
Abstract
Background
Multiple endocrine neoplasia (MEN) type 2, a cancer syndrome inherited in the dominant fashion, is defined by the occurrence of medullary thyroid carcinoma (MTC), either as a singular lesion (familial medullary thyroid carcinoma, FMTC) or with the variable expression of pheochromocytoma, hyperparathyroidism (MEN 2A), ganglioneuromas, buccal neuromas and Marfanoid-like phenotype (MEN 2B).
Discussion
Germline mutations of the RET proto-oncogene, localized on chromosome 10q11.2, have been identified as the underlying genetic cause of the disorder. In the majority of patients with MEN 2A/FMTC missense mutations at exon 10 or exon 11 are identifiable. Cysteine to arginine exchange at codon 634 is the mutation most frequently found. In MEN 2B approximately 95% of patients present with a mutation at codon 918 (exon 16). Additionally, less frequent mutations in other codons have been found in both syndromes. The DNA-based genotype analysis enables the identification of gene carriers at risk of developing MTC and offer them prophylactic thyroidectomy prior to development of any thyroid pathologies. Prophylactic surgery is generally recommended for MEN 2A/FMTC gene carriers at the age of 4–6 years. Due to the aggressiveness of the MEN 2B syndrome gene carriers should be operated by the age of 1 year. Presumably some less virulent mutations allow postponement of the prophylactic treatment to the second to fourth decade of life.
Conclusions
Compared to standard presymptomatic biochemical screening, genetic testing and consecutive prophylactic treatment contribute to better outcome of individuals at risk for MTC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple endocrine neoplasia (MEN) type 2, an autosomal-dominant inherited syndrome with complete gene penetrance, is characterized by synchronous or metachronous occurrence of various endocrine tumors in variable clinical expression. Features of MEN 2A (Sipple syndrome) include medullary thyroid carcinoma (MTC), pheochromocytoma, and parathyroid neoplasia. MEN 2B (Gorlin syndrome) encompasses MTC, pheochromocytoma, mucosal neuromas, ganglioneuromatosis of the gastrointestinal tract, and Marfanoid habitus. In some rare families cutaneous lichen amyloidosis or Hirschsprung's disease is an additional feature of the MEN 2A syndrome. Familial MTC (FMTC) is a variant of the syndrome in which MTC is the only lesion in the absence of further endocrinopathies. Generally the clinical course of MEN 2B and of FMTC is more aggressive than that of MEN 2A.
MTC acts as the hallmark of the MEN 2 syndrome since it is the only malignant component and is thus of significant prognostic value. The carcinoma secretes calcitonin, which serves as a particularly valuable tumor marker. C-cell hyperplasia has been considered a precursor lesion of MTC. In the past the diagnosis of MTC in family members at risk was based on elevated basal and/or stimulated levels of calcitonin and subsequent histological conformation. Although of enormous clinical importance, the presymptomatic calcitonin screening entailed several disadvantages. All individuals at risk of developing MTC had to undergo annual calcitonin testing until at least the age of 30 years, even though in one-half of them the screening was theoretically unnecessary since they were not gene carriers for the disease. Although accurately screened, some family members already presented lymph node metastases at the time of initial tumor diagnosis and had increased risk of morbidity and mortality [1]. In contrast, unnecessary thyroidectomies were performed in some borderline cases or falsely interpreted calcitonin testing results.
Since it was shown that specific germline mutations in the RET proto-oncogene are the underlying cause of the disorder, molecular genetic diagnostics based on direct DNA analysis with the aim of detecting specific RET proto-oncogene mutations have become the gold standard of presymptomatic MEN 2 screening. Furthermore, genetic testing significantly influenced the clinical management and surgical treatment of MEN 2 family members. Those identified as gene carriers are offered prophylactic therapy with the intent of cancer prevention and those tested as non-carriers may be omitted from further screening procedures.
Genetic mapping of the gene responsible for MEN 2 development
In 1987 Simpson et al. [2] and Mathew et al. [3] mapped the susceptibility locus for MEN 2A to the pericentromeric region of chromosome 10. Subsequent linkage studies led to localization of the RET proto-oncogene on chromosomal segment 10q11.2 as the MEN 2 causative gene and to the identification of MEN 2 specific mutations [4, 5]. The RET (rearranged during transfection) proto-oncogene plays a key role in the development and differentiation of various tissues derived from neural crest. MEN 2 arises as a result of unstrained activation of the mutated RET proto-oncogene, a phenomena uncommon among human inherited cancer syndromes, which are generally caused by inactivation of a tumor suppressor gene.
