
Abstract
Obesity-induced endoplasmatic reticulum (ER) stress has been demonstrated to underlie the induction of obesity-induced JNK and NF-κB activation inflammatory responses, and generation of peripheral insulin resistance. On the other hand, exercise has been used as a crucial tool in obese and diabetic patients, and may reduce inflammatory pathway stimulation. However, the ability of exercise training to reverse endoplasmatic reticulum stress in adipose and hepatic tissue in obesity has not been investigated in the literature. Here, we demonstrate that exercise training ameliorates ER stress and insulin resistance in DIO-induced rats. Rats were fed with standard rodent chow (3,948 kcal kg−1) or high-fat diet (5,358 kcal kg−1) for 2 months. After that rats were submitted to swimming training (1 h per day, 5 days for week with 5% overload of the body weight for 8 weeks). Samples from epididymal fat and liver were obtained and western blot analysis was performed. Our results showed that swimming protocol reduces pro-inflammatory molecules (JNK, IκB and NF-κB) in adipose and hepatic tissues. In addition, exercise leads to reduction in ER stress, by reducing PERK and eIF2α phosphorylation in these tissues. In parallel, an increase in insulin pathway signaling was observed, as confirmed by increases in IR, IRSs and Akt phosphorylation following exercise training in DIO rats. Thus, results suggest that exercise can reduce ER stress, improving insulin resistance in adipose and hepatic tissue.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity is associated with chronic low-grade inflammation (Bray 2004; Hotamisligil 2008). Activation of serine kinases in obesity and their involvement in insulin action illustrates the close relationship between metabolic and immune pathways; in particular, c-jun N-terminal kinase (JNK) (Hotamisligil 2008) and I kappa β kinase (IKKβ) (Hirosumi et al. 2002; Shoelson et al. 2003). Together, JNK and IKKβ might serve as an inhibitor of insulin signaling, by inducing inhibitory serine 307 (Ser307) phosphorylation of IRS1 (Aguirre et al. 2000). IKK is a serine kinase; and its activation phosphorylates I kappa β kinase (IKKβ)/inhibitor of κB (IκB). After phosphorylation, IκB is ubiquitinated and degraded in the proteaosome, releasing nuclear factor κB (NF-κB) for the translocation to the nucleus and activation of gene expression (Cai et al. 2004). It has been proposed that increased NF-κB activation may play an important role in the pathogenesis of insulin resistance (Ragheb et al. 2008).
Recent studies provide an intriguing link between metabolic inflammation, endoplasmic reticulum (ER) stress and insulin resistance. It has been proposed that JNK and IKKβ activation was associated with altered endoplasmatic reticulum function and insulin resistance (Ozcan et al. 2004; Zhang et al. 2008). The two major molecules/markers of ER stress are PERK and eIF2α. Ozcan et al. (2004) showed that, in obesity, the damage to ER function results in insulin resistance and type 2 diabetes, which are dependent upon JNK activation. In contrast, the enhancement of ER function in transgenic mice or the use of chemical chaperones protects against diet-induced insulin resistance. In addition, the forced activation of IKKβ/NF-κB increased ER stress and interrupted central insulin/leptin signaling (Zhang et al. 2008). Thus, strategies to reduce the aberrant activation of inflammatory signaling and ER stress are of great interest to improve insulin action and glucose homeostasis.
Physical exercise has been linked to improved glucose homeostasis and enhanced insulin sensitivity in humans (Zierath 2002; Frosig et al. 2007) and rodents (Ropelle et al. 2006; Pauli et al. 2008). The improvement in insulin sensitivity was associated with the reduction in JNK activity in the skeletal muscle in humans after a single bout of exercise (Schenk and Horowitz 2007). JNK phosphorylation was reduced after resistance exercise in old men (Williamson et al. 2003). Several studies have examined the effects of exercise on inflammatory signaling. Sriwijitkamol and colleagues showed that 8 weeks of aerobic exercise training reduced IκB/NF-κB signaling in muscle from subjects with type 2 diabetes (Sriwijitkamol et al. 2006). However, the impact of exercise on ER stress remains to be better understood. Thus, the current study was designed to investigate the effects of exercise training on inflammatory signaling, ER and insulin resistance in the adipose, and hepatic tissue of obese rats.
