
Abstract
The effect of hyperventilation-induced hypocapnic alkalosis (HYPO) and prior heavy-intensity exercise (HVY) on pulmonary O2 uptake \( (\dot{V}{\text{O}}_{{ 2 {\text{p}}}}) \) kinetics were examined in young adults (n = 7) during moderate-intensity exercise (MOD). Subjects completed leg cycling exercise during (1) normal breathing (CON, PETCO2 ~ 40 mmHg) and (2) controlled hyperventilation (HYPO, PETCO2 ~ 20 mmHg) throughout the protocol, with each condition repeated on four occasions. The protocol consisted of two MOD transitions (MOD1, MOD2) to 80% estimated lactate threshold with MOD2 preceded by HVY (Δ50%); each transition lasted 6 min and was preceded by 20 W cycling. \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was measured breath-by-breath and concentration changes in oxy- and deoxy-hemoglobin/myoglobin (Δ[HHb]) of the vastus lateralis muscle were measured by near-infrared spectroscopy. Adjustment of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) and Δ[HHb] were modeled using a mono-exponential equation by non-linear regression. During MOD1, the phase 2 time constant (τ) for \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \,(\tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} ) \) was greater (P < 0.05) in HYPO (45 ± 24 s) than CON (28 ± 17 s). During MOD2, \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was reduced (P < 0.05) in both conditions (HYPO: 24 ± 7 s, CON: 20 ± 8 s). The Δ[HbTOT] and Δ[O2Hb] were greater (P < 0.05) prior to and throughout MOD2. The Δ[HHb] mean response time was similar in MOD1 and MOD2, and between conditions, however, the MOD1 Δ[HHb] amplitude was greater (P < 0.05) in HYPO compared to CON, with no differences between conditions in MOD2. These findings suggest that the speeding of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics after prior HVY in HYPO was related, in part, to an increase in microvascular perfusion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Following a sudden increase in exercise intensity (and thus muscle ATP requirement), muscle O2 utilization and, after a brief delay, the fundamental component of pulmonary O2 uptake \( (\dot{V}{\text{O}}_{{ 2 {\text{p}}}} ) \) increase exponentially towards a new steady-state level. Whether the delay in reaching the required \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response resides in a relatively slow activation of rate-limiting enzymes and provision of substrates (other than O2) to the mitochondrial tricarboxylic acid (TCA) cycle and electron transport chain (ETC), to impaired convective and/or diffusive delivery of O2 to the terminal oxidase in the mitochondria, or to a combination of these factors is unresolved (Poole et al. 2008; Tschakovsky and Hughson 1999).
Voluntary hyperventilation resulting in a hypocapnic alkalosis (HYPO, i.e., respiratory alkalosis) has been shown to slow the adjustment of pulmonary \( \dot{V}{\text{O}}_{ 2} \, (\dot{V}{\text{O}}_{{ 2 {\text{p}}}} ) \) (Chin et al. 2007; Hayashi et al. 1999) and the breakdown of muscle phosphocreatine (PCr) (Forbes et al. 2007) [both measures reflecting the kinetics of muscle O2 utilization (Grassi et al. 1996; Krustrup et al. 2009; Rossiter et al. 1999)], and to delay the activation of the mitochondrial pyruvate dehydrogenase (PDH) enzyme complex (LeBlanc et al. 2002) during the transition to moderate-intensity exercise. Hayashi et al. (1999) attributed the slowed \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) on-kinetics to impaired O2 off-loading from hemoglobin as a consequence of an alkalosis-induced leftward-shift in the oxyhemoglobin dissociation curve. Alternatively, based on near-infrared spectroscopy (NIRS)-derived measures of total- and deoxy-hemoglobin/myoglobin changes in the vastus lateralis muscle during exercise, Chin et al. (2007) suggested that reduced muscle microvascular perfusion, in part, was responsible for the slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics observed during the on-transition to exercise in respiratory alkalosis. Similarly Forbes et al. (2007) using simultaneous measures of phosphorus magnetic resonance spectroscopy (31P-MRS) and NIRS, suggested that impaired microvascular perfusion and O2 delivery was responsible for a slowed adjustment of muscle O2 utilization which resulted in the greater and slower rate of muscle PCr breakdown reported in that study.