The RET proto-oncogene
The RET-proto-oncogene comprises 21 exons which encode the protein RET, a membrane bound receptor tyrosine kinase (RTK) of 170 kDa. Animal studies reveal that RET is developmentally highly regulated. Highest expression is found early during kidney morphogenesis and during cell maturation of neural crest derived tissues such as thyroid and adrenal gland [6]. Additional physiological functions of the RET proto-oncogene include enteric and autonomic neuronal development and differentiation of spermatogonia [7]. RTK transduce the extracellular signals for processes as diverse as cell differentiation, survival, growth, and apoptosis. Generally RTKs are activated by homodimeric or heterodimeric complex formation. In this repect the RET RTK is unique as it requires a multicomplex formation to induce activation and stimulation of downstream signaling [8]. Ligands for RET activation are distantly related to the transforming growth factor β superfamily. Four neuronal survival factors belonging to the glial cell line-derived neurotropic factor (GDNF) family have been identified to date, including GDNF, neurturin, persephin, and artemin [9, 10]. These ligands do not bind RET RTK directly but interact with at least four membrane-bound coreceptors. These adaptor molecules do not have an intracellular domain but are linked to the cell-surface by a glycosyl-phosphatidylinositol anchor [10, 11]. GDNF family receptor (GFR) α1 interacts with GDNF while neurturin binds preferentially to GFRα2, artemin shows high affinity to GFRα3, and GFRα4 interacts with persephin [8, 11, 12]. While each of different GDNF and GFRα family members show individual expression patterns and are involved in distinct developmental roles, their common denominator is complex formation and activation of RET. Strikingly, both c-RET −/− and GDNF−/− knock-out mice share the same phenotype with early death after birth, lack of neurons in the entire gastrointestinal tract, and kidney agenesis [6]. The GDNF/GFRα complex interact with RET RTK through the cysteine-rich region, inducing RET dimerization and thereby triggering autophosphorylation which activates the protein tyrosine kinase RET (Fig. 1) [9, 13].

Interaction leading to RET proto-oncogene activation. GDNF binds with GRFα and triggers the dimerization of the receptor
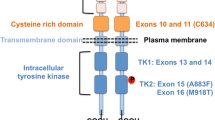
In addition to RTK activation by RET ligand/coreceptor complex formation, novel studies demonstrate RET-independent mechanisms of GFRα signal transduction. However, these observations reveal that RET-independent signaling of GFRα1 is ligand-specific and occurs only with GDNF [14]. RET RTK contains four structural domains: the extracellular ligand binding domain, a cysteine-rich domain, a dynamic transmembrane domain, and the intracellular tyrosine kinase domain. The extracellular sequence contains regions with homology to the cadherin family of cell adhesion molecules. This domain is involved in recognition and binding of its ligands and coreceptors as well as calcium-dependent cell-cell adhesion [15, 16]. The highly conserved large cysteine-rich region near the transmembrane domain forms intramolecular covalent disulfide bonds. It is believed that this mechanism is crucial for RET tertiary structure and thus ligand binding as well as dimerization of RET-RTK by induction of intracellular cross-phosphorylation within the tyrosine kinase domain. Of the 28 cysteine sites at this domain 27 are conserved between species implicating its fundamental role for the tertiary ultrastructure of the receptor [15]. The tyrosine kinase domain lies intracellularly and is responsible for phosphorylation of key tyrosine residues and activation of downstream signaling pathways.
RET downstream signaling
Both ligand-stimulated RET wild type and constitutive active oncogenic RET mutants are autophosphorylated at one of the 18 specific cytoplasmatic tyrosine residues within the intracellular tyrosine kinase domain. Alternative splicing of 3′ exons results in three different protein isoforms with distinct C-terminal sequences (RET 9, RET 43, RET 51) that contribute to different downstream target activation [15, 17]. Studies have now identified 9 of these 18 tyrosine sites to be phosphorylated upon RET activation and in turn may contribute to the downstream signaling mechanism. Five autophosphorylation sites (Y687, Y826, Y864, Y905, Y952) can be found within the kinase domain, and another four are located within the COOH-terminal tail (Y1015, Y1029, Y1062, Y1096). More detailed downstream signaling mechanisms have been identified for six of these sites.
Upon RET activation the autophosphorylated tyrosine residues Y905, Y1015, Y1062, and Y1096 act as docking sites for Src homology 2 (SH2) domain containing target molecules which in turn activate distinct intracellular signaling cascades. RET associates with the transduction proteins Grb2/Grb7/Grb10/Grb14, phospholipase Cγ (PLC-γ) and Shc/Enigma. One major pathway shown to be activated by RET is the Ras/mitogen-activated protein kinase (MAPK) pathway [18]. The Ras superfamily comprises around 50 related genes encoding membrane bound GTPases. Activated by ligand binding, they in turn activate serine/threonine protein kinases that are involved in control of cell growth and differentiation. The tyrosine residues Y864 and Y952 are crucial for transforming activity of mutations associated with MEN 2B [19]. They contribute to conformational changes within the kinase domain. The tyrosine residues Y1090 and Y1096 are found only in among the intermediate (RET 43) and long (RET 51) isoforms, respectively [15]. Interaction with Y1096 involves the adapter protein GRB2 and subsequent stimulation of the RAS-mediated activation of the MAPK signaling pathway that is involved in neuronal survival and differentiation.
The last amino acid common to all isoforms is Y1062. Y1062 acts as a multifunctional docking site, which is a binding site for SHC adaptor proteins and is crucial for both RAS/MAPK and phosphatidylinositol 3-kinase (PI3-K)/AKT signaling pathways [20]. Through this mechanism Y1062 functions as a major binding site for adapter molecules that are involved in cell transformation. Depending on the protein isoform, Y1062 is found in context with distinct C-terminal amino acids that affect the binding of adaptor proteins. In the short isoform RET 9 Y1062 provides a docking site for the SH2 domain of SHC, while for Y1062 in association with RET 51 (long isoform) C-terminal sequence a preferred binding with the SHC phosphotyrosine-binding domain has been described.