Materials and methods
Experimental animals and diet
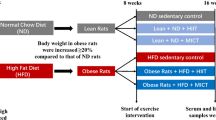
Male Wistar rats bred in the University of Southern Santa Catarina were used in the investigation. All experiments were approved by the Ethics Committee of the University of Southern Santa Catarina, UNESC. The 4-week-old Wistar rats were divided into four groups: control rats (C) fed on standard rodent chow, control rats submitted to 8-week-endurance training with workload (C + ET), obese rats fed on an obesity-inducing diet for 2 months (DIO) (Table 1), and DIO rats submitted to 8-week endurance training with workload (DIO + ET). The control rats were maintained sedentary throughout the experimental period and the trained rats were submitted to 1 h daily swimming sessions in water at 32°C, with an attached weight corresponding to 5% of body weight. Training occurred 5 days/week for 8 weeks. The rats were allowed free access to standard rodent chow or high-fat diet and water.
Fasting glucose, insulin tolerance test (ITT), serum insulin and TNFα quantification
After 24 h of the last exercise session and 6 h of fasting, eight (8) rats per group were submitted to an insulin tolerance test (ITT; 1.5 U/kg). Briefly, 1.5 IU/kg of human recombinant insulin (Humulin R) from Eli Lilly (Indianapolis, IN, USA) was injected intraperitoneally in anesthetized mice and blood samples were collected from the tail at 0, 5, 10, 15, 20, 25 and 30 min for serum glucose determination. The rate constant for plasma glucose disappearance (K ITT) was calculated using the formula 0.693/biological half life (t 1/2) (Bonora et al. 1989). Plasma glucose level was determined by a colorimetric method using a glucose meter (Advantage, Boehringer Mannheim, Irvine, USA). Plasma was separated by centrifugation (1,500g) for 15 min at 4°C and stored at −80°C until assay. Radioimmunoassay (RIA) was employed to measure serum insulin, according to a previous description (Scott et al. 1981). Serum TNFα concentration was measured by ELISA (Pierce Biotechnology, Rockford, IL, USA), following the manufacturer’s recommendations.
Killing of the animals
The rats were anesthetized with an intraperitoneal (i.p.) injection of sodium thiopental (40 mg kg−1). In all experiments, the appropriateness of anesthesia depth was tested by evaluating pedal and corneal reflexes, throughout the experimental procedure. Following the experimental procedures, the rats were killed under anesthesia (thiopental 200 mg kg−1), following the recommendations of the NIH publication no. 85–23.