A prior priming bout of heavy-intensity exercise (HVY) has been shown to be effective at speeding \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics during a subsequent bout of MOD, especially in older adults (DeLorey et al. 2004; Scheuermann et al. 2002) and in young adults presenting with slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics (Gurd et al. 2005, 2006). Although a speeding of the \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response in MOD is not seen consistently with prior HVY (Burnley et al. 2000; DeLorey et al. 2004; Gerbino et al. 1996; Scheuermann et al. 2002), it is evident that the likelihood of observing a measureable decrease in the phase 2 \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) time constant \( \left( {\tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} } \right) \) following priming exercise may be dependent on the relative “sluggishness” of the unprimed response (e.g., see Fig. 2 in Gurd et al. 2005 and Fig. 4 in Gurd et al. 2006). After HVY exercise, heart rate (DeLorey et al. 2004; Gurd et al. 2005; Scheuermann et al. 2002) and NIRS-derived measures of oxy- and total hemoglobin/myoglobin [reflecting greater local (microvascular) perfusion] (DeLorey et al. 2007; DeLorey et al. 2004; Gurd et al. 2005, 2006) remained elevated prior to and throughout the subsequent bout of exercise, while conduit artery muscle (i.e., bulk) blood flow remained elevated at baseline and during the immediate onset of exercise (DeLorey et al. 2007; Endo et al. 2005; Fukuba et al. 2004; Hughson et al. 2003; Jones et al. 2006; MacDonald et al. 2001; Paterson et al. 2005). Based on these findings, it was argued that the speeding of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics seen after HVY in these individuals is due, in part, to the removal of the constraint imposed by an inadequate local microvascular blood flow distribution and O2 delivery during the exercise on-transition.
Therefore, the purpose of the present study was to examine the effect of hyperventilation-induced hypocapnic alkalosis (HYPO) and prior heavy-intensity exercise on \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics during the transition to moderate-intensity exercise. It was hypothesized that the slowed \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics associated with HYPO would become faster when preceded by a priming bout of heavy-intensity exercise and that this would be due, in part, to improved perfusion within the muscle [as assessed by a greater concentration change in the NIRS-derived oxy- and total hemoglobin/myoglobin, and less reliance on fractional O2 extraction (as assessed by the NIRS-deoxygenation signal)].
Methods
Subjects
Seven healthy male subjects [age 23 ± 3 years, \( \dot{V}{\text{O}}_{{ 2\,{\text{peak}}}} \) 49 ± 10 ml/(kg min), mean ± SD] were given verbal and written explanation of the experimental protocol and possible associated risks and discomforts before volunteering to participate in this study. Informed consent was obtained from each subject prior to any data collection. The protocol was approved by The University of Western Ontario Ethics Committee for Research on Human Subjects, in accordance with the Declaration of Helsinki. All subjects were non-smokers and free of known respiratory, cardiovascular and metabolic disease.
Pre-experimental protocol
Each subject completed a ramp incremental exercise test (20 W/min) to volitional fatigue on an electromagnetically braked cycle ergometer (Lode, model H- 300-R) to estimate their lactate threshold \( \left( {\hat{\theta }_{\text{L}} } \right) \) using gas exchange indices, and to measure their peak \( \dot{V}{\text{O}}_{ 2} \,(\dot{V}{\text{O}}_{{ 2\,{\text{peak}}}} ). \) The \( \hat{\theta }_{\text{L}} \) was defined as the \( \dot{V}{\text{O}}_{ 2} \) at which \( \dot{V}{\text{CO}}_{ 2} \) (CO2 output) began to increase out of proportion to the increase of \( \dot{V}{\text{O}}_{ 2} \) with a rise similarly observed with the ventilatory equivalent for \( \dot{V}{\text{O}}_{ 2} \, (\dot{V}_{\text{E}} /\dot{V}{\text{O}}_{ 2} ) \) and end-tidal PO2 (PETO2) with no systematic increase in the ventilatory equivalent for \( \dot{V}{\text{CO}}_{ 2} \, (\dot{V}_{\text{E}} /\dot{V}{\text{CO}}_{ 2} ) \) or fall in end-tidal PCO2 (PETCO2) (Beaver et al. 1986; Whipp et al. 1986). \( \dot{V}{\text{O}}_{{ 2\;{\text{peak}}}} \) was determined as the average \( \dot{V}{\text{O}}_{ 2} \) from the final 20 s of the ramp incremental test. Results from the ramp test also allowed determination of the work rate (WR) that elicited ~80% of the \( \dot{V}{\text{O}}_{ 2} \) at \( \hat{\theta }_{\text{L}} \) (moderate-intensity exercise, MOD) and ~50% of the difference (Δ50%) between the \( \dot{V}{\text{O}}_{ 2} \) at \( \hat{\theta }_{\text{L}} \) and \( \dot{V}{\text{O}}_{{ 2\;{\text{peak}}}} \) (heavy-intensity exercise, HVY).