Furthermore, phosphorylation-independent binding of Enigma to Y1062 has been reported [21]. Enigma, a 470 amino acid protein containing one PDZ and three LIM domains, belongs to a protein family (PDZ-LIM family) that acts as an adapter protein between kinases and cytoskeleton. PDZ and LIM domains are modular protein-interaction domains. The names derives from the first three proteins in which the PDZ domain was identified (PSD-95, DLG, ZO-1) and the first three transcription factors (Lin-1, Isl-1, Mec-3) in which LIM domain was found.
While the LIM3 domain interacts with the multifunctional docking site Y1062 of proto-RET, the PDZ domains anchors the protein to the cell surface [21]. Recent studies have revealed a novel adaptor protein for the multifunctional docking site Y1062 [22]. SNT/FRS2 is a lipid anchored docking protein that contains an amino-terminal myristylation signal, followed by a phosphotyrosine-binding domain and a carboxy-terminal region with multiple tyrosine residues. Unlike SHC bound to RET, which is associated with GRB2 and Grb2-associated binder (GAB) 1 proteins, SNT/FRS2 is associated with GRB2 proteins only, thus involved mainly in the activation of the RAS/MAPK pathway but not the PI3-K/AKT pathway [22]. As outlined above, adaptor molecules binding to autophosporylated RET tyrosine residues contain mainly SH2 domains. The adapter proteins GRB7 and GRB10 have been identified as ligands for tyrosine residue 905 (Y905). As with SHC/GRB2, this mechanism has been shown to be linked to the RAS/MAPK pathway. Activation of PI3-K is one key mechanism for RET downstream signaling [18]. The activation of this kinase probably occurs by interaction of SHC, GRB2, and GAB1 with the 85-kDa (p85) regulatory subunit of PI3-K. PI3-K acts as a switchboard for regulating cell interactions by phosphorylation of proteins linked to cell-cell interaction, such as the gap junction protein connexin 43 and cell adhesion molecules such as paxillin, focal adhesion kinase, and p130cas. Furthermore PI3-K induces protein kinase B and AKT signaling pathway, which play a crucial role in cell survival and proliferation [18, 23]. Overexpression of AKT markedly enhances the transforming ability of MEN 2 mutations. RET has been shown to activate the c-Jun NH2-terminal protein kinase (JNK) pathway through a cdc42/RAC1 small GTPase. The detailed signaling pathway has not yet been established; however, data from other systems suggest JNK to lie downstream of PI3-K [24]. Tyrosine residue Y1015 is associated with stimulation of PLC-γ. While the mechanism of interaction has not yet been clarified, activation of PLC-γ triggers changes in intracellular Ca2+ levels and thereby contributes to mitogenesis and transformation associated with RET activation (Fig. 2).

RET downstream signaling mechanism activated by GDNF-RET-GFRα
RET proto-oncogene mutations in MEN 2 and FMTC: correlations between genotype and phenotype
MEN Germline missense mutations in exon 10 (codons 609, 611, 618, and 620) or exon 11 (codons 630 and 634) that encode part of the extracellular cysteine-rich domain can be found in the vast majority of MEN 2A gene carriers and in a significant portion of FMTC families [25, 26, 27]. Mutations in these codons lead to an alteration of a critical cysteine residue with subsequent dimerization and constitutive activation of downstream signal transduction. The most frequent mutation in MEN 2A affects codon 634, causing a cysteine to arginine exchange (C634R). In FMTC mutations are more evenly distributed among codons 618, 620, and 634, with lower frequencies in codons 609, 611, 630, and 631 [28, 29, 30, 31]. At codon 634 the most common alteration is C634Y. Noncysteine mutations have been identified in a few FMTC families and recently also in some MEN 2A families at exon 13 (codon 768, 790, and 791), exon 14 (codon 804, 844) and exon 15 (codon 891) [31, 32, 33, 34, 35, 36]. The effects of these mutations are not yet clear in detail; however, it is thought that they may induce kinase activity by changing the substrate specificity or ATP-binding capacity of the receptor. So far mutations at codon 790 and codon 791 (exon 13) have been identified exclusively in Germany, Austria, France, and the former Yugoslavia [37, 38, 39]. Reasons why these mutations with still unknown detailed functional mechanism have not been found outside of Europe needs to be elucidated.
MEN 2B is generally associated with a single base substitution (ATG→ACG) at codon 918 (exon 16) and accounts for more than 95% of identified missense mutations in this subgroup of MEN 2 [40]. The MEN 2B specific codon is located in the substrate recognition pocket of the intracellular tyrosine kinase domain. RET mutations within this codon are thought to alter specificity towards substrates such as c-src and c-abl and support the involvement of down-stream signaling proteins not usually activated via RET [41]. In some clinically clear MEN 2B families without codon 918 mutation analysis of neighboring RET regions has revealed mutations in codon 883 [42].
In addition to hot-spot mutations described above, a few novel, unusual mutations have recently been identified: in a FMTC family a double RET mutation (codon 804 GTG→ATG, codon 844 CGC→TTG) [43] and in a MEN 2A family triple mutation of exon 11 (codon 631 GAC→TAC) and 14 (codon 819 AGC→ATC, codon 843 GAG→GAT) [44]. Rarely MEN 2A is associated with in-frame duplications of 9 and 12 bp in exon 11 [45, 46]. In a de novo MEN 2A case two new mutations were identified at codon 634 and 640 on the same RET allele [47]. Furthermore, there is a single reported FMTC family with a 9-bp duplication at exon 8.