Protein analysis by immunoblotting
As soon as anesthesia was assured by the loss of pedal and corneal reflexes, the abdominal cavity was opened, the cava vein exposed, and 0.2 mL of normal saline (−) (5 rats per group) or insulin (10−6 mol L−1) (+) were injected (5 rats per group). After insulin injection (only for insulin pathway analysis), hepatic and adipose tissue fragments were excised. The tissues were homogenized immediately in extraction buffer (mM) (1% Triton X-100, 100 mM Tris, pH 7.4, containing 100 mM sodium pyrophosphate, 100 mM sodium fluoride, 10 EDTA, 10 sodium vanadate, 2 PMSF and 0.1 mg of aprotinin/ml) at 4°C with a Polytron PTA 20S generator (Brinkmann Instruments, Westbury, New York, USA) operated at maximum speed for 30 s. The extracts were centrifuged at 11,000 rpm and 4°C in a Beckman 70.1 Ti rotor (Palo Alto, CA, USA) for 40 min to remove insoluble material, and the supernatants of these tissues were used for protein quantification, using the Bradford method (Bradford 1976). Aliquots of the resulting supernatants containing 2.0 mg of total protein were used for immunoprecipitation with antibodies against IR, IRS1, IRS2 at 4°C overnight, followed by SDS-PAGE, transfer to nitrocellulose membranes and blotting with anti-phosphotyrosine (PY). In direct immunoblot experiments, 0.2 mg of protein extracts were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked, probed and developed, as described previously De Souza et al. 2005). Antibodies used for immunoblotting were anti-phospho-PERK, anti-phospho-eIF-2α (Cell Signaling Technology, Beverly, MA, USA), anti-IR, anti-IRS1, anti-IRS2, anti-Phosphotyrosine (PY), anti-phospho-JNK, anti-NF-κB, anti-phospho-IκB, and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Chemiluminescent detection was performed with horseradish peroxidase-conjugate secondary antibodies. Visualization of protein bands was performed by exposure of membranes to RX-films. The protein loading of immunoblots was always evaluated by two methods: before blotting, nitrocellulose membranes were stained with Coomassie blue, and after blotting membranes were reprobed with a β-actin antibody.
Statistical analysis
All numeric results are expressed as the mean ± standard error of mean (SEM) of the indicated number of experiments. The results of blots are presented as direct comparisons of bands or spots in autoradiographs and quantified by optical densitometry (Scion Image). Statistical analysis was performed using the ANOVA test with Tukey post test. Significance level was established as p < 0.05. Data were analyzed using the Statistical Package for the Social Sciences (SPSS) version 18.0 for Windows.
Results
Physiological and metabolic parameters
Table 2 shows comparative data for control and DIO rats, when sedentary and after training exercise. The DIO group demonstrated a significant increase in body weight, epididymal fat and fasting serum insulin; however, fasting glucose was not altered. No significant variations were found in body weight, epididymal fat and fasting glucose in DIO rats after exercise training. However, the rate constant for plasma glucose disappearance and fasting serum insulin were increased (95%) and decreased (52.8%), respectively, in the exercise training group, when compared with DIO sedentary group. In addition, the levels of insulin were reduced after exercise in DIO rats (53%) compared with DIO sedentary group. The obesity-induced high-fat diet caused a significant increase in serum TNFα levels (35.15%, p = 0.02), when compared with C group. On the other hand, the exercise-trained DIO group showed a reduction of 21.9% in TNFα levels (p = 0.04), when compared with sedentary DIO.
Effects exercise training on JNK and IkBα phosphorylation and NF-κB expression
An increase in JNK phosphorylation in the adipose (53%) and hepatic (120%) tissue was observed in DIO rats, when compared with control rats (Fig. 1a, d). In the DIO trained group, JNK phosphorylation decreased in both adipose (48%) and hepatic (40%) tissue, when compared with DIO sedentary (Fig. 1a, d). No difference was observed in JNK protein level in either the adipose or the hepatic tissue.

Effects of training exercise on NF-κB protein levels and JNK and IkBα phosphorylation. Adipose and hepatic extracts from rats were prepared as described in “Materials and methods”. a, d Adipose and hepatic (respectively) extracts were IB with anti-phospho JNK. b, e adipose and hepatic (respectively) extracts were IB with anti-NF-κB. c, f Adipose and hepatic (respectively) extracts were IB with anti-phospho IκBα. Bars represent mean ± SEM of n = 5 rats. *p < 0.05, DIO rats versus control groups; # p < 0.05, DIO + ET group versus DIO sedentary rats
Subsequently, we examined the IKK/NF-κB pathway in the liver and adipose tissues of lean and obese rats after the exercise protocol. In the DIO group, we observed an increase in IκBα phosphorylation in the adipose (75%) and hepatic (100%) tissues, when compared with control rats (Fig. 1b, e, respectively). In the adipose tissue of DIO + ET rats, IκBα phosphorylation decreased by 65% (Fig. 1b), while a 48% decrease was observed in hepatic tissue (Fig. 1e), when compared to DIO sedentary. The high-fat diet-increased NF-κB expression in adipose (104%) and hepatic (107%) tissues, when compared with the control (Fig. 1c, f, respectively). On the other hand, exercise training in the DIO group decreased NF-κB expression in adipose (40%) and hepatic (41%) tissues, when compared with DIO sedentary (Fig. 1c, f, respectively). No difference was observed in IκB protein level in either the adipose or hepatic tissue.
Exercise training reduces phospho PERK and phospho eIF2α
We determined the phosphorylation status of PERK (Thr980) and eIF2α (Ser51) in adipose and hepatic tissues. The DIO group demonstrated increases of 80% and 135% in PERK phosphorylation, when compared to the C group (Fig. 2a, c adipose and hepatic tissues, respectively). In the DIO group, exercise training reduced PERK phosphorylation by 58 and 47% (Fig. 2a, c adipose and hepatic tissues, respectively). No difference was observed in PERK expression in either the adipose or hepatic tissue.

Effects of exercise training on reticulum stress. Liver and adipose extracts from rats were prepared as described in “Materials and methods”. a Adipose tissue extracts were immunoblotted (IB) with anti-phospho PERK or anti β-actin antibodies (upper and lower panels, respectively). b Adipose tissue extracts were immunoblotted (IB) with anti-phospho eIF2α or anti-β-actin antibodies (upper and lower panels, respectively). c Liver extracts were immunoblotted (IB) with anti-phospho PERK or anti-β-actin antibody (upper and lower panels, respectively). d Liver extracts were immunoblotted (IB) with anti-phospho eIF2α or anti-β-actin antibodies (upper and lower panels, respectively). β-Actin was used as protein load. The results of scanning densitometry are expressed as arbitrary units. Bars represent mean ± SEM of n = 5 rats. *p < 0.05, DIO group versus sedentary control rats; # p < 0.05, DIO + ET versus DIO sedentary rats
The DIO sedentary rats demonstrated increases of 98 and 138% in eIF2α phosphorylation, when compared with the control group (Fig. 2b, d adipose and hepatic tissues, respectively). Exercise training in DIO rats reduced eIF2α phosphorylation by 64 and 58% (Fig. 2b, d adipose and hepatic tissues, respectively). No difference was observed in eIF2α expression in either the adipose or hepatic tissue. In all experiments, β-actin was used as a loading protein (data not shown).
Improve insulin signaling by exercise training
In adipose tissue, insulin induced increases in IR tyrosine phosphorylation in both the C and C + ET groups, when compared with basal (B)/saline injection group (Fig. 3a). Similar results were observed in the hepatic tissue (Fig. 3d). In the DIO group, IR phosphorylation was reduced by 105% in the adipose and 98% in the hepatic tissues, respectively, when compared with the respective C+ group (Fig. 3a, d). In the adipose and hepatic tissues of DIO + ET, IR phosphorylation increased by 72 and 70%, respectively, when compared to the respective DIO sedentary group (Fig. 3a, d). There were no differences in basal levels of IR tyrosine phosphorylation between the groups (data not shown), and IR protein levels did not differ between the groups (Fig. 3a, d lower panels).

Insulin signaling. Liver and adipose extracts from rats, injected with saline (B, Basal group) or insulin (+), were prepared as described in “Materials and methods”. a, d Adipose and hepatic (respectively) tissue extracts were immunoprecipitated (IP) with anti-IR antibody and blotted (IB) with anti-PY antibody (upper panels) or anti-IR antibody (lower panels). b, e Adipose and hepatic (IRS1 and IRS2, respectively) tissue extracts were IP with anti-IRS1 antibody and IB with anti-PY (upper panels) and anti-IRS1 antibodies (lower panels) for adipose tissue, and IP with anti-IRS2 antibody and IB with anti-PY (upper panels) and anti-IRS2 antibodies (lower panels) for hepatic tissue. c, f Adipose and hepatic tissue extracts were IB with anti-phospho Akt or anti-Akt antibody (upper and lower panels, respectively). The results of scanning densitometry are expressed as arbitrary units. Bars represent means ± SEM of n = 5 rats. *p < 0.05, control and control plus exercise training stimulated-insulin (+) versus Basal [no insulin control (−)]; # p < 0.05 DIO rats versus control sedentary rats; and & p < 0.05, DIO + ET group versus DIO sedentary rats
IRS1 phosphorylation in the adipose tissue and IRS2 phosphorylation in the hepatic tissue increased after insulin injection in both C and C + ET groups, when compared with group B (Fig. 3b, e, respectively). In the DIO group, IRS1 tyrosine phosphorylation was reduced by 56% in adipose tissue and IRS2 was reduced by 60% in hepatic tissue, when compared with the respective control group (Fig. 3b, e). In the adipose tissue of DIO + ET, IRS1 phosphorylation increased by 57%, while IRS2 phosphorylation increased by 60% in the hepatic tissue, as compared with DIO groups (Fig. 3b, e). There was no difference in the basal levels of IRS1 and IRS2 tyrosine phosphorylation between the groups (data not shown). The IRS1 and IRS2 protein levels did not differ between the groups (Fig. 3b, e lower panels).
Insulin induced increases in Akt serine phosphorylation in both the C and C + ET groups, when compared with group B (Fig. 3c). Similar results were observed in hepatic tissue (Fig. 3f). In the DIO group, Akt phosphorylation was reduced by 61% in the adipose and 50% in the hepatic tissues, when compared with the respective control group (Fig. 3c, f). In the adipose and hepatic tissues of DIO + ET, Akt phosphorylation increased by 44 and 59%, respectively, in comparison with the DIO group (Fig. 3c, f). There was no difference in basal levels of IR tyrosine phosphorylation between the groups (data not shown). The Akt protein levels did not differ between the groups (Fig. 3c, f lower panels).
Discussion
The reports indicate that ER stress, induced by obesity, is associated with inflammation and established a link between inflammatory responses, particularly through the JNK and IKK signaling pathways (Hu et al. 2006; Hotamisligil 2008), with abnormal insulin action (Ozcan et al. 2004; Wellen and Hotamisligil 2005). Ozcan and colleagues found that obesity is associated with the induction of chronic ER stress, predominantly in the liver and adipose tissues (Ozcan et al. 2004). The molecular mechanisms linking ER stress to glucose intolerance in trained obese animals has not been investigated yet. Therefore, this study sought to verify whether exercise training reduces ER stress in hepatic and adipose tissues of obese animals and whether this is relational with a reduction of insulin resistance of these tissues.
In this study, we demonstrate that high-fat diet lead to an increase in JNK protein level. It is interesting to note that chronic exercise reverses these parameters in parallel with a reduction in JNK activity and IκBα degradation. In accordance with this, our results show increased phosphorylation of PERK and eIF2α in animals fed with a high-fat diet. However, the activity of these proteins was observed to drop in animals submitted to exercise training. This decrease was accompanied by increased insulin sensitivity and correlates with the increase in phosphorylation of IR, IRS1 and IRS2 and serine phosphorylation Akt in liver and adipose tissues. More importantly, the changes occur in the absence of changes in adiposity or body weight.
Several mechanisms may be involved in the pathogenesis of insulin resistance in peripherals tissues. Nevertheless, it is well known that some of the molecular mechanisms leading to insulin resistance involve alterations in key molecules due to their intracellular signaling pathways, such as JNK and NF-κB (Aguirre et al. 2000; Hirosumi et al. 2002; Lee et al. 2003). Moreover, current evidence has demonstrated that one of the pathological mechanisms of insulin resistance is derived from impairment of endoplasmic reticulum function, a process known as ER stress.
JNK has been linked to the regulation of insulin signaling by several studies (Aguirre et al. 2000; Hirosumi et al. 2002; Lee et al. 2003; Gao et al. 2002; Cai et al. 2004; Ragheb et al. 2008). In addition, recent works linked JNK activation, ER stress and insulin resistance (Ozcan et al. 2004; Zhang et al. 2008). Thus, the role of exercise in JNK activation is more important, and deserves to be investigated in more detail. Several studies suggest that the activity of JNK intracellular signaling cascade is increased following prolonged running exercise (Boppart et al. 2000; Thompson et al. 2003). In contrast, JNK phosphorylation was reduced after resistance exercise in elderly men (Williamson et al. 2003). In this study, we observed that exercise training inhibited high-fat diet-induced JNK activity.
Activation of inflammatory signaling, including of the IκB–NF-κB pathway may also contribute to mediate ER stress (Gao et al. 2002). However, a few studies have examined the effect of physical exercise on the IκB–NF-κB pathway. In rats, exercise activates IκB–NF-κB signaling in muscle (Ji et al. 2004), and acute fatiguing exercise in humans reduces NF-κB activity. Similarly, a recent study showing that 8 weeks of aerobic exercise training reduced IκB–NF-κB signaling in vastus lateral is muscle from subjects with type 2 diabetes (Sriwijitkamol et al. 2006), our results show that animals submitted with a high-fat diet has pronounced reduction of IκBα in liver and adipose tissue. This finding is an indication of IKK activation and suggests that endurance exercise training is able to reduce IKK activation and restore IκBα expression.
Recently, it has been shown that obesity induces chronic ER stress, which, in turn, plays a central role in the development of insulin resistance and diabetes (Ozcan et al. 2004). The phosphorylation status of PERK and eIF2α is, therefore, a key indicator of the presence of ER stress. In this study we found an increase in these indicators of ER stress in liver and adipose tissues of obese rats. Thus, we suggest that a high-fat diet-induced endoplasmic reticulum stress. In hepatic cells, ER stress inhibits the action of insulin (Ozcan et al. 2004); however, there are no reports demonstrating that exercise training decreases ER stress in liver and adipose tissues in DIO-induced obesity. In the present study, we showed that exercise training reduces phosphorylation of PERK and eIF2, indicating a decrease in ER stress and an improvement in insulin signal transduction. Although the present study does not produce direct evidence that changes in the PERK pathway are linked to changes in insulin signaling, many works in the literature have observed this outcome (Ozcan et al. 2004; Wellen and Hotamisligil 2005; Hu et al. 2006; Hotamisligil 2008).
Elegant reports indicate that ER stress, induced by obesity, is associated with inflammation and established a link between inflammatory responses, particularly through the JNK and IKK signaling pathways (Hu et al. 2006; Hotamisligil 2008), with abnormal insulin action (Ozcan et al. 2004; Wellen and Hotamisligil 2005). Thus, we suggest that exercise training reduces markers of ER stress by changing inflammatory pathways. Hu and colleagues reported that ER stress is induced in a NF-κB-dependent manner. In addition, the inhibition of NF-κB suppresses ER stress-induced cell death in MCF-7 cells (Hu et al. 2006). Jiang and colleagues showed that, during ER stress, phosphorylation of the alpha subunit of eIF2 by PERK is required for activation of NF-κB (Jiang et al. 2003). A cause-effect relationship between inflammatory pathways and ER stress is not totally clear. In the present study, NF-κB expression was elevated in obese rats, and reduced in trained obese rats. In addition, the activity of JNK was also increased and appears to be involved in ER stress (for review see Hotamisligil 2008). On the order hand, our data show that the activity of JNK was largely decreased in trained obese rats. Thus, these effects may reduce ER stress.
In this context, cytokines contribute to the activation of intracellular inflammatory signal transduction. Furthermore, our group has reported that obese animals present high cytokine levels (De Souza et al. 2005; Barbuio et al. 2007; Cintra et al. 2008; Pauli et al. 2008; Prada et al. 2009; Romanatto et al. 2009). Thus, we evaluated serum TNFα concentrations. We observed increased TNFα levels in DIO rats, which may explain the modification of pro-inflammatory molecules (JNK, IκB and NF-κB) induced by high-fat diet (as previously demonstrated). After endurance exercise training, DIO rats demonstrated a reduction in TNFα levels.
In this sense, the liver should, therefore, be considered as a working organ during exercise and thus some of the beneficial effects of physical activity on insulin sensitivity and metabolism occur in the liver. Insulin signaling plays an important role in controlling gluconeogenic gene expression, including that of phosphoenolpyruvate carboxykinase (PEPCK), which catalyzes the rate-limiting step of hepatic gluconeogenesis (Sutherland et al. 1996). More specifically, in the hepatic tissue this improved insulin signaling may decrease glucose production by liver. Collectively, our data provide evidence that exercise training improves insulin signaling and this mechanism involved a decrease in inflammation and ER stress in liver and adipose tissue of diet-induced obesity rats.
Unfortunately, the present study has limitations, especially, of secondary proof and isolated tissues analysis. Currently, we are unable, in our laboratory, to use 4-phenylbutyric acid (for inhibition ER stress) or tunicamycin (for induction ER stress). In addition, it is not yet clear whether exercise training improves ER stress by reducing the inflammatory pathway or by a direct action on endoplasmic reticulum stress. Although the literature shows that a reduction in ER stress results in improved insulin resistance, we could not demonstrate this in the present study, and the two events may occur in parallel. However, while this is a descriptive study, the discovery that exercise training reduces markers of ER stress may be one possible explanation, among several others, as how exercise has beneficial effects on obesity and diabetes.
References
Aguirre V, Uchida T, Yenush L, Davis R, White MF (2000) The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275(12):9047–9054
Barbuio R, Milanski M, Bertolo MB, Saad MJ, Velloso LA (2007) Infliximab reverses steatosis and improves insulin signal transduction in liver of rats fed a high-fat diet. J Endocrinol 194:539–550
Bonora E, Moghetti P, Zancanaro C, Cigolini M, Querena M, Cacciatori V, Corgnati A, Muggeo M (1989) Estimates of in vivo insulin action in man: comparison of insulin tolerance tests with euglycemic and hyperglycemic glucose clamp studies. J Clin Endocrinol Metab 68:374–378
Boppart MD, Asp S, Wojtaszewski JF, Fielding RA, Mohr T, Goodyear LJ (2000) Marathon running transiently increases c-Jun NH2-terminal kinase and p38 activities in human skeletal muscle. J Physiol 526(Pt 3):663–669
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Bray GA (2004) Medical consequences of obesity. J Clin Endocrinol Metab 89:2583–2589
Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE (2004) IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119:285–298
Cintra DE, Pauli JR, Araujo EP, Moraes JC, De Souza CT, Milanski M, Morari J, Gambero A, Saad MJ, Velloso LA (2008) Interleukin-10 is a protective factor against diet-induced insulin resistance in liver. J Hepatol 48:628–637
De Souza CT, Araújo EP, Prada PO, Saad MJ, Boschero AC, Velloso LA (2005) Short-term inhibition of peroxisome proliferator-activated receptor gamma coactivator-1alpha expression reverses diet-induced diabetes mellitus and hepatic steatosis in mice. Diabetologia 48:1860–1871
Frosig C, Rose AJ, Treebak JT, Kiens B, Richter EA, Wojtaszewski JF (2007) Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes 56:2093–2102
Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J (2002) Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem 277(50):48115–48121
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Hotamisligil GS (2008) Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond) 32:S52–S54
Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH (2006) Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol 8:3071–3084
Ji LL, Gomez-Cabrera MC, Steinhafel N, Vina J (2004) Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in rat skeletal muscle. FASEB J 18(13):1499–1506
Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC (2003) Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-KappaB in response to diverse cellular stresses. Mol Cell Biol 16:5651–5663
Lee YH, Giraud J, Davis RJ, White MF (2003) c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 278(5):2896–2902
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Pauli JR, Ropelle ER, Cintra DE, Carvalho-Filho MA, Moraes JC, De Souza CT, Velloso LA, Carvalheira JB, Saad MJ (2008) Acute physical exercise reverses S-nitrosation of the insulin receptor, insulin receptor substrate 1 and protein kinase B/Akt in diet-induced obese Wistar rats. J Physiol 586(2):659–671
Prada PO, Ropelle ER, Mourão RH, De Souza CT, Pauli JR, Cintra DE, Schenka A, Rocco SA, Rittner R, Franchini KG, Vassalo J, Velloso LA, Carvalheira JB, Saad MJ (2009) EGFR tyrosine kinase inhibitor (PD153035) improves glucose tolerance and insulin action in high-fat diet fed mice. Diabetes 58:2910–2919
Ragheb R, Medhat AM, Shanab GM, Seoudi DM, Fantus IG (2008) Links between enhanced fatty acid flux, protein kinase C and NFkappaB activation, and apoB-lipoprotein production in the fructose-fed hamster model of insulin resistance. Biochem Biophys Res Commun 370:134–139
Romanatto T, Roman EA, Arruda AP, Denis RG, Solon C, Milanski M, Moraes JC, Bonfleur ML, Degaspari GR, Picardi PK, Hirabara S, Boschero AC, Curi R, Velloso LA (2009) Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J Biol Chem 284:36213–36222
Ropelle ER, Pauli JR, Prada PO, de Souza CT, Picardi PK, Faria MC, Cintra DE, Fernandes MF, Flores MB, Velloso LA, Saad MJ, Carvalheira JB (2006) Reversal of diet-induced insulin resistance with a single bout of exercise in the rat: the role of PTP1B and IRS-1 serine phosphorylation. J Physiol 577:997–1007
Schenk S, Horowitz JF (2007) Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest 117:1690–1698
Scott MD, Atwater I, Rojas E (1981) A method for the simultaneous measurement of insulin release and B cell membrane potential in single mouse islets of Langerhans. Diabetologia 21:470–475
Shoelson SE, Lee J, Yuan M (2003) Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord 27:S49–S52
Sriwijitkamol A, Christ-Roberts C, Berria R, Eagan P, Pratipanawatr T, DeFronzo RA, Mandarino LJ, Musi N (2006) Reduced skeletal muscle inhibitor of kappaB beta content is associated with insulin resistance in subjects with type 2 diabetes: reversal by exercise training. Diabetes 55:760–767
Sutherland C, O’Brien RM, Granner DK (1996) New connections in the regulation of PEPCK gene expression by insulin. Philos Trans R Soc Lond B Biol Sci 351(1336):191–199
Thompson HS, Maynard EB, Morales ER, Scordilis SP (2003) Exercise-induced HSP27, HSP70 and MAPK responses in human skeletal muscle. Acta Physiol Scand 178(1):61–72
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 5:1111–1119
Williamson D, Gallagher P, Harber M, Hollon C, Trappe S (2003) Mitogen-activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle. J Physiol 547:977–987
Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135:61–73
Zierath JR (2002) Invited review: exercise training-induced changes in insulin signaling in skeletal muscle. J Appl Physiol 93:773–781
Acknowledgments
This study was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). We thank Dr Nicola Conran for English language editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Klaas R Westerterp.
Rights and permissions
About this article
Cite this article
da Luz, G., Frederico, M.J.S., da Silva, S. et al. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur J Appl Physiol 111, 2015–2023 (2011). https://doi.org/10.1007/s00421-010-1802-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-010-1802-2