Before any actual data collection, subjects familiarized themselves with the hyperventilation maneuver by establishing the degree of hyperventilation needed to attain the target end-tidal PCO2 (PETCO2) of ~20 mmHg from a normal PETCO2 of ~40 mmHg (Chin et al. 2007; LeBlanc et al. 2002).
Exercise protocol
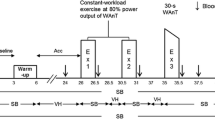
The protocol consisted of 5 min baseline with subjects sitting quietly on the cycle ergometer and breathing normally, followed by an “accommodation” period lasting 20 min where subjects either continued with normal breathing (CON; PETCO2 ~ 40 mmHg) or began voluntarily hyperventilating to induce a hypocapnic alkalosis (HYPO; PETCO2 ~ 20 mmHg). During HYPO, subjects hyperventilated throughout the MOD-HVY-MOD protocol, thereby maintaining PETCO2 at ~20 mmHg for the entire exercise protocol. PETCO2 was displayed (ADInstruments Inc., PowerLab Chart v4.2, Colorado Springs, CO, USA) and continuously monitored by the subjects, with adjustments made to breathing frequency and/or tidal volume as required to maintain PETCO2 at the target level.
Exercise consisted of two moderate-intensity step-transitions in work rate (MOD1, MOD2) that were separated by a bout of heavy-intensity (HVY) exercise. Each step-transition was preceded and followed by 6 min of 20 W baseline exercise and all steps were 6 min in duration except for HVY which lasted 5 min. Changes in WR were made instantaneously and without warning. Subjects performed four repetitions of the exercise protocol in each of the CON and HYPO conditions on separate days.
Materials
Inspired and expired airflow and volumes were measured throughout the exercise protocol by a low dead space (90 mL) bi-directional turbine (Alpha Technologies, VMM-110, Laguna Hills, CA, USA). The volume turbine was calibrated prior to each test using a syringe of known volume (3.0 L; Hans Rudolph, Kansas City, MO, USA). Respired gases were sampled continuously (1 mL/s) at the mouth and analyzed for the fractional concentrations of O2, CO2 and N2 by mass spectrometry (Innovision, AMIS 2000, Lindvedvej, Denmark) following calibration with precision-analyzed gas mixtures. Inspired and expired volumes were time-aligned with changes in gas concentrations by measuring the time delay for a bolus of gas to travel through a capillary line from the turbine transducer and to be detected by the mass spectrometer. The algorithms of Beaver et al. (1981) were used to calculate breath-by breath alveolar gas exchange.
An electrocardiogram (Life Pulse, HME Ltd., Hertfordshire, UK) was used to monitor beat-by-beat heart rate (HR) by a three-lead arrangement. Data were recorded and stored for further analysis on a separate computer (PowerLab Chart v4.2).
Changes in local muscle oxy- (Δ[O2Hb]), deoxy- (Δ[HHb]) and total (Δ[HbTOT]) hemoglobin/myoglobin concentrations were measured continuously by near-infrared spectroscopy (NIRS; Hamamatsu Photonics KK, NIRO 300, Japan). Optodes were housed in an optically dense rubber holder (optode separation, 5 cm) and placed on the vastus lateralis muscle of the right leg, midway between the lateral epicondyle and greater trochanter of the femur. A black vinyl sheet was taped to the skin surface to cover the optode assembly and to minimize the loss of near-infrared transmitted light from the region of interrogation and reduce the intrusion of extraneous light. To further secure the position of the optodes during the exercise protocol, an elastic bandage was wrapped around the leg to prevent any movement of the optode assembly while still permitting freedom of movement for leg cycling. The NIRS-signals were monitored until steady-state baseline levels were established at which time the signals were “zeroed” such that further changes in the signals were relative to this “zero” value (i.e., “delta”). Baseline NIRS-signals were monitored for an additional 5 min while the subjects breathed normally and this was followed by 20 min accommodation period (see above).
A detailed explanation of the principle and theory of this NIRS device is described by Elwell (1995). Briefly, four laser diodes produce different wavelengths (775, 810, 850, and 910 nm) that are pulsed in rapid succession and transmitted through fiber optic bundles to the tissue of interest. The transmitted light returns through a separate fiber optic bundle to a photomultiplier tube where the light intensities are coupled with the relevant specific extinction coefficient and optical path length to give rise to changes in Δ[O2Hb], Δ[HHb] and Δ[HbTOT] relative to resting, pre-accommodation values. As the differential path length in the quadriceps muscle at rest and in exercise is presently unknown, the NIRS-derived measures are reported in arbitrary units (a.u.). Changes in light intensities were monitored and recorded continuously at 2 Hz and the raw attenuation signals were transferred and stored on a computer for later analysis.
Analysis of data
\( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) data for each individual trial were initially filtered to remove any aberrant breaths that lay outside four standard deviations of the local mean as they do not conform to a Gaussian distribution as described by Lamarra et al. (1987). Data were then interpolated into 1-s intervals and time-aligned to correspond to the onset of the MOD1 transition (time = 0). The four repetitions within a condition were ensemble-averaged and further time-averaged into 10 s bins to yield a single response profile for each subject in each condition. The on-transient response for \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was modeled using nonlinear, least squares regression fitting techniques (Origin, OriginLab Corp., Northampton, MA, USA) with a mono-exponential function of the form:
where Y (t) represents \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) as a function of time (t) throughout the exercise transient; Y(BSL) is the baseline \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) during 20 W cycling prior to the step increase in WR; Amp is the amplitude of the increase in \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) above the baseline value; τ is the time constant (i.e., time taken to reach 63% of the steady-state response); and TD is the time delay. The fit for both MOD1 and MOD2 began at the phase 1–phase 2 transition (as previously described by Rossiter et al. (1999) and Gurd et al. (2005)), to the end of the exercise transition (i.e., 360 s).
Beat-by-beat heart rate data were edited and averaged in a fashion similar to that described above for the \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) data. Using the exponential model in Eq. 1, TD was constrained to “zero” and the on-transient HR response was fit from the onset of the exercise transition through to the end of each exercise response.
The NIRS-derived Δ[O2Hb], Δ[HHb] and Δ[HbTOT] data were time-aligned to the onset of MOD1 (time = 0) and ensemble-averaged into 5 s bins to yield a single response for each subject for both CON and HYPO. The time delay for the Δ[HHb] response (i.e., TDΔ[HHb]) at the onset of MOD1 and MOD2 was determined using second-by-second data for each transition and defined as the first increasing point that consistently remained above the nadir of the signal. This was performed on every trial for all subjects, and averaged to yield a single TDΔ[HHb] for CON and HYPO for every subject. To determine the time course for muscle deoxygenation, the Δ[HHb] data were modeled using an exponential function as in Eq. 1 from the TDΔ[HHb] to 90 s of the WR step. Visual inspection of the NIRS-derived Δ[HHb] profile and minimal variation of residuals around the Y axis (Y = 0) suggests this fitting procedure provides a reasonable estimate of the time course of muscle deoxygenation (i.e., τΔ[HHb]). The overall time course for muscle deoxygenation, which reflects the period of time corresponding to the adjustments of both muscle O2 utilization and microvascular blood flow, was calculated as the mean response time (MRT = TDΔ[HHb] + τΔ[HHb]). Analysis of the Δ[O2Hb] and Δ[HbTOT] signals were restricted to the steady-state baseline and end-exercise values since these signals do not exhibit an exponential-like response.
Statistical analysis
The parameter estimates for \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} , \) HR, Δ[O2Hb], Δ[HHb], and Δ[HbTOT] were analyzed using a two-way analysis of variance (ANOVA) for repeated measures with the main effects of condition and moderate level. A significant F ratio was analyzed using Tukey’s post hoc analysis with statistical significance accepted at P < 0.05. All values were expressed as mean ± SD.
Results
Respiratory measures
The gas exchange profile for a representative subject is shown in Fig. 1, with the mean values of several respiratory measures presented in Fig. 2. The hyperventilation maneuver in HYPO resulted in higher (P < 0.05) CO2 output \( (\dot{V}{\text{CO}}_{ 2} ) \) values but similar \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) values throughout the protocol compared to CON. Subjects were able to maintain a lower (P < 0.05) PETCO2 of ~20 mmHg throughout the entire HYPO protocol (as shown in Fig. 2) with ventilation \( (\dot{V}_{\text{E}} ) \) during HYPO being approximately twice as great (P < 0.05) than in CON. This was achieved primarily through a 50–113% increase (P < 0.05) in the frequency of breathing (Freq) during HYPO, as the higher tidal volume (V t) in HYPO (by 6–34%) was not significant between conditions.

The mean respiratory response of a representative subject during the CON (open circles) and HYPO (closed circles) exercise protocol. Dashed lines indicate an event intervention (i.e., start of hyperventilation) or an exercise transition

The mean respiratory response from all subjects at specific events of the experimental protocol for CON (open circles) and HYPO (closed circles). Error bars are ±SD. *Significant difference (P < 0.05) from control, †significant difference (P < 0.05) from pre-accommodation (Pre-acc); ‡Significant difference (P < 0.05) from post-accommodation (Post-acc), #significant difference (P < 0.05) from baseline 1 (BSL1), §significant difference (P < 0.05) from MOD1, $significant difference (P < 0.05) from baseline 2 (BSL2)
\( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics
The moderate-intensity exercise bout in the present study was performed at 79% \( \hat{\theta }_{\text{L}} \) (±5%) (\( \dot{V}{\text{O}}_{{ 2\;{\text{peak}}}} \): 56 ± 3%). The mean normalized \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response profiles for all subjects with model fit and associated residuals during the transition to MOD1 and MOD2 in CON and HYPO are presented in Fig. 3. The parameter estimates for the \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response and the individual phase 2 time constants \( (\tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} ) \) for each condition are presented in Table 1 and Fig. 4, respectively. Baseline \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) for MOD1 was not different between HYPO (0.96 ± 0.08 L/min) and CON (0.97 ± 0.06 L/min), however, \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was elevated (P < 0.05) prior to MOD2 in both conditions and was higher (P < 0.05) in HYPO (1.15 ± 0.04 L/min) than CON (1.10 ± 0.06 L/min). A higher (P < 0.05) end-exercise \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was observed in MOD2 compared to MOD1, with no differences between conditions (Table 1). The \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) amplitude was similar in CON and HYPO between conditions, although lower (P < 0.05) in MOD2 than MOD1. During MOD1 the \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was greater (P < 0.05) in HYPO (45 ± 23 s) than CON (28 ± 17 s), and was reduced (P < 0.05) in MOD2 during HYPO (24 ± 7 s). Of note, all seven subjects presented with slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics during HYPO and all showed faster kinetics after HVY (Fig. 4b) with no overlap in the 95% confidence interval (C95) (Table 1). While \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) tended to be lower in MOD2 of CON (20 ± 8 s), the difference was not significant from CON in MOD1, even though a lower \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was observed in all seven subjects during MOD2. However, given this observation, if only data in CON are considered (using a paired t test) the \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was lower (P = 0.02) in MOD2 than in MOD1.

The mean normalized \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response profile of all subjects with model fit and associated residuals for CON (open circles, solid gray line) and HYPO (closed circles, solid black line). Dashed lines indicate an exercise transition to MOD1 (a) and MOD2 (b)

The \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) time constant \( (\tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} ) \) of each subject (open circles) in MOD1 and MOD2, with mean values (closed squares) for CON (a) and HYPO (b). Error bars are ±SD. *Significant difference (P < 0.05) from control, †significant difference (P < 0.05) from MOD1
Near-infrared spectroscopy (NIRS)
The averaged profile with mean values and parameter estimates for Δ[HHb], Δ[O2Hb] and Δ[HbTOT] are shown in Fig. 5 and Table 2, respectively. No difference was observed in the steady-state Δ[HbTOT] or Δ[HHb] between conditions at any time during the protocol (Fig. 5). Baseline and end-exercise Δ[HbTOT] were higher (P < 0.05) in MOD2 and the Δ[HbTOT] amplitude was smaller (P < 0.05) compared to MOD1 (Table 2). Baseline and end-exercise Δ[O2Hb] also were higher in MOD2 than in MOD1, but both were lower (P < 0.05) in HYPO than in CON throughout the exercise protocol (Fig. 5); the decrease in Δ[O2Hb] during exercise (i.e., Δ[O2Hb] amplitude) was greater (P < 0.05) in MOD2 than in MOD1 (Table 2) in both conditions.

The averaged response and mean values of total- (Δ[HbTOT]; a Oxy- (Δ[O2Hb], b and deoxy- (Δ[HHb], c hemoglobin/myoglobin for all subjects for CON (open circles, solid gray line) and HYPO (closed circles, solid black line). Error bars are ±SD. Dashed lines indicate an event intervention (i.e., start of hyperventilation) or an exercise transition. *Significant difference (P < 0.05) from control, †significant difference (P < 0.05) from pre-accommodation (Pre-acc), ‡significant difference (P < 0.05) from post-accommodation (Post-acc); #significant difference (P < 0.05) from baseline 1 (BSL1), §significant difference (P < 0.05) from MOD1, $significant difference (P < 0.05) from baseline 2 (BSL2)
Parameter estimates for Δ[HHb] and the averaged Δ[HHb] response profile for all subjects are presented in Table 2 and Fig. 6, respectively. No difference in any of the parameter estimates were observed when the fitting window was extended to 360 s compared to 90 s, therefore the values reported here are based on a 90 s fitting window which provides a “tighter” fit at the on-transient of MOD. Baseline and the end of 90 s fit for Δ[HHb] were not different between HYPO and CON throughout the protocol, however, at the end of 90 s Δ[HHb] was higher (P < 0.05) in MOD2 than MOD1 (Table 2). The Δ[HHb] amplitude in MOD1 was greater (P < 0.05) in HYPO (9.2 ± 4.7 a.u.) compared to CON (7.6 ± 4.4 a.u.); after HVY, the Δ[HHb] amplitude was similar in HYPO (10.8 ± 4.6 a.u.) and CON (10.3 ± 4.4 a.u.). The TDΔ[HHb] was shorter (P < 0.05) after HVY, but remained similar between HYPO and CON (Table 2). There were no differences in τΔ[HHb] or Δ[HHb] mean response time (i.e., MRT = TDΔ[HHb] + τΔ[HHb]) between conditions or between MOD steps (Table 2).

The mean Δ[HHb] response profile for all subjects presented as normalized values with model fit to 90 s for CON (open circles, solid gray line) and HYPO (closed circles, solid black line). Dashed lines indicate an exercise transition to MOD1 (a) and MOD2 (b)
Heart rate kinetics
A summary of the parameter estimates for HR is presented in Table 1. Baseline and end-exercise HR were higher (P < 0.05) in MOD2 than in MOD1 in both conditions, although end-exercise HR in HYPO was lower (P < 0.05) compared to CON in both moderate steps. The τHR was not affected by hyperventilation or prior HVY. The amplitude of HR in MOD2 was lower (P < 0.05) than in MOD1, and was lower (P < 0.05) in HYPO for both MOD1 and MOD2.
Discussion
Hyperventilation-induced hypocapnic alkalosis (HYPO) was shown to slow the adjustment of pulmonary \( \dot{V}{\text{O}}_{ 2} \, (\dot{V}{\text{O}}_{{ 2 {\text{p}}}} ) \) kinetics (considered a proxy measure for muscle O2 utilization) during the transition to moderate-intensity (MOD) exercise, such that the time to reach a new steady-state was increased from ~120 s (CON) to ~190 s (HYPO) (Chin et al. 2007). The present study examined the effect of a “priming” bout of heavy-intensity exercise (HVY) in relieving the constraint imposed on \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics during the HYPO maneuver. In agreement with our previous findings (Chin et al. 2007), we observed that during the transition to MOD in the “unprimed” state (i.e., MOD1) (1) \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was greater in HYPO than in CON, reflecting slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in this condition; (2) the time course of muscle deoxygenation (τΔ[HHb], MRTΔ[HHb]) was not different between conditions, and the Δ[HHb] amplitude (and \( \Updelta \left[ {\text{HHb}} \right]/\Updelta \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) ratio) was greater in HYPO, despite slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics, reflecting a greater reliance on fractional O2 extraction, likely a consequence of an attenuated microvascular blood flow response (i.e., greater ratio of muscle O2 utilization-to-blood flow) in this condition. The novel findings in this study were that during the transition to MOD after HVY priming exercise (i.e., MOD2) (1) \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was reduced both in HYPO and in CON such that no difference was observed between conditions after the HVY priming bout, which reflected a speeding of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in both conditions; (2) Δ[HHb] kinetics and Δ[HHb] amplitude were not different from MOD1 despite faster \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in HYPO and CON, reflecting a greater muscle perfusion after HVY and thus less reliance on microvascular O2 extraction. The elevated HR and NIRS-derived Δ[HbTOT] and Δ[O2Hb] prior to and throughout MOD2 also are consistent with greater local muscle (microvascular) perfusion in MOD2 compared to MOD1.
Collectively, these results suggest that the slowed adjustment of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) during the transition to MOD1 observed in HYPO (and possibly in CON) may be related, in part, to a slow or inadequate distribution of blood flow to muscle, especially in the microvascular units supplying the recruited muscle fibers, as improved muscle perfusion after heavy-intensity exercise (as assessed by NIRS-derived Δ[HbTOT] and Δ[O2Hb]) was associated with faster \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in MOD2. A slow microvascular blood flow response relative to the instantaneous increase in muscle ATP requirement that occurs at exercise onset would be expected to increase the muscle O2 utilization-to-microvascular blood flow ratio, requiring a greater O2 extraction to support the provision of O2 required by oxidative ATP synthesis. A reliance on O2 extraction relative to blood flow adjustment would result in a greater lowering of the microvascular PO2, which, according to Fick’s Law of Diffusion, would reduce the pressure gradient between the microvasculature and mitochondria, slow O2 diffusive delivery and constrain the rise in muscle O2 utilization and thus pulmonary O2 uptake \( (\dot{V}{\text{O}}_{{ 2 {\text{p}}}} ). \) This fall in microvascular PO2 would be exacerbated when coupled to an already lower PO2 consequent to a leftward-shift of the oxyhemoglobin dissociation curve. Also, although not measured in this present study, HYPO is associated with a slower activation of the mitochondrial PDH complex which is rate-limiting for delivery of carbohydrate-derived substrate into the mitochondrial TCA cycle and ETC for use in oxidative phosphorylation (LeBlanc et al. 2002). Although a role for PDH activation (or other oxidative rate-limiting enzymes) in constraining the activation of muscle O2 utilization and adjustment of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) is equivocal (Poole et al. 2008), the time course of enzyme activation most likely assumes more importance in those conditions in which PDH activity is attenuated (as in HYPO) as provision of other oxidative substrates (acetyl CoA; reduced coenzymes) becomes inadequate to support the required rate of oxidative phosphorylation. This stenosis is relieved by prior HVY exercise and also can contribute to faster muscle O2 utilization and \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics (Gurd et al. 2006).
HYPO without prior heavy-intensity exercise (MOD1)
The slower adjustment of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) to MOD1 in HYPO in the present study is consistent with our previous finding (Chin et al. 2007) and that of Hayashi et al. (1999), but not with that of Ward et al. (1983) where although the half-time (t 1/2) of the \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response tended to be greater with hyperventilation (39 s compared to 31 s in control), differences were not significant.
A factor contributing to the different findings in the literature may be related to the duration of the hyperventilation maneuver during the pre-exercise accommodation. Removal of CO2 from body stores at rest (Brandi and Clode 1969) and systemic circulatory changes (Richardson et al. 1972) with hyperventilation occur in a time-dependent manner. In the present and in a previous study (Chin et al. 2007) subjects hyperventilated for ~26 min prior to the initial transition to MOD1, and continued to hyperventilate for the remainder of the exercise protocol, thereby maintaining PETCO2 at ~20 mmHg, and ensuring equilibration of CO2 between the various CO2 storage compartments within the body. Indeed the lower arterial plasma PCO2 and [H+] (Chin et al. 2007) and intramuscular [H+] (Forbes et al. 2007) measured in previous studies that used a similar sustained hