of cofilin bound to di-C8 PIP2 has been solved by nuclear magnetic resonance [50]. It reveals rich mechanistic details about how cofilin interacts with PIP2 and suggests a model in which the interplay between cofilin inactivation by PIP2 and activation by dephosphorylation can specify the spatial and temporal regulation of cofilin at the PM.
Cloning of PIP5Ks
Yeast has a single PIP5K gene, while mammals have three. In 1996, two mammalian PIP5K isoforms were cloned simultaneously from human and mice [64, 85]. These isoforms were independently named α and β, but unfortunately, the human and mouse isoform designations were reversed. That is, human PIP5Kα is equivalent to mouse β, and human PIP5Kβ is equivalent to mouse α. This disparate nomenclature has generated much confusion in the literature. In this review, we will use the human isoform designation exclusively (Table 1). In 1998, a third isoform, named PIP5Kγ, was cloned [65], and it has splice variants that are functionally distinct [47, 65]. The EST database suggests that the α and β isoforms may also have alternative splice variants [85, 138], but they have not been characterized functionally.
The three PIP5K isoforms have a highly conserved central kinase homology domain which has approximately 80% sequence identity (Fig. 1). The PIP5Ks are functionally similar in many respects. In vitro, they have similar enzyme kinetics and they are activated by phosphatidic acid (PA) [65], which is generated by phospholipase D (PLD) [126] or DAG kinase [86]. In addition, all PIP5K isoforms are activated by ser/thr dephosphorylation [102, 145] and by small GTPases such as RhoA [24], Rac1 [141], and Arf6 [61]. When overexpressed, all can potentially cause trapping of membrane in recycling endosomes [18, 124], form endosomal tubules [124], inhibit phagosome closure [119], generate actin comet tails [112], and prime secretory granules for exocytosis [138].

The domain structure of human PIP5K isoforms and their phosphorylation sites. PIP5Kγ has an 87-kDa isoform and a 90-kDa isoform which has a 28 amino acid extension at its C-terminus (tail). Ser257 in PIP5Kα (equivalent to ser214 in β and ser264 in γ) is constitutively phosphorylated by PKA or autophosphorylation, and it is dephosphorylated by a PKC-dependent pathway [66, 102]. Dephosphorylation activates the lipid kinases. In addition, the PIP5Kγ90 tail has two tandem phosphorylation sites (tyr649 and ser650) which are mutually exclusive. Tyr649 phosphorylation activates PIP5Kγ90, while ser650 phosphorylation inhibits activity and blocks tyr649 phosphorylation. Tyr649 is phosphorylated by Src [83] and dephosphorylated by Shp-1 [10]. In neurons and neuroendocrine cells, PIP5Kγ90’s ser650 is phosphorylated by Cdk5 and dephosphorylated by calcineurin in response to K+-induced depolarization [79, 99]
In addition, the PIP5K isoforms have divergent amino and carboxyl terminal extensions (Fig. 1). These regions are likely to be important for generating isoform-specific function and regulation [36, 37, 82, 92, 99, 116, 139, 145]. In this review, we will summarize what is known about the role of each PIP5K isoform and highlight their isoform-specific roles.
PIP5K localization
PIP5Ks are cytosolic proteins that associate with the membrane as peripheral proteins. PIP5Kα, as well as the yeast and drosophila orthologs, is also present in the nucleus [16]. The mammalian PIP5K isoforms associate with the PM to a different extent [138], and membrane association is regulated by multiple stimuli [21, 40], especially Arf6, RhoA, and Rac [115, 149]. In addition, isoform-specific binding partners that promote site-specific targeting have also been identified. Some examples are talin for PIP5Kγ90 [37, 82], Ajuba [69], and Bruton’s tyr kinase [114] for PIP5Kα.
The PIP5K kinase homology domain is necessary for PM association [4], and the minimal targeting motif has been identified. These include two invariant lys residues in the specificity loop, the conserved glu that specifies PI(4)P recognition [76], and two tandem basic residues at the C-terminus of the kinase domain [4]. These five residues are conserved in all known PIP5Ks from yeast to human, suggesting that they may be the universal PIP5K PM targeting code.
Non-mammalian PIP5Ks
PIP5Ks are found in yeast, arabidopsis, drosophila, and Caenorhabditis elegans. Mss4p, the Saccharomyces cerevisiae PIP5K, is particularly enriched at the PM [8, 60]. It clusters at sites of dynamic cortical assembly that are distinct from cortical actin patches, and the formation of these clusters is independent of actin filaments [127]. Mss4p recruitment to the PM is dependent on normal sphingolipid biosynthesis, although it is not located in raft microdomains per se [70]. Mss4 mutants are unable to form actin cables, have abnormal distribution of actin patches, irregular cell shape, aberrant deposition of cell wall material, and decreased viability [34, 60]. These results establish that PIP2 has important roles in S. cerevisiae, including the regulation of the actin cytoskeleton. The Schizosaccharomyces pombe PIP5K, Its3p, is also enriched at the PM. It regulates cell wall integrity of the fission yeast through a PLC-mediated pathway [33], and it is concentrated at the septum during cytokinesis [150].
Drosophila and C. elegans each has three putative PIP5K genes, but most of them have not been characterized. The partially characterized drosophila Sktl is required for cell viability, germline development, and bristle morphology [54]. It has a nuclear localization signal, which shuttles Sktl between the nucleus and cytoplasm [22]. Its nuclear localization suggests that it might be an ortholog of PIP5Kα, which is also found in the nucleus [16]. Furthermore, as Sktl is not required for neurotransmitter release [54], it is not likely to be equivalent to mammalian PIP5Kγs, which have been strongly implicated in synaptic functions [37] (see below).
Mammalian PIP5Kβ
PIP5Kβ is ubiquitously expressed [64, 85] and exists as a soluble protein in the cytoplasm, in association with punctate cytoplasmic structures and the PM [101, 123]. Initially, much of the information about PIP5Kβ was obtained using transient transfection of WT and KD PIP5Kβ plasmids in cultured cells. There was no systemic attempt to distinguish between the function of individual PIP5K isoform. In some cases, the level of overexpression was unphysiologically high, overwhelming the normal mechanisms for specifying unique isoform functions. In addition, some of the putative KD mutants are not actually kinase dead, and most are not consistently dominant-negative (Table 2). Nevertheless, in spite of these caveats, there is convincing evidence that PIP5Kβ has a major role in actin regulation. Recently, RNA interference (RNAi) and gene knockout by homologous recombination have provided definitive evidence for its isoform-specific function.
Regulation of actin polymerization
PIP5Kβ overexpression induces actin polymerization, but the type of actin filaments formed is dependent on the cells studied and the extent of overexpression [9, 90, 123, 135, 146]. For example, PIP5Kβ overexpression induces the formation of pine needle-like actin structures in COS-7 cells [123] and robust stress fibers in CV1 cells [146]. The stress fibers are formed in a RhoA-dependent manner by changing the activity of several PIP2-sensitive actin regulatory proteins including gelsolin, profilin, cofilin, and erzin [146]. PIP5Kβ is also implicated in RhoA-dependent ezrin recruitment to microvilli in HeLa cells [90] and Rac1-dependent recruitment to cell junctions [9].
Frequently, overexpression of PIP5Kβ, or the other PIP5K isoforms, induces the formation of actin comet tails that propel vesicles enriched with PIP5K and PIP2 [112]. Comet formation is WASP- and Arp2/3-dependent. These comets may be used for vesicle trafficking of “raft”-associated cargoes from the TGN to the PM of nonpolarized cells [112] and selectively to the apical PM of polarized MDCK cells [52].
PIP5Kβ has also been implicated in neurite retraction downstream of RhoA and its effector, ROCK. PIP5Kβ overexpression in neuroblastoma N1E-115 cells induces neurite retraction even when ROCK is inactive; while overexpression of the PIP5Kβ KD mutant induces spontaneous neurite extension [136, 147]. PIP5Kβ may also be important for the phagocytosis of Yersinia [143], which binds to the host integrin β1 receptor to activate Rac1. Overexpression of either PIP5Kβ or Arf6 bypasses the Rac1 requirement, suggesting that they act downstream of Rac1 during Yersinia phagocytosis. As FcγR-mediated phagocytosis is also regulated by Rac and Arf6 [51], the possibility that PIP5Kβ is also involved merits investigation.
The importance of PIP5Kβ in actin regulation is supported by gene knockout. Mast cells from the PIP5Kβ−/− (mouse PIP5Kα−/−) mice have 35% less PIP2 and less polymerized actin at the cell cortex [116]. They have abnormally robust responses to Fcɛ receptor I cross-linking, and hyperresponsiveness is supported by the finding that the PIP5Kβ−/− mice exhibit enhanced passive cutaneous and systemic anaphylaxis. Latrunculin, which depolymerizes actin, increases degranulation and cytokine generation in WT mast cells, establishing that PIP5Kβ−/− phenotype can be directly attributed to decreased actin polymerization. Taken together, these results indicate that PIP5Kβ negatively regulates mast cell functions by maintaining a cortical actin network that dampens the dynamics of Fcɛ receptor I signaling and downstream responses. Paradoxically, although in vitro and in vivo evidence suggest that PIP5Kβ has an important role in actin regulation, the PIP5Kβ−/− mice have no other reported phenotype. They are viable and develop normally [116]. Furthermore, no compensatory change in the expression of the other PIP5Ks is detected.
Receptor-mediated endocytosis
PIP5Kβ also has roles that are not related to actin regulation. For example, PIP5Kβ overexpression promotes receptor-mediated endocytosis in HeLa cells. These are manifested by an increase in transferrin uptake, the number of nascent clathrin-coated pits at the PM, and the amount of membrane-associated clathrin adaptor protein AP-2 complexes [101]. Cytochalasin D or latrunculin A, agents that depolymerize actin by different mechanisms, does not block the PIP5Kβ effects. Significantly, PIP5Kβ depletion by RNAi inhibits transferrin uptake in HeLa cells, while depletion of the other PIP5Ks has little effect [101]. Taken together, these results establish that PIP5Kβ is the primary regulator of receptor-mediated endocytosis in HeLa cells. Neurons use another PIP5K isoform to regulate endocytosis, although the mechanism for increased AP-2 recruitment may be similar [11, 99].
Regulation by Rho and Arf family small GTPases
There is now extensive evidence to suggest that PIP5Ks are regulated by RhoA, Rac1, and Arf6. RhoA and Rac1 have potent, and sometimes opposite, effects on the actin cytoskeleton, and their ability to recruit and activate PIP5Ks provides a very attractive model for how they may regulate the actin cytoskeleton. This relation has been reviewed extensively [100,115,149] and will not be discussed further here.
Arf6, which regulates membrane trafficking between endosomes and the PM, has also been implicated in the regulation of cell motility and the actin cytoskeleton through PIP5Ks. Overexpression of the constitutively active Arf6 mutant or PIP5K induces trapping of PM-derived PIP2-rich vesicles in the recycling endosome compartment by polymerized actin [18]. Honda et al. [61] reported that PIP5Kβ (as well as PIP5Kα) is recruited to membrane ruffles by Arf6 and that recombinant Arf6-GTP binds PIP5Kβ and stimulates its lipid kinase activity. Surprisingly, Rac1 and RhoA do not stimulate PIP5Kβ activity under similar conditions, although they are reported to do so in other studies [24,133, 134,141]. The relation between Arf6 and PIP5K is further consolidated by the finding that Arf6 also recruits PLD2 to membrane ruffles [61,126]. PLD has been intimately linked to PIP5Ks because it generates the PIP5K activator, PA [61,126] and, in addition, it is itself activated by PIP2 [39,118]. Thus, Arf6 recruitment of both PIP5Ks and PLD to membrane ruffles establishes a positive feedback loop between PIP5K and PLD to synergistically amplify the initial Arf6 signal. Now there is also evidence that Arf6 regulates PIP5Kγ in neurons and chromaffin cells to promote membrane trafficking and vesicle priming for exocytosis [1, 73] (see below). In addition, Arf6 induces formation of endosomal tubules that contain PIP5Ks [124].
Although Arf6 is likely to be a primary regulator of PIP5K at the PM, some of the Arf6 responses may be coordinated with the activity of Rho GTPases because Arf6 may act upstream or downstream of RhoA and Rac, depending on the cellular context. This web of interactions could involve elaborate positive and negative feedback loops that are only beginning to be understood.
Regulation by phosphorylation/dephosphorylation
-
a.
Ser/thr phosphorylation
Unexpectedly, the PIP5Ks kinase core, which has no identifiable homology to any known protein kinase [15], can phosphorylate itself on ser/thr residues in vitro, and phosphorylation inhibits lipid kinase activity [66]. Although there is no evidence that PIP5K autophosphorylates in the context of the cell, this could explain why PIP5Ks are constitutively phosphorylated and provide a mechanism to dampen basal PIP2 generation under resting conditions. PIP5Ks’ phosphorylation is also regulated by conventional kinases and phosphatases. The cAMP-dependent protein kinase A (PKA) phosphorylates PIP5Kβ in vitro [102], and the PKA phosphorylation consensus is located in the kinase homology domain. Mutation of ser214 within the consensus (Fig. 1) to ala decreases basal PIP5Kβ phosphorylation by 60% [102].
Stimuli that activate PIP5Kβ by ser/thr dephosphorylation have also been identified. Lysophosphatidic acid, which activates RhoA, induces PIP5K dephosphorylation in a PKC-dependent manner in NIH 3T3 cells [102]. Hypertonicity, which increases PIP2 level and induces actin assembly in many types of cells, activates PIP5Kβ by dephosphorylation [145]. In addition, hypertonicity also promotes PIP5Kβ association with the PM. As neither actin disruption nor stabilization by pharmacological agents blocks PIP5Kβ dephosphorylation, PIP5Kβ is dephosphorylated upstream of actin remodeling. The RhoA effector, ROCK, is not involved because the PIP5Kβ response is not blocked by a ROCK inhibitor. Significantly, PIP5Kα and γ, which are also constitutively ser/thr phosphorylated under isotonic conditions, are not dephosphorylated by hypertonicity. These results clearly establish that PIP5Kβ is regulated by a balance between protein kinase and phosphatase activity in response to hypertonic stress and that the PIP5Ks have isoform-specific function and distinct modes of regulation.
-
b.
Tyr phosphorylation
It has been known for some time that tyr phosphorylation is involved in the control of PIP2 homeostasis. Pervanadate, a potent tyr phosphatase inhibitor, increases PIP2 in HEK293 and REF52 cells [112,113], while oxidative stress, which activates multiple tyr kinases, decreases PIP2 generation by isolated cardiac PM [93] and by HeLa cells [53]. The paradox of how stimuli that promote tyr phosphorylation have opposite effects on cellular PIP2 can be explained. First, PIP5K isoforms are expressed at different levels in different types of cells, and second, tyr phosphorylation appears to have different effects on the PIP5Ks. PIP5Kγ90 is activated by Src kinases [82] (see below), while preliminary evidence suggests that PIP5Kβ is inhibited [53]. PIP5Kβ inhibition is inferred from the finding that the PIP5Ks immunoprecipitated from H2O2-treated HeLa cells are less active in vitro than those from control cells [53]. Although this antibody recognizes all PIP5K isoforms, PIP5Kβ should dominate in the immunoprecipitate because it accounts for most of the PIP2 in HeLa cells [138]. Immunofluorescence studies also show that oxidative stress dissociates PIP5Kβ from the PM [53]. Thus, the large decrease in PM PIP2 in oxidant-stressed cells may be due to PIP5Kβ inactivation and dissociation from the PM. This decrease may be an early signal for apoptosis because PIP5Kβ overexpression, which prevents oxidant-induced PIP2 decrease, protects cells from apoptosis [53].
Mammalian PIP5Kγ
Unlike PIP5Kβ knockout, PIP5Kγ knockout mice die within a day of birth [36]. The primary cause of death has not been determined, but may be due to generalized neuronal defects (see below) which are manifested in the inability to suckle and move normally. Humans have at least two major PIP5Kγ splice variants: a short 87-kDa protein (PIP5Kγ87) and a slightly longer one that has 28 additional amino acids at its C-terminus (PIP5Kγ90) [65] (Fig. 1). PIP5Kγ87 is more abundant than PIP5Kγ90 in most cells [10, 49, 139], while PIP5Kγ90 dominates in the brain [37, 142]. Mice have an additional brain-specific 93-kDa splice variant which has not yet been described in humans [47]. As all three splice variants are knocked out in the currently available mouse model [36], it is difficult to attribute a defect to the knockout of a particular splice variant.
Focal adhesion dynamics
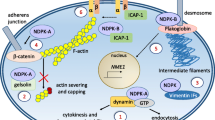
PIP2 has long been implicated in the regulation of FAs, which are sites of actin filament attachment to the extracellular matrix through integrin receptors and mediators of bidirectional integrin signaling [63]. The PIP2 level increases transiently during cell attachment to extracellular matrix, and PIP2 activates several key FA components including vinculin, α-actinin, and talin. Although vinculin mutants that do not bind PIP2 are recruited to FA normally, their FAs are static and turnover slowly [20, 117]. These results suggest that vinculin is a sensor of PIP2 in the FA and that it promotes dynamic FA assembly and disassembly. Talin, which binds vinculin, actin, and integrin, has a key role in coupling the cytoskeleton to integrins [29]. An early study suggests that PIP2 promotes talin/integrin interaction [87]. Now, there is strong evidence that talin has a direct role in increasing PIP2 at FA because it binds to the carboxyl-terminal tail of PIP5Kγ90, and binding recruits PIP5Kγ90 to FA [37, 82].
Talin/PIP5Kγ90 interaction is reciprocally regulated by phosphorylation of the tandem tyr649 and ser650 residues in PIP5Kγ90’s talin binding tail (WVYSPL) [79, 82] (Fig. 1). Under basal conditions, ser650 is constitutively phosphorylated, and as a result, lipid kinase activity and talin binding are suppressed. Integrin signaling via FAK and Src promotes tyr649 phosphorylation, either directly [82] or indirectly, by suppressing phosphorylation of the adjacent ser650 [79]. Tyr649 phosphorylation promotes interaction with talin and stimulates lipid kinase activity [82]. The model that emerges is that integrin signaling increases PIP2 synthesis at FA by recruiting and activating PIP5Kγ90, and the localized increase in PIP2 activates talin, which then binds and further activates integrins. The signal is turned off by PIP5Kγ90 dephosphorylation, and Shp-1 tyr phosphatase, which has been previously implicated in the regulation of FA dynamics, dephosphorylates PIP5Kγ90 [10]. FA turnover is therefore dynamically regulated via the reciprocal actions of multiple FA components by Src and Shp-1.
Unlike PIP5Kγ90, PIP5Kγ87 is not found in FA and does not bind talin [37, 82]. Nevertheless, it has also been implicated in integrin adhesion via a PLD2-mediated, but actin-independent, mechanism [106]. PIP5Kβ overexpression has no effect on spreading. These results raise the possibility that PIP5Kγ90 and 87 may be involved in different stages of FA formation or turnover, while PIP5Kβ is not.
Synaptic vesicle physiology
PIP2 level is increased in neurons in response to K+-induced depolarization [36], and it regulates synaptic transmission by multiple mechanisms. PIP2 is the immediate precursor of PLC-generated InsP3 and DAG, which activate Ca2+ signaling and PKC, respectively. PIP2 also directly regulates exocytosis and endocytosis by binding to clathrin adaptors and other endocytic proteins, by priming exocytic vesicles, by promoting membrane fusion and fission, and by regulating the actin cytoskeleton. Synaptojanin, which dephosphorylates PIP2, is also required for normal synaptic vesicle cycling and actin dynamics at the synapse [28].
PIP5Kγ90 overexpression in chromaffin cells increases the amount of PM PIP2 as well as the number of vesicles in the docked releasable pool [94]. The importance of PIP5Kγ in vesicle trafficking is confirmed by gene knockout. Synaptosomes prepared from the brains of the PIP5Kγ−/− mice have 40% less PIP2 than WT, and they do not generate PIP2 in response to K+ depolarization. Although primary cultures of cortical neurons develop normally in vitro in spite of the lack of PIP5Kγ, they have severe defects in synaptic transmission, which correlate with abnormal exocytosis and clathrin-mediated endocytosis [36]. Likewise, chromaffin cells isolated from these mice have defective vesicle priming and fusion dynamics [49].
Talin, which regulates PIP5Kγ90 in FA, is also present in synapses. Disruption of talin/PIP5Kγ90 interaction induces actin depolymerization and decreases clathrin-mediated synaptic vesicle endocytosis [95]. These results suggest that PIP5Kγ90 is regulated by talin in neurons using mechanisms similar to those in FAs. Likewise, neuronal PIP5Kγ90 is also reciprocally regulated by ser and tyr phosphorylation. In neurons, PIP5Kγ90 is constitutively phosphorylated on ser650 by p35/Cdk5 and MAPKs, and it is dephosphorylated during K+-induced depolarization by calcineurfin [1,142]. Ser650 dephosphorylation activates PIP5Kγ90 and facilitates tyr649 phosphorylation by Src. Significantly, K+-induced depolarization also promotes PIP5Kγ90 interaction with Arf6, which would further promote PIP2 synthesis [61, 73]. Thus, PIP5Kγ is activated through a confluence of different and interrelated signals during neuronal stimulation.
Interaction with clathrin adaptor protein complexes
The clathrin adaptor protein complex AP-2 is activated by PIP2 to bind its transport cargoes at the PM [62]. Now, there is evidence that AP-2 directly participates in increasing PIP2 synthesis at the nascent endocytic site by binding and activating PIP5Ks. Therefore, it is attractive to hypothesize that the coincidence detection of membrane cargoes and PIP2 by AP-2, together with AP-2 activation of PIP5Ks, specify the site-specific generation of a local PIP2 pool that is dedicated to clathrin/AP-2-dependent endocytosis [72].
The details of how this occurs remain to be explored. One study reports that the μ subunit of AP-2 binds all PIP5Ks [74]. Thus, AP-2 binding to PIP5Kβ may explain how PIP5Kβ promotes endocytosis in HeLa cells [101] (see above). However, other studies show that the interaction with AP-2 is mediated primarily through the PIP5Kγ90 tail, which is not present in the other PIP5Ks. Tail binding to AP-2 potently stimulates PIP5Kγ90’s lipid kinase activity [11, 99]. Overexpression of the PIP5Kγ90 tail, which competes with endogenous PIP5Kγ90 for AP-2, decreases AP-2 recruitment and synaptic vesicle endocytosis [99]. The trafficking abnormalities are similar to those described in PIP5Kγ−/− neurons [36], suggesting that PIP5Kγ90 binding to AP-2 is physiologically relevant and that the neuronal defects in PIP5Kγ−/− mice can be attributed at least partly to the lack of PIP5Kγ90.
PIP5Kγ has been implicated in membrane trafficking in non-neuronal cells as well. Overexpression of WT PIP5Kγ90 in MDCK cells increases transferrin uptake, while overexpression of KD PIP5Kγ90 inhibits [11]. These effects are specific for the long splice variant of PIP5Kγ because PIP5Kγ87 overexpression has little effect [11].
Recently, the PIP5Kγ90 tail has been reported to bind to another clathrin adaptor AP-1 [81, 99]. This interaction is proposed to be critical for E-cadherin trafficking and adhesion junction formation [81]. As AP-1 is located primarily at the TGN and it binds PI(4)P instead of PIP2 [55, 140], this interaction is perplexing. Furthermore, PIP5Kγ87, which does not have the tail, has also been implicated in cadherin and actin-enriched cell/cell adhesion in the epithelial A431 cells [2].
InsP3-mediated Ca2+ signaling
PIP2 is critical to intracellular Ca2+ signaling because it is the obligatory precursor of InsP3. RNAi studies show that although depletion of both PIP5Kγ90 and 87 isoforms together (using siRNA directed against a common sequence) decreases total PIP2 by less than 15% in HeLa cells, it blocks histamine-induced, heterotrimeric G-protein-activated InsP3 generation by more than 70% [139]. Ca2+ flux is also inhibited. However, depletion of PIP5Kγ90 with the unique tail-specific siRNA has no effect. Therefore, these results suggest that PIP5Kγ87 is important for G-protein-coupled receptor signaling. Remarkably, depletion of PIP5Kβ or α, which individually accounts for a larger fraction of total PIP2 than PIP5Kγ in HeLa cells, has almost no effect on InsP3 generation. Single cell immunofluorescence imaging shows that the PIP5Kγ- and PIP5Kβ-depleted HeLa cells have a similar drop in PM PIP2, but that the latter has more PIP2 loss in internal membranes as well. The exquisitely selective effect of PIP5Kγ87 depletion on Ca2+ signaling suggests that the PM PIP2 pools generated by PIP5Kγ87 and β are functionally compartmentalized [139].
This finding again raises the important question of how the PIP2 pools existing on the same PM can have distinct functions. One possibility is that PIP5Kγ87 is part of a supramolecular PLCβ signaling scaffold that specifies rapid local Ca2+ generation and propagation [31], while PIP5Kβ is not. In this model, the PIP2 generated by PIP5Kγ87 would be immediately available for hydrolysis by PLCβ within the signaling scaffold, thus, behaving like the previously proposed agonist-sensitive, de novo synthesized PIP2 pool [71, 98]. In contrast, the PIP2 generated by PIP5Kβ may represent a preexisting agonist-insensitive pool that maintains the PM’s status quo. The two PIP2 pools may physically segregated from the other PIP2 pool by interaction with scaffolding proteins. Studies using heart sarcolemma support the existence of agonist-sensitive and -insensitive PIP2 pools [23, 96].
Mammalian PIP5Kα
PIP5Kα is ubiquitously expressed [64, 85] and found in multiple cell compartments. Like other PIP5Ks, it is partly cytosolic and partly PM-associated. However, PIP5Kα is also found in the nucleus [16]. The PIP5Kα−/− (mouse PIP5Kβ−/−) mouse has not been described in the literature.
Membrane ruffling
The PM ruffles in response to many types of stimuli and requires remodeling of cortical actin networks downstream of the activation of Rac [110]. PIP2 has long been implicated in this process because Rac and Arf6 regulate PIP5Ks and induce ruffling. In MG-63 fibroblasts, PIP5Kα, but not β, translocates to the PM after PDGF stimulation [41]. Overexpressed PIP5Kα promotes the formation of actin foci formation when Rac1 is inhibited, but stimulates ruffle formation when Rac1 is activated. These results suggest that PIP5Kα promotes actin assembly and that additional inputs from Rac1 are required to generate active ruffles. The LIM protein ajuba, which is a component of the integrin-mediated adhesive complex and a Rac activator, is a potential effector [69]. It promotes PIP5Kα localization to membrane ruffles and leading lamellipodia.
B cell and platelet activation
Upon the engagement of B cell receptors, PIP5Kα is recruited to the PM by Bruton’s tyr kinase which has a PH domain [114]. Membrane fractionation shows that PIP5Kα is translocated to lipid rafts where PIP2 is converted to second messengers including PIP3, InsP3, and DAG. Thrombin activation of platelets induces recruitment of PIP5Kα to the PM in a Rho and ROCK-mediated, but Rac1-independent, manner [148]. RhoA also recruits PIP5Kα (and β, but not γ) to the cleavage furrow during cytokinesis [43].
Phagocytosis
Actin remodeling during FcγR-mediated phagocytosis is regulated by a highly orchestrated series of events [17, 26]. One of the initial changes is a localized increase in PIP2 at the nascent phagocytic cup. PIP5Kα is recruited to the phagocytic cup, and overexpression of a PIP5Kα KD mutant blocks actin remodeling and PIP2 accumulation there [17, 26]. It is not known at present whether PIP5Kβ or γ is also important for this type of phagocytosis, although PIP5Kβ has been implicated in integrin-mediated Yersinia phagocytosis [143]. The PIP2 increase is critical for actin modeling at the phagocytic cup. PIP2 promotes actin assembly by recruiting WASP family proteins to induce Arp2/3-dependent actin nucleation, and PIP2 also inhibits gelsolin to prevent actin severing during the ingestion phase [5].
Receptor-mediated endocytosis
Like PIP5Kβ and γ, PIP5Kα has also been implicated in receptor-mediated endocytosis. Overexpression of a PIP5Kα truncation mutant (Table 2), which has little kinase activity and is not recruited to the PM, inhibits the endocytosis of epidermal growth factor receptor [14] and mutated colony stimulating factor-1 receptor which is endocytosed more rapidly [30].
Apoptosis
PIP5Kα, but not PIP5Kβ or γ, is cleaved by caspase 3 during apoptosis, and overexpression of PIP5Kα protects cells against apoptosis by inhibiting caspase activity [92]. Protection is dependent on PIP2 generation because KD PIP5Kα shows no protection. The mechanism of protection is, however, different from that ascribed to PIP5Kβ, which is not cleaved by caspase 3 and which prevents upstream of caspase activation [53].
Nuclear PIP2
There is increasing evidence that the nucleus has its own phosphatidylinositol machinery [19]. PIP2 is found in nuclear speckles, which contain mRNA-processing components that are used for chromatin remodeling [16]. The PDZ domain-containing protein syntenin-2 is targeted to the nuclear speckles by binding PIP2, and syntenin-2 depletion by RNAi disrupts PIP2 nuclear speckles and impairs cell survival [97]. Nuclear PIP2 has also been implicated in mRNA processing, transcriptional regulation, and stress responses [19]. PIP5Kα and PIP4K, which synthesize PIP2 using different routes, are both found in the nucleus, and type III PI4Kα, which generates the PIP5K substrate PI(4)P, has been identified there as well [68]. PIP5Kα does not have a recognizable nuclear localization signal, and the mechanism for its shuttling between the nucleus and cytoplasm remains to be identified. The S. cerevisiae and Drosophila PIP5Ks are also partially nuclear localized, and they have a nuclear localizing signal [7, 22].
References
Aikawa Y, Martin TFJ (2003) ARF6 regulates a plasma membrane pool of phosphatidylinositol(4,5)bisphosphate required for regulated exocytosis. J Cell Biol 162:647–659
Akiyama C, Shinozaki-Narikawa N, Kitazawa T, Hamakubo T, Kodama T, Shibasaki Y (2005) Phosphatidylinositol-4-phosphate 5-kinase gamma is associated with cell–cell junction in A431 epithelial cells. Cell Biol Int 29:514–520
Aoyagi K, Sugaya T, Umeda M, Yamamoto S, Terakawa S, Takahashi M (2005) The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J Biol Chem 280:17346–17352
Arioka M, Nakashima S, Shibasaki Y, Kitamoto K (2004) Dibasic amino acid residues at the carboxy-terminal end of kinase homology domain participate in the plasma membrane localization and function of phosphatidylinositol 5-kinase gamma. Biochem Biophys Res Commun 319:456–463
Arora PD, Chan MW, Anderson RA, Janmey PA, McCulloch CA (2005) Separate functions of gelsolin mediate sequential steps of collagen phagocytosis. Mol Biol Cell 16:5175–5190
Astle MV, Horan KA, Ooms LM, Mitchell CA (2007) The inositol polyphosphate 5-phosphatases: traffic controllers, waistline watchers and tumour suppressors? Biochem Soc Symp 74:161–181
Audhya A, Emr SD (2003) Regulation of PI4,5P2 synthesis by nuclear-cytoplasmic shuttling of the Mss4 lipid kinase. EMBO J 22:4223–4236
Audhya A, Loewith R, Parsons AB, Gao L, Tabuchi M, Zhou H, Boone C, Hall MN, Emr SD (2004) Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. EMBO J 23:3747–3757
Auvinen E, Kivi N, Vaheri A (2007) Regulation of ezrin localization by Rac1 and PIPK in human epithelial cells. Exp Cell Res 313:824–833
Bairstow SF, Ling K, Anderson RA (2005) Phosphatidylinositol phosphate kinase type Igamma directly associates with and regulates Shp-1 tyrosine phosphatase. J Biol Chem 280:23884–23891
Bairstow SF, Ling K, Su X, Firestone AJ, Carbonara C, Anderson RA (2006) Type Igamma661 phosphatidylinositol phosphate kinase directly interacts with AP2 and regulates endocytosis. J Biol Chem 281:20632–20642
Balla T, Bondeva T, Varnai P (2000) How accurately can we image inositol lipids in living cells? Trends Pharmacol Sci 21:238–241
Balla T, Varnai P (2002) Visualizing cellular phosphoinositide pools with GFP-fused protein-modules. Sci STKE 2002:PL3
Barbieri MA, Heath CM, Peters EM, Wells A, Davis JN, Stahl PD (2001) Phosphatidylinositol-4-phosphate 5-kinase-1beta is essential for epidermal growth factor receptor-mediated endocytosis. J Biol Chem 276:47212–47216
Boronenkov IV, Anderson RA (1995) The sequence of phosphatidylinositol-4-phosphate 5-kinase defines a novel family of lipid kinases. J Biol Chem 270:2881–2884
Boronenkov IV, Loijens JC, Umeda M, Anderson RA (1998) Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol Biol Cell 9:3547–3560
Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, Grinstein S (2000) Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol 151:1353–1368
Brown FD, Rozelle AL, Yin HL, Balla T, Donaldson JG (2001) Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J Cell Biol 154:1007–1017
Bunce MW, Bergendahl K, Anderson RA (2006) Nuclear PI(4,5)P(2): a new place for an old signal. Biochim Biophys Acta 1761:560–569
Chandrasekar I, Stradal TE, Holt MR, Entschladen F, Jockusch BM, Ziegler WH (2005) Vinculin acts as a sensor in lipid regulation of adhesion-site turnover. J Cell Sci 118:1461–1472
Chatah NE, Abrams CS (2001) G-protein-coupled receptor activation induces the membrane translocation and activation of phosphatidylinositol-4-phosphate 5-kinase I alpha by a Rac- and Rho-dependent pathway. J Biol Chem 276:34059–34065
Cheng MK, Shearn A (2004) The direct interaction between ASH2, a Drosophila trithorax group protein, and SKTL, a nuclear phosphatidylinositol 4-phosphate 5-kinase, implies a role for phosphatidylinositol 4,5-bisphosphate in maintaining transcriptionally active chromatin. Genetics 167:1213–1223
Cho H, Kim YA, Yoon JY, Lee D, Kim JH, Lee SH, Ho WK (2005) Low mobility of phosphatidylinositol 4,5-bisphosphate underlies receptor specificity of Gq-mediated ion channel regulation in atrial myocytes. Proc Natl Acad Sci USA 102:15241–15246
Chong LD, Traynor-Kaplan A, Bokoch GM, Schwartz MA (1994) The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell 79:507–513
Condeelis J (2001) How is actin polymerization nucleated in vivo? Trends Cell Biol 11:288–293
Coppolino MG, Dierckman R, Loijens J, Collins RF, Pouladi M, Jongstra-Bilen J, Schreiber AD, Trimble WS, Anderson R, Grinstein S (2002) Inhibition of phosphatidylinositol-4-phosphate 5-kinase Ialpha impairs localized actin remodeling and suppresses phagocytosis. J Biol Chem 277:43849–43857
Corbett-Nelson EF, Mason D, Marshall JG, Collette Y, Grinstein S (2006) Signaling-dependent immobilization of acylated proteins in the inner monolayer of the plasma membrane. J Cell Biol 174:255–265
Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99:179–188
Critchley DR (2005) Genetic, biochemical and structural approaches to talin function. Biochem Soc Trans 33:1308–1312
Davis JN, Rock CO, Cheng M, Watson JB, Ashmun RA, Kirk H, Kay RJ, Roussel MF (1997) Complementation of growth factor receptor-dependent mitogenic signaling by a truncated type I phosphatidylinositol 4-phosphate 5-kinase. Mol Cell Biol 17:7398–7406
Delmas P, Crest M, Brown DA (2004) Functional organization of PLC signaling microdomains in neurons. Trends Neurosci 27:41–47
DeMali KA, Wennerberg K, Burridge K (2003) Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol 15:572–582
Deng L, Sugiura R, Ohta K, Tada K, Suzuki M, Hirata M, Nakamura S, Shuntoh H, Kuno T (2005) Phosphatidylinositol-4-phosphate 5-kinase regulates fission yeast cell integrity through a phospholipase C-mediated protein kinase C-independent pathway. J Biol Chem 280:27561–27568
Desrivieres S, Cooke FT, Parker PJ, Hall MN (1998) MSS4, a phosphatidylinositol-4-phosphate 5-kinase required for organization of the actin cytoskeleton in Saccharomyces cerevisiae. J Biol Chem 273:15787–15793
Di Paolo G, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657
Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431:415–422
Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P (2002) Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature 420:85–89
Divecha N, Irvine RF (1995) Phospholipid signaling. Cell 80:269–278
Divecha N, Roefs M, Halstead JR, D’Andrea S, Fernandez-Borga M, Wakelam MJO, D’Santos C (2000) Interaction of type Ialpha PIPkinase with phospholipase D: a role for the local generation of phosphatidylinositol 4,5-bisphosphate in the regulation of PLD2 activity. EMBO J 19:5440–5449
Doughman RL, Firestone AJ, Anderson RA (2003) Phosphatidylinositol phosphate kinases put PI4,5P(2) in its place. J Membr Biol 194:77–89
Doughman RL, Firestone AJ, Wojtasiak ML, Bunce MW, Anderson RA (2003) Membrane ruffling requires coordination between type Ialpha phosphatidylinositol phosphate kinase and Rac signaling. J Biol Chem 278:23036–23045
Dressman MA, Olivos-Glander IM, Nussbaum RL, Suchy SF (2000) Ocrl1, a PtdIns(4,5)P(2) 5-phosphatase, is localized to the trans-Golgi network of fibroblasts and epithelial cells. J Histochem Cytochem 48:179–190
Emoto K, Inadome H, Kanaho Y, Narumiya S, Umeda M (2005) Local change in phospholipid composition at the cleavage furrow is essential for completion of cytokinesis. J Biol Chem 280:37901–37907
Franca-Koh J, Kamimura Y, Devreotes PN (2007) Leading-edge research: PtdIns(3,4,5)P3 and directed migration. Nat Cell Biol 9:15–17
Funaki M, DiFransico L, Janmey PA (2006) PI 4,5-P2 stimulates glucose transport activity of GLUT4 in the plasma membrane of 3T3-L1 adipocytes. Biochim Biophys Acta 1763:889–899
Galiano FJ, Ulug ET, Davis JN (2002) Overexpression of murine phosphatidylinositol 4-phosphate 5-kinase type Ibeta disrupts a phosphatidylinositol 4,5 bisphosphate regulated endosomal pathway. J Cell Biochem 85:131–145
Giudici ML, Emson PC, Irvine RF (2004) A novel neuronal-specific splice variant of type I phosphatidylinositol 4-phosphate 5-kinase isoform gamma. Biochem J 379:489–496
Golub T, Caroni P (2005) PI(4,5)P2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. J Cell Biol 169:151–165
Gong L-W, Di Paolo G, Diaz E, Cestra G, Diaz M-E, Lindau M, De Camilli P, Toomre D (2005) Phosphatidylinositol phosphate kinase type I{gamma} regulates dynamics of large dense-core vesicle fusion. Proc Natl Acad Sci USA 102:5204–5209
Gorbatyuk VY, Nosworthy NJ, Robson SA, Bains NP, Maciejewski MW, Dos Remedios CG, King GF (2006) Mapping the phosphoinositide-binding site on chick cofilin explains how PIP2 regulates the cofilin–actin interaction. Mol Cell 24:511–522
Greenberg S (1999) Modular components of phagocytosis. J Leukoc Biol 66:712–717
Guerriero CJ, Weixel KM, Bruns JR, Weisz OA (2006) Phosphatidylinositol 5-kinase stimulates apical biosynthetic delivery via an Arp2/3-dependent mechanism. J Biol Chem 281:15376–15384
Halstead JR, van Rheenen J, Snel MH, Meeuws S, Mohammed S, D’Santos CS, Heck AJ, Jalink K, Divecha N (2006) A role for PtdIns(4,5)P2 and PIP5Kalpha in regulating stress-induced apoptosis. Curr Biol 16:1850–1856
Hassan BA, Prokopenko SN, Breuer S, Zhang B, Paululat A, Bellen HJ (1998) Skittles, a Drosophila phosphatidylinositol 4-phosphate 5-kinase, is required for cell viability, germline development and bristle morphology, but not for neurotransmitter release. Genetics 150:1527–1537
Heldwein EE, Macia E, Wang J, Yin HL, Kirchhausen T, Harrison SC (2004) Crystal structure of the clathrin adaptor protein 1 core. Proc Natl Acad Sci USA 101:14108–14113
Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T (2006) PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314:1458–1461
Higgs HN, Pollard TD (2001) Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu Rev Biochem 70:649–676
Hilgemann DW, Feng S, Nasuhoglu C (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE 2001:RE19
Hilpela P, Vartiainen MK, Lappalainen P (2004) Regulation of the actin cytoskeleton by PI(4,5)P2 and PI(3,4,5)P3. Curr Top Microbiol Immunol 282:117–163
Homma K, Terui S, Minemura M, Qadota H, Anraku Y, Kanaho Y, Ohya Y (1998) Phosphatidylinositol-4-phosphate 5-kinase localized on the plasma membrane is essential for yeast cell morphogenesis. J Biol Chem 273:15779–15786
Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, Kanaho Y (1999) Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99:521–532
Honing S, Ricotta D, Krauss M, Spate K, Spolaore B, Motley A, Robinson M, Robinson C, Haucke V, Owen DJ (2005) Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol Cell 18:519–531
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687
Ishihara H, Shibasaki Y, Kizuki N, Katagiri H, Yazaki Y, Asano T, Oka Y (1996) Cloning of cDNAs encoding two isoforms of 68-kDa type I phosphatidylinositol-4-phosphate 5-kinase. J Biol Chem 271:23611–23614
Ishihara H, Shibasaki Y, Kizuki N, Wada T, Yazaki Y, Asano T, Oka Y (1998) Type I phosphatidylinositol-4-phosphate 5-kinases. Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J Biol Chem 273:8741–8748
Itoh T, Ishihara H, Shibasaki Y, Oka Y, Takenawa T (2000) Autophosphorylation of type I phosphatidylinositol phosphate kinase regulates its lipid kinase activity. J Biol Chem 275:19389–19394
Janmey PA, Stossel TP (1987) Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 325:362–364
Kakuk A, Friedlander E, Vereb G Jr, Kasa A, Balla A, Balla T, Heilmeyer LM Jr, Gergely P, Vereb G (2006) Nucleolar localization of phosphatidylinositol 4-kinase PI4K230 in various mammalian cells. Cytometry A 69:1174–1183
Kisseleva M, Feng Y, Ward M, Song C, Anderson RA, Longmore GD (2005) The LIM protein Ajuba regulates phosphatidylinositol 4,5-bisphosphate levels in migrating cells through an interaction with and activation of PIPKI{alpha}. Mol Cell Biol 25:3956–3966
Kobayashi T, Takematsu H, Yamaji T, Hiramoto S, Kozutsumi Y (2005) Disturbance of sphingolipid biosynthesis abrogates the signaling of Mss4, phosphatidylinositol-4-phosphate 5-kinase, in yeast. J Biol Chem 280:18087–18094
Koreh K, Monaco ME (1986) The relationship of hormone-sensitive and hormone-insensitive phosphatidylinositol to phosphatidylinositol 4,5-bisphosphate in the WRK-1 cell. J Biol Chem 261:88–91
Krauss M, Haucke V (2007) Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep 8:241–246
Krauss M, Kinuta M, Wenk MR, De Camilli P, Takei K, Haucke V (2003) ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type I{gamma}. J Cell Biol 162:113–124
Krauss M, Kukhtina V, Pechstein A, Haucke V (2006) Stimulation of phosphatidylinositol kinase type I-mediated phosphatidylinositol (4,5)-bisphosphate synthesis by AP-2mu-cargo complexes. Proc Natl Acad Sci USA 103:11934–11939
Kunz J, Fuelling A, Kolbe L, Anderson RA (2002) Stereo-specific substrate recognition by phosphatidylinositol phosphate kinases is swapped by changing a single amino acid residue. J Biol Chem 277:5611–5619
Kunz J, Wilson MP, Kisseleva M, Hurley JH, Majerus PW, Anderson RA (2000) The activation loop of phosphatidylinositol phosphate kinases determines signaling specificity. Mol Cell 5:1–11
Lassing I, Lindberg U (1985) Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature 314:472–474
Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P (2000) GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol 149:1455–1472
Lee SY, Voronov S, Letinic K, Nairn AC, Di Paolo G, De Camilli P (2005) Regulation of the interaction between PIPKI gamma and talin by proline-directed protein kinases. J Cell Biol 168:789–799
Lemmon MA (2003) Phosphoinositide recognition domains. Traffic 4:201–213
Ling K, Bairstow SF, Carbonara C, Turbin DA, Huntsman DG, Anderson RA (2007) Type I{gamma} phosphatidylinositol phosphate kinase modulates adherens junction and E-cadherin trafficking via a direct interaction with {micro}1B adaptin. J Cell Biol 176:343–353
Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA (2002) Type I[gamma] phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 420:89–93
Ling K, Doughman RL, Iyer VV, Firestone AJ, Bairstow SF, Mosher DF, Schaller MD, Anderson RA (2003) Tyrosine phosphorylation of type I gamma phosphatidylinositol phosphate kinase by Src regulates an integrin–talin switch. J Cell Biol 163:1339–1349
Logan MR, Mandato CA (2006) Regulation of the actin cytoskeleton by PIP2 in cytokinesis. Biol Cell 98:377–388
Loijens JC, Anderson RA (1996) Type I phosphatidylinositol-4-phosphate 5-kinases are distinct members of this novel lipid kinase family. J Biol Chem 271:32937–32943
Luo B, Prescott SM, Topham MK (2004) Diacylglycerol kinase zeta regulates phosphatidylinositol 4-phosphate 5-kinase Ialpha by a novel mechanism. Cell Signal 16:891–897
Martel V, Racaud-Sultan C, Dupe S, Marie C, Paulhe F, Galmiche A, Block MR, Albiges-Rizo C (2001) Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem 276:21217–21227
Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K (2007) PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128:383–397
Martin TF (2001) PI(4,5)P(2) regulation of surface membrane traffic. Curr Opin Cell Biol 13:493–499
Matsui T, Yonemura S, Tsukita S, Tsukita S (1999) Activation of ERM proteins in vivo by Rho involves phosphatidyl-inositol 4-phosphate 5-kinase and not ROCK kinases. Curr Biol 9:1259–1262
McLaughlin S, Murray D (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438:605–611
Mejillano M, Yamamoto M, Rozelle AL, Sun HQ, Wang X, Yin HL (2001) Regulation of apoptosis by phosphatidylinositol 4,5-bisphosphate inhibition of caspases, and caspase inactivation of phosphatidylinositol phosphate 5-kinases. J Biol Chem 276:1865–1872
Mesaeli N, Tappia PS, Suzuki S, Dhalla NS, Panagia V (2000) Oxidants depress the synthesis of phosphatidylinositol 4,5-bisphosphate in heart sarcolemma. Arch Biochem Biophys 382:48–56
Milosevic I, Sorensen JB, Lang T, Krauss M, Nagy G, Haucke V, Jahn R, Neher E (2005) Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J Neurosci 25:2557–2565
Morgan JR, Di Paolo G, Werner H, Shchedrina VA, Pypaert M, Pieribone VA, De Camilli P (2004) A role for talin in presynaptic function. J Cell Biol 167:43–50
Morris JB, Huynh H, Vasilevski O, Woodcock EA (2006) Alpha1-adrenergic receptor signaling is localized to caveolae in neonatal rat cardiomyocytes. J Mol Cell Cardiol 41:17–25
Mortier E, Wuytens G, Leenaerts I, Hannes F, Heung MY, Degeest G, David G, Zimmermann P (2005) Nuclear speckles and nucleoli targeting by PIP2–PDZ domain interactions. EMBO J 24:2556–2565
Nakanishi S, Catt KJ, Balla T (1995) A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc Natl Acad Sci USA 92:5317–5321
Nakano-Kobayashi A, Yamazaki M, Unoki T, Hongu T, Murata C, Taguchi R, Katada T, Frohman MA, Yokozeki T, Kanaho Y (2007) Role of activation of PIP5Kgamma661 by AP-2 complex in synaptic vesicle endocytosis. EMBO J 26:1105–1116
Oude Weernink PA, Schmidt M, Jakobs KH (2004) Regulation and cellular roles of phosphoinositide 5-kinases. Eur J Pharmacol 500:87–99
Padron D, Wang YJ, Yamamoto M, Yin H, Roth MG (2003) Phosphatidylinositol phosphate 5-kinase I{beta} recruits AP-2 to the plasma membrane and regulates rates of constitutive endocytosis. J Cell Biol 162:693–701
Park SJ, Itoh T, Takenawa T (2001) Phosphatidylinositol 4-phosphate 5-kinase type I is regulated through phosphorylation response by extracellular stimuli. J Biol Chem 276:4781–4787
Pearson MA, Reczek D, Bretscher A, Karplus PA (2000) Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 101:259–270
Perera RM, Zoncu R, Lucast L, De Camilli P, Toomre D (2006) Two synaptojanin 1 isoforms are recruited to clathrin-coated pits at different stages. Proc Natl Acad Sci USA 103:19332–19337
Pike LJ, Miller JM (1998) Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J Biol Chem 273:22298–22304
Powner DJ, Payne RM, Pettitt TR, Giudici ML, Irvine RF, Wakelam MJ (2005) Phospholipase D2 stimulates integrin-mediated adhesion via phosphatidylinositol 4-phosphate 5-kinase Igamma b. J Cell Sci 118:2975–2986
Rao VD, Misra S, Boronenkov IV, Anderson RA, Hurley JH (1998) Structure of type IIbeta phosphatidylinositol phosphate kinase: a protein kinase fold flattened for interfacial phosphorylation. Cell 94:829–839
Raucher D, Stauffer T, Chen W, Shen K, Guo S, York JD, Sheetz MP, Meyer T (2000) Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell 100:221–228
Rhee SG (2001) Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70:281–312
Ridley AJ (2001) Rho family proteins: coordinating cell responses. Trends Cell Biol 11:471–477
Roth MG (2004) Phosphoinositides in constitutive membrane traffic. Physiol Rev 84:699–730
Rozelle AL, Machesky LM, Yamamoto M, Driessens MH, Insall RH, Roth MG, Luby-Phelps K, Marriott G, Hall A, Yin HL (2000) Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr Biol 10:311–320
Rumenapp U, Schmidt M, Olesch S, Ott S, Eichel-Streiber CV, Jakobs KH (1998) Tyrosine-phosphorylation-dependent and rho-protein-mediated control of cellular phosphatidylinositol 4,5-bisphosphate levels. Biochem J 334:625–631
Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, Scharenberg A, Rawlings DJ, Kinet J-P, Carpenter CL (2003) BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19:669–677
Santarius M, Lee CH, Anderson RA (2006) Supervised membrane swimming: small G-protein lifeguards regulate PIPK signalling and monitor intracellular PtdIns(4,5)P2 pools. Biochem J 398:1–13
Sasaki J, Sasaki T, Yamazaki M, Matsuoka K, Taya C, Shitara H, Takasuga S, Nishio M, Mizuno K, Wada T, Miyazaki H, Watanabe H, Iizuka R, Kubo S, Murata S, Chiba T, Maehama T, Hamada K, Kishimoto H, Frohman MA, Tanaka K, Penninger JM, Yonekawa H, Suzuki A, Kanaho Y (2005) Regulation of anaphylactic responses by phosphatidylinositol phosphate kinase type I {alpha}. J Exp Med 201:859–870
Saunders RM, Holt MR, Jennings L, Sutton DH, Barsukov IL, Bobkov A, Liddington RC, Adamson EA, Dunn GA, Critchley DR (2006) Role of vinculin in regulating focal adhesion turnover. Eur J Cell Biol 85:487–500
Schmidt M, Rumenapp U, Nehls C, Ott S, Keller J, Von Eichel-Streiber C, Jakobs KH (1996) Restoration of clostridium difficile toxin-B-inhibited phospholipase D by phosphatidylinositol 4,5-bisphosphate. Eur J Biochem 240:707–712
Scott CC, Dobson W, Botelho RJ, Coady-Osberg N, Chavrier P, Knecht DA, Heath C, Stahl P, Grinstein S (2005) Phosphatidylinositol-4,5-bisphosphate hydrolysis directs actin remodeling during phagocytosis. J Cell Biol 169:139–149
Sechi AS, Wehland J (2000) The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J Cell Sci 113(Pt 21):3685–3695
Shaw AS (2006) Lipid rafts: now you see them, now you don’t. Nat Immunol 7:1139–1142
Sheetz MP, Sable JE, Dobereiner HG (2006) Continuous membrane-cytoskeleton adhesion requires continuous accommodation to lipid and cytoskeleton dynamics. Annu Rev Biophys Biomol Struct 35:417–434
Shibasaki Y, Ishihara H, Kizuki N, Asano T, Oka Y, Yazaki Y (1997) Massive actin polymerization induced by phosphatidylinositol-4-phosphate 5-kinase in vivo. J Biol Chem 272:7578–7581
Shinozaki-Narikawa N, Kodama T, Shibasaki Y (2006) Cooperation of phosphoinositides and BAR domain proteins in endosomal tubulation. Traffic 7:1539–1550
Shyng SL, Barbieri A, Gumusboga A, Cukras C, Pike L, Davis JN, Stahl PD, Nichols CG (2000) Modulation of nucleotide sensitivity of ATP-sensitive potassium channels by phosphatidylinositol-4-phosphate 5-kinase. Proc Natl Acad Sci USA 97:937–941
Skippen A, Jones DH, Morgan CP, Li M, Cockcroft S (2002) Mechanism of ADP ribosylation factor-stimulated phosphatidylinositol 4,5-bisphosphate synthesis in HL60 cells. J Biol Chem 277:5823–5831
Stefan D, Baird D, Ling Y, Audhya A, Emr S (2006) Regulation of phosphoinositide kinase signaling at the plasma membrane. Mol Cell Biol 17 (Suppl):2493 (CD-ROM)
Stephens LR, Hughes KT, Irvine RF (1991) Pathway of phosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils. Nature 351:33–39
Stossel TP, Fenteany G, Hartwig JH (2006) Cell surface actin remodeling. J Cell Sci 119:3261–3264
Suh BC, Hille B (2005) Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol 15:370–378
Suh BC, Inoue T, Meyer T, Hille B (2006) Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314:1454–1457
Toker A, Cantley LC (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387:673–676
Tolias KF, Cantley LC, Carpenter CL (1995) Rho family GTPases bind to phosphoinositide kinases. J Biol Chem 270:17656–17659
Tolias KF, Couvillon AD, Cantley LC, Carpenter CL (1998) Characterization of a Rac1- and RhoGDI-associated lipid kinase signaling complex. Mol Cell Biol 18:762–770
Tolias KF, Hartwig JH, Ishihara H, Shibasaki Y, Cantley LC, Carpenter CL (2000) Type Ialpha phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr Biol 10:153–156
van Horck FP, Lavazais E, Eickholt BJ, Moolenaar WH, Divecha N (2002) Essential role of type I(alpha) phosphatidylinositol 4-phosphate 5-kinase in neurite remodeling. Curr Biol 12:241–245
Varnai P, Thyagarajan B, Rohacs T, Balla T (2006) Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol 175:377–382
Wang L, Li G, Sugita S (2005) A central kinase domain of type I phosphatidylinositol phosphate kinases is sufficient to prime exocytosis: isoform specificity and its underlying mechanism. J Biol Chem 280:16522–16527
Wang YJ, Li WH, Wang J, Xu K, Dong P, Luo X, Yin HL (2004) Critical role of PIP5KI{gamma}87 in InsP3-mediated Ca(2+) signaling. J Cell Biol 167:1005–1010
Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin HL (2003) Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114:299–310
Weernink PAO, Meletiadis K, Hommeltenberg S, Hinz M, Ishihara H, Schmidt M, Jakobs KH (2004) Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J Biol Chem 279:7840–7849
Wenk MR, Pellegrini L, Klenchin VA, Di Paolo G, Chang S, Daniell L, Arioka M, Martin TF, De Camilli P (2001) PIP kinase Igamma is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron 32:79–88
Wong KW, Isberg RR (2003) Arf6 and phosphoinositol-4-phosphate-5-kinase activities permit bypass of the Rac1 requirement for beta1 integrin-mediated bacterial uptake. J Exp Med 198:603–614
Wymann MP, Marone R (2005) Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol 17:141–149
Yamamoto M, Chen MZ, Wang YJ, Sun HQ, Wei Y, Martinez M, Yin HL (2006) Hypertonic stress increases phosphatidylinositol 4,5-bisphosphate levels by activating PIP5KIbeta. J Biol Chem 281:32630–32638
Yamamoto M, Hilgemann DH, Feng S, Bito H, Ishihara H, Shibasaki Y, Yin HL (2001) Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J Cell Biol 152:867–876
Yamazaki M, Miyazaki H, Watanabe H, Sasaki T, Maehama T, Frohman MA, Kanaho Y (2002) Phosphatidylinositol 4-phosphate 5-kinase is essential for ROCK-mediated neurite remodeling. J Biol Chem 277:17226–17230
Yang S-A, Carpenter CL, Abrams CS (2004) Rho and Rho-kinase mediate thrombin-induced phosphatidylinositol 4-phosphate 5-kinase trafficking in platelets. J Biol Chem 279:42331–42336
Yin HL, Janmey PA (2003) Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol 65:761–789
Zhang Y, Sugiura R, Lu Y, Asami M, Maeda T, Itoh T, Takenawa T, Shuntoh H, Kuno T (2000) Phosphatidylinositol 4-phosphate 5-kinase Its3 and calcineurin Ppb1 coordinately regulate cytokinesis in fission yeast. J Biol Chem 275:35600–35606
Acknowledgments
This work is supported by NIH R01GM06110, NIH P05GM21681, and a Welch Foundation grant to HLY.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mao, Y.S., Yin, H.L. Regulation of the actin cytoskeleton by phosphatidylinositol 4-phosphate 5 kinases. Pflugers Arch - Eur J Physiol 455, 5–18 (2007). https://doi.org/10.1007/s00424-007-0286-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-007-0286-3