The hypothesis of genotype-phenotype correlation was validated by the data of the International RET Mutation Consortium which analyzed 477 unrelated MEN 2 families. A significant association was found between the presence of any amino acid exchange at codon 634 and the manifestation of pheochromocytoma and hyperparathyroidism. Codon 918 mutation, leading to a threonine for methionine exchange, was specific to MEN 2B. In the case of association between MEN 2A and Hirschsprung's disease all affected individuals presented either with codon 620 or codon 618 mutation. Codon 634 mutation was found exclusively in the small portion of MEN 2 families with cutaneous lichen amyloidosis [27]. It was recently observed in FMTC families that index cases with noncysteine mutations were older when diagnosed than those with exon 10 mutations. In gene carriers noncysteine mutations were associated with a lower incidence of MTC than in those carrying a mutation in one of the codons at exon 10 (Fig. 3) [38].

RET proto-oncogene mutations associated with MEN 2/FMTC. Regions containing different mutations are indicated
Consequences of genetic information obtained by predictive testing
Genetic screening for specific RET proto-oncogene mutations in MEN 2 families enables identification of gene carriers at risk of MTC. The ability to identify early a predisposition to the development of a malignant disorder is of particular importance for infants born to an affected parent. In accordance with ethical requirements for molecular genetic testing of children, the screening offers immediate medical benefit [48]. In addition, the costs per life saved in MEN 2 gene carriers by performing RET analysis has been calculated to be insignificant when compared to the costs per life saved under condition of standard biochemical screening [49]. Due to the high accuracy of DNA-based genetic testing and inconsideration of worse treatment results when MTC is diagnosed only in the clinical stage, it is generally accepted that family members who inherit a MEN 2 specific RET mutation should be offered prophylactic thyroidectomy prior to any pathological changes in serum calcitonin level. The importance of this strategy was stressed by the observation of late tumor recurrences in 15–20% of patients thyroidectomized in childhood for C-cell hyperplasia or MTC at an early stage [50]. The first large series on prophylactic thyroidectomy in patients at risk for MEN 2A was presented by Wells et al. [51, 52] in 1994. Of 21 family members 13 identified as gene carriers underwent surgery: 6 with normal (age 6–14 years) and 7 with pathological calcitonin levels (age 8 – 20 years). Histological examination revealed C-cell hyperplasia or/and MTC in each thyroid specimen; however, there was no evidence of lymph node metastases. At follow-up 3 years postoperatively all patients were tumor free and showed normal calcitonin levels. The French Calcitonin Tumors Study Group reported findings from 71 MEN 2/FMTC gene carriers aged 10 months–20 years (mean 12.5±5), in 61 of whom the thyroid was removed prophylactically, combined with lymph node dissection [38]. Preoperative basal and/or stimulated calcitonin was within the pathological range in all patients, congruent with histological finding of C-cell hyperplasia or MTC in all cases. Lymph node status was positive in six patients aged 16 (FMTC) to 20 years. Biochemical cure was not achieved in six patients; in five there was either with lymph node metastasis or an insufficient lymph node dissection, or no lymphadenectomy performed. Postoperative deaths occurred in two cases of transient hypoparathyroidism and in one case of permanent hypoparathyroidism. A multicentric study conducted in Germany and Austria included 75 juvenile MEN 2 gene carriers in whom prophylactic thyroidectomy was performed, in 76% of cases combined with cervical lymph node dissection. Upon surgery 61% of them were found to have MTC (youngest patient 4 years) and in 39% C-cell hyperplasia. Of the three patients with lymph node metastases the youngest was 14 years old. Postoperative findings were transient hypoparathyroidism (n=20), permanent hypoparathyroidism (n=5), and permanent unilateral recurrent nerve injury (n=1). Biochemical cure, confirmed by normal stimulated calcitonin levels, was achieved in 96% of the patients [53]. The results of these and of several other studies [54, 55, 56, 57] undoubtedly demonstrate the impact of prophylactic thyroidectomy on the prognosis of patients with hereditary MTC. At the same time however, they disclose the weakness of calcitonin screening since in a significant portion of gene carriers MTC or extensive C-cell hyperplasia were already present despite normal biochemical testing. In these individuals the thyroidectomy performed was in fact therapeutic and not prophylactic.
The crucial question of the age at which prophylactic thyroidectomy should be carried out to remove thyroid tissue prior to the initiation of malignant transformation has not yet been unequivocally answered. Based on the data obtained in the initial studies, the age of 4–6 years is generally considered appropriate for surgery in MEN 2A gene carriers [51, 52, 53, 56, 58]. Several centers, however, follow their own policy due to experience with their specific population. The French Calcitonin Tumors Study Group recommends operation within the first 2 years of age, since in their series the youngest child with MTC was 2 years old, and several gene carriers with histological tumor evidence were under the age of 6 [36]. Similar experience has been reported by van Heuren et al. [59] in a family with MEN 2A, in which three children aged 2, 3, and 6 years with elevated basal calcitonin levels were found to have MTC upon thyroidectomy, forcing the Group to suggest 2 years of age as appropriate for prophylactic treatment. The importance of a very early intervention was also underscored by Sanso et al. [60] who found a 0.2-cm MTC in a diffuse C-cell hyperplasia in a 17-month-old infant of a MEN 2A kindred. In contrast to these recommendations, some groups do not accept the general need for aggressive treatment of MEN 2A since they fear that this would entail too many unnecessary operations being performed at an age in which thyroid removal might be associated with higher morbidity or problems related to external thyroxin supplementation [61, 62]. Since several of the reported juvenile gene carriers with MTC or C-cell hyperplasia have shown basal and/or stimulated calcitonin a pathological range, they recommend a more liberal policy that includes determination of gene carrier status immediately after birth, regular pentagastrin testing, and immediate surgery when stimulated calcitonin exceeds 10 pg/ml.
The more uniform opinion that MEN 2B gene carriers should undergo very early prophylactic thyroidectomy reflects the well-recognized aggressiveness of this form of hereditary MTC. Several institutions have reported MEN 2B affected infants in whom MTC already involving regional lymph nodes was diagnosed within the first 12 months after birth [63, 64]. Since in most cases the disorder originates from a de novo mutated paternal allele, it is advisable to perform RET analysis in every newborn that presents with MEN 2B characteristic features regardless of family history to avoid missing the appropriate time for prophylactic surgical intervention.
Rather rare RET abnormalities have been identified at codons 804 and 790/791 in some families with the MEN 2A/FMTC phenotype in which DNA study results were negative at the time of an initial screening of exons 10 and 11. The low number of patients with these mutations makes it difficult to suggest the proper therapeutic strategy for asymptomatic carriers. Due to delayed manifestation of C-cell hyperplasia in the presence of codon 790/791 mutation the risk of MTC occurrence in juvenile gene carriers is presumably rather low in this genotype. Thyroidectomy may be delayed until the age of 10 years [65], but is unavoidable when pentagastrin testing becomes pathological [36]. For a RET codon 804 mutation the recommendations concerning treatment are more controversial. While some groups associate this genotype with an aggressive clinical course and recommend for early thyroidectomy [35, 66], others take a more conservative approach due to favorable tumor behavior documented in their families [67, 68].
The finding of lymph node metastases in children subjected to preventative thyroidectomy raises the question of whether lymphadenectomy is necessary at the time of thyroidectomy, and the question of the extent to which lymph nodes should be removed. In addition to its oncological impact, this issue is important since surgical morbidity is related mainly to lymph node cleavage [38, 53]. Cervical lymph node dissection should be an integral part of the operation in MEN 2B gene carriers who are at higest risk. No consensus has been reached in regard to MEN 2A/FMTC gene carriers. The French Calcitonin Tumors Study Group stresses the importance of cervical lymph node dissection since in their series 4 of 71 gene carriers younger than 20 years presented with nodal involvement [38]. Based on the German and Austrian experience lymphadenectomy in the cervical central compartment should be performed in cases of elevated calcitonin levels and in patients older than 10 years. Patients older than 15 years with elevated calcitonin levels should undergo bilateral lymph node resection [53]. Gene carriers with codon 790/791 mutation might not need lymph node dissection during the first four decades of life [65].
Since its introduction only 10 years ago presymptomatic genetic screening for hereditary MTC has largely replaced the standard biochemical screening. The procedure has gained worldwide acceptance [55, 69]. Close surveillance of patients prophylactically thyroidectomized during childhood will be of great importance in the future for evaluating the effectiveness of the procedure. Pentagastrin testing and screening for specific nonmalignant features of the MEN 2 syndrome are advisable on a biannual basis. Gene carriers in whom normal thyroid specimens were removed may be of particular interest since these individuals would be the only ones offered truly prophylactic surgery. In addition to the health aspects, the medical community involved in managing hereditary diseases suitable for prophylactic treatment must focus on economic issues since cost has become a factor that obviously influences our decision making. A recent nationwide study in the United States evaluated present coverage policies for prophylactic thyroidectomy and found that 12% of private plans and 50% of government carriers provide no coverage for gene carriers at risk for thyroid cancer [70].
References
Frilling A, Röher H-D, Ponder BAJ (1994) Presymptomatic screening for medullary thyroid carcinoma in patients with multiple endocrine neoplasia type 2A. World J Surg 18:557–582
Simpson NE, Kidd KK, Goodfellow PJ, McDermid H, Myers S, Kid JR, Jackson CE, Duncan AM, Farrer LA, Brasch K (1987) Assignment of multiple endocrine neoplasia type 2a to chromosome 10 by linkage. Nature 328:528–530
Matthew CGP, Chin KS, Easton DF, Thorpe K, Carter C, Ilou GI, Fong S-L, Bridges CDB, Haak H, Nieuwenhuijzen Kruseman AC, Schifter S, Hansen HH, Telenius H, Telenius-Berg M, Ponder BAJ (1987) A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature 328:527–528
Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA (1993) Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC Hum Mol Genet 2:851–856
Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, Ponder MA, Telenius H, Tunnacliffe A, Ponder BAJ (1993) Germ-line mutation of RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363:458–460
Schuchardt A, Dàgati V, Larsson-Blomberg L, Costantini F, Pachnis V (1994) Defects in the kidney and enteric nervous sytem of mice lacking the tyrosine kinase receptor Ret. Nature 367:380–383
Hansford JR, Mulligan LM (2000) Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. J Med Genet 37:817–827
Treanor JJS, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahas M, Vandlen R, Henderson CE, Rosenthal A (1996) Characterization of a multicomponent receptor for GDNF. Nature 382:80–83
Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Gruezo E, Pachnis V, Arumae U (1996) Functional receptor for GDNF encoded by the c-RET proto-oncogene. Nature 381:785
Takahashi M (2001) The GDNF/RET signaling pathway and human disease. Cytokine Growth Factor Rev 12:361–373
Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Cupples R. Louis JC, Hu S, Altrock BW, Fox GM (1996) GDNF-induce activation of the RET protein tyrosine kinase is mediated by GDNFR-a, a novel receptor for GDNF. Cell 85:1113–1124
Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM Jr, Milbrandt J (1998) Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRa2-RET receptor complex. Neuron 21:1291–1302
Klein RD, Sherman D, Ho WH, Stone D, Bennett GL, Moffat B, Vandlen R, Simmons L, Gu Q, Hongo JA, Devaux B, Poulsen K, Armanini M, Nozaki C, Asai N, Goddard A, Phillips H, Henderson CE, Takahashi M, Rosenthal A (1998) A gpi-linked protein that interacts with RET to form a candidate neurturin receptor. Nature 392:210
Trupp M, Scott R, Whittemore SR, Ibanez CF (1999) Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signalling in neuronal cells. J Biol Chem 274:20885–20894
Takahashi M, Buma Y, Iwamoto T, Inaguma Y, Ikeda H, Hiai H (1988) Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene 3:571–578
Anders J, Kjar S, Ibanez CF (2001) Molecular modelling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domain and calcium-binding site. J Biol Chem 276:35808–35817
Myers SM, Eng C, Ponder BAJ, Mulligan LM (1995) Characterization of RET proto-oncogene 3'splicing variants and polyadenylation sites: a novel C terminus for RET. Oncogene 11:2039–2045
Segouffin-Cariou C, Billaud M (2000) Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase/AKT signaling pathway. J Biol Chem 275:3568–3576
Hansford JR, Mulligan LM (2000) Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. J Med Genet 37:817–827
Hayashi H, Ichihara M, Iwashita T, Murakami H, Shimono Y, Kumi K, Kurokawa K, Yoshiki M, Imai T, Funahashi H, Nakao A, Takahashi M (2000) Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene 19:4469–4475
Borrello MG, Mercalli E, Perego C, Degl Innocenti D, Ghizzoni S, Arighi E, Eroini B, Rizzetti BG, Pierotti MA (2002) Differential interaction of Enigma protein with the two RET isoforms. Biochem Biophys Res Commun 296:515–522
Kurokawa K, Iwashita T, Murakami H, Hayashi H, Kawai K, Takahashi M (2001) Identification of SNT/FRS2 docking site on RET receptor tyrosine kinase and its role for signal transduction. Oncogene 20:1929–1938
Murakami H, Iwashita T, Asai N, Shimono Y, Iwata Y, Kawai K, Takahashi M (1999) Enhanced phosphatidylinositol 3-kinase activity and high phosphorylation state of its downstream signaling molecules mediated by Ret with the MEN 2B mutation. Biochem Biophys Res Commun 262:68–75
Chiariello M, Visconti R, Carlomagno F, Melillo MM, Bucci C, de Franciscis V, Fox GM, Jing S, Coso OA, Gutkind JS, Fusco A, Santoro M (1998) Signaling of the RET receptor tyrosine kinase through the c-Jun NH2-terminal protein kinase (JNKS): evidence for a divergence of the ERKs and JNKs pathways induced by Ret. Oncogene 16:2435–2445
Donis-Keller H (1995) The RET proto-oncogene and cancer. J Int Med 238:319–325
Mulligan LM, Marsh JD, Robinson BG, Schuffenecker I, Zedenius J, Lips CJ, Gagel RF, Takai S-I, Noll WW, Fink M, Raue F, Lacroix A, Thibodeau SN, Frilling A, Ponder BAJ, Eng C (1995) Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J Intern Med 238:343–346
Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, Ploos van Amste HK, Lips CJM, Nishisho I, Takai S-I, Marsh DJ, Robinson BG, Frank-Raue K, Raue F, Xue F, Noll WW, Romei C, Pacini F, Fink M, Niederle B, Zedenius J, Nordenskjöld M, Komminoth P, Hendy GN, Gharib H, Thibodeau SN, Lacroix A, Frilling A, Ponder BAJ, Mulligan LM (1996) The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276:1575–1579
Mulligan LM, Eng C, Attie T, Lyonnet S, Marsh DJ, Hyland VJ, Robinson BG, Frilling A, Verellen-Dumoulin C, Safar A, Venter DJ, Munnich A Ponder BAJ (1994) Diverse phenotypes associated with exon 10 mutations of the RET proto-oncogene. Hum Mol Genet 3:2163–2167
Frank-Raue K, Höppner W, Frilling A, Kotzerke J, Dralle H, Haas R, Mann K, Seif F, Kirchner R, Rendl J, Deckart HF, Ritter MM, Hampel R, Klempa J, Scholz GH, Group GMTCS (1996) Mutations of the RET proto-oncogene in German MEN families: relation between genotype and phenotype. J Clin Endocrinol Metab 81:1780–1783
Schuffenecker I, Billaud M, Calender A, Chambe B, Ginet N, Calmettes C, Modigliani E, Lenoir GM (1994) RET proto-oncogene mutations in French MEN 2A and FMTC families. Hum Mol Genet 3:1939–1943
Eng C, Smith DP, Mulligan LM, Healey CS, Zvelebil MJ, Stonehouse TJ, Ponder MA, Jackson CE, Waterfield MD, Ponder BA (1995) A novel point mutation in the tyrosine kinase domain of the RET proto-oncogene in sporadic medullary thyroid carcinoma and in a family with FMTC. Oncogene 10:509–513
Berndt I, Reuter M, Saller B, Frank-Raue K, Groth, Grussendorf M, Raue F, Ritter MM, Höppner W (1998) A new hot spot for mutations in the RET proto-oncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 83:770–774
Bolino A, Schuffenecker I, Yin L, Seri M, Silengo M, Tocco T, Chabrier G, Houdent C, Murat A, Schlumberger M, Tourniaire J, Lenoir GM, Romeo G (1995) RET mutations in exons 13 and 14 of FMTC patients. Oncogene 10:2415–2419
Fattoruso O, Quadro L, Libroia A, Verga U, Lupoli G, Cascone E, Colantuoni V (1998) A GTG to ATG novel point mutation at codon 804 in exon 14 of the RET proto-oncogene in two families affected by familial medullary thyroid carcinoma. Hum Mutat 1:167–171
Feldmann GL, Edmonds MW, Ainsworth PJ, Schuffenecker I, Lenoir GM, Saxe AW, Talpos GB, Roberson J, Petrucelli N, Jackson CE (2000) Variable expression of familial medullary thyroid carcinoma (FMTC) due to a RET V804 M (CTG→ATG) mutation. Surgery 128:93–98
Niccoli-Sire P, Murat A, Rohmer V, Franc S, Chabrier G, Baldet L, Maes B, Frederique S, Giraud S, Bezieau S, Kottler ML, Morange S, Conte-Devolx B, The French Tumor Study Group (GETC) (2001) Familial medullary thyroid carcinoma with noncysteine RET mutations: phenotype-genotype relationship in a large series of patients. J Clin Endocrinol Metab 86:3746–3753
Gimm O, Marsh DJ, Andrew SD, Frilling A, Dahia PLM, Mulligan LM, Zajac JD, Robinson BG, Eng C (1997) Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J Clin Endocrinol Metab 82:3902–3904
Niccoli-Sire P, Murat A, Baudin E, Henry JF, Proye C, Bigorgne JC, Bstandig B, Modigliani E, Morange S, Schlumberger M, Conte-Devoix B, The French Tumor Study Group (GETC) (1999) Early or prophylactic thyroidectomy in MEN 2/FMTC gene carriers: results in 71 thyroidectomized patients. Eur J Endocrinol 141:468–474
Fitze G, Scherz M, Bredow J, Saeger HD, Roesner D, Schackert HK (2002) Various penetrance of familial medullary thyroid carcinoma in patients with RET protooncogene codon 790/791 germline mutations. Ann Surg 236:570–575
Iwashita T, Kato M, Murakami H, Asai N, Ishiguro Y, Ito S, Iwata Y, Kawai K, Asai M, Kurokawa K, Kajta H, Takahiashi M (1999) Biological and biochemical properties of Ret with kinase domain mutations identified in multiple endocrine neoplasia type 2b and familial medullary thyroid carcinoma. Oncogene 18:3919–3922
Bocciardi R, Mograbi B, Pasini B, Borello MG, Pierotti MA, Bourget I, Fischer S, Romeo Giovanni, Rossi B (1997) The multiple endocrine neoplasia type 2B point mutation switches the specificity of the Ret tyrosine kinase towards cellular substrates that are susceptible to interact with Crk and Nck. Oncogene 15:2257–2265
Smith DP, Houghton C, Ponder BAJ (1997) Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene 15:1213–1217
Bartsch DK, Hasse C, Schug C, Barth P, Rothmund M, Höppner W (2000) A RET double mutation in the germline of a kindred with FMTC. Exp Clin Endocrinol Diabetes 108:128–132
Koch CA, Huang S, Vortmeyer AO, Zhuang Z, Chrousos GP, Pacak K (2000) A patient with MEN 2 and multiple mutations of RET in the germline. Exp Clin Endocrinol Diabetes 108:493
Höppner W, Ritter MM (1997) A duplication of 12 bp in the critical cysteine rich domain of the RET proto-oncogene results in a distinct phenotype of multiple endocrine neoplasia type 2A. Hum Mol Genet 6:587–590
Höppner W, Dralle H, Brabant G (1998) A duplication of 9 base pairs in the critical cysteine rich domain of the RET proto-oncogene causes multiple endocrine neoplasia type 2A. Hum Mutat 1:128–130
Tessitore A, Sinisi AA, Pasquali D, Cardone M, Vitale D, Bellastella A, Collantuoni V (1999) A novel case of multiple endocrine neoplasia type 2A associated with two de novo mutations of the RET proto-oncogene. J Clin Endocrinol Metab 84:3522–3527
Committee on Genetics (2000) Molecular genetic testing in pediatric practice: a subject review. Pediatrics 106:1494–1497
Delbridge L, Robinson B (1998) Genetic and biochemical screening for endocrine disease. III. Costs and logistics. World J Surg 22:1212–1217
Skinner MA, De Benedetti MK, Moley JF, Norton JA, Wells SA (1996) Medullary thyroid carcinoma in children with multiple endocrine neoplasia types 2A and 2B. J Pediatr Surg 1:177–181, discussion 181–182
Wells SA Jr, Chi DD, Toshima K, Dehner LP, Coffin CM, Dowton B, Ivanovich JL, DeBenedetti MK, Dilley WG, Moley JF, Norton JA, Donis-Keller H (1994) Predictive DNA testing and prophylactic thyroidectomy in patients at risk for multiple endocrine neoplasia type 2A. Ann Surg 220:237–250
Wells SA Jr, Skinner MA (1998) Prophylactic thyroidectomy, based on direct genetic testing, in patients at risk for multiple endocrine neoplasia type 2 syndromes. Exp Clin Endocrinol Diabetes 106:29–34
Dralle H, Gimm O, Simon D, Frank-Raue K, Görtz G, Niederle B, Wahl RA, Koch B, Walgenbach S, Hampel R, Ritter MM, Spelsberg F, Heiss A, Hinze R, Höppner W (1998) Prophylactic thyroidectomy in 75 children and adolescents with hereditary medullary thyroid carcinoma: German and Austrian experience. World J Surg 22:744–751
Rodriquez Gonzales JM, Balsalobre MD, Pomares F, Torregrosa NM, Rios A, Carbonell P, Glower G, Sola J, Tebar J, Parrilla P (2002) Prophylactic thyroidectomy in MEN 2A syndrome: experience in a single center. J Am Coll Surg 195:159–166
Learoyd DL, Marsh DJ, Richardson AL, Twigg SM, Delbridge L, Robinson BG (1997) Genetic testing for familial cancer. Arch Surg 132:1022–1025
Lallier M, St-Vil D, Giroux M, Huot C, Gabourcy L, Oligny L, Desjardins JG (1998) Prophylactic thyroidectomy for medullary thyroid carcinoma in gene carriers of MEN 2 syndrome. J Pediatr Surg 33:846–848
Klein I, Esik O, Homolya V, Szeri F, Varadi A (2001) Molecular genetic diagnostic program of multiple endocrine neoplasia type 2A and familial thyroid carcinoma syndromes in Hungary. J Endocrinol 170:661–666
Pacini F, Romei C, Miccoli P, Elisei R, Molinaro E, Mancusi F (1995) Early treatment of hereditary medullary thyroid carcinoma after attribution of multiple endocrine neoplasia type 2 gene carrier status by screening for ret gene mutations. Surgery 118:1031–1035
van Heurn LWE, Schaap C, Sie G, Haagen AAM, Gerver WJ, Freling G, van Amstel PHK, Heinemann E (1999) Predictive DNA testing for multiple endocrine neoplasia 2: a therapeutic challenge of prophylactic thyroidectomy in very young children. J Pediatr Surg 34:568–571
Sanso GE, Domene HM, Garcia Rudaz MC, Pusiol E, de Mondino AK, Ronue M, Ring A, Perinetti H, Elsner B, Iorcansky S, Barotini M (2002) Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children. Cancer 94:323–330
La-Quaglia MP, Telander RL (1997) Differentiated and medullary thyroid cancer in childhood and adolescences. Semin Pediatr Surg 6:42–49
Iihara M, Yamashita T, Okamoto T, Kanabe M, Yamazaki K, Egawa S (1997) A nationwide clinical survey of patients with multiple endocrine neoplasia type 2 and familial medullary carcinoma in Japan. Jpn J Clin Oncol 27:128–134
O'Rioardan DS, O'Brien T, Crotty TB, Gharib H, Grant CS, van Heerden JA (1995) Multiple endocrine neoplasia type 2B: more than an endocrine disorder. Surgery 118:936–942
Torre M, Martucciello G, Ceccherini I, Lerone M, Aicardi M, Gambini C, Jasonni V (2002) Diagnostic and therapeutic approach to multiple endocrine neoplasia type 2B ind pediatric patients. Pediatr Surg Int 18:378–383
Gimm O, Niederle BE, Weber T, Bockhorn M, Ukkat J, Brauckhof M, Thanh PN, Frilling A, Klar E, Niederle B, Dralle H (2002) RET proto-oncogene mutations affecting codon 790/791: a mild form of multiple endocrine neoplasia 2A? Surgery 132:952–959
Frohnauer MK, Decker RA (2000) Update on the MEN 2A c804 RET mutation: is prophylactic thyroidectomy indicated? Surgery 128:1052–1058
Lecube A, Hernandez C, Oriola J, Galard R, Gemar E, Mesa J, Simo R (2002) V804 M RET mutation and familial medullary thyroid carcinoma: report of a large family with expression of the disease only in the homozygous gene carriers. Surgery 131:509–514
Lombardo F, Baudin E, Chiefari E, Arturi F, Bardett S, Caillou B, Conte C, Dallapiccola B, Guiffrida D, Bidart JM, Schlumberger M, Filetti S (2002) Familial medullary thyroid carcinoma: clinical variability and low aggressiveness associated with RET mutation at codon 804. J Clin Endocrinol Metab 87:1674–1680
Hansen HS, Terring H, Godballe C, Jäger AC, Nielsen FC (2000) Is thyroidectomy necessary in RET mutations carriers of the familial medullary thyroid carcinoma syndrome? Cancer 89:863–867
Dackiw AP, Kuerer HM, Clark OH (2002) Current national health insurance policies for thyroid cancer prophylactic surgery in the United States. World J Surg 26:903–906
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Frilling, A., Weber, F., Tecklenborg, C. et al. Prophylactic thyroidectomy in multiple endocrine neoplasia: the impact of molecular mechanisms of RET proto-oncogene. Langenbecks Arch Surg 388, 17–26 (2003). https://doi.org/10.1007/s00423-003-0368-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00423-003-0368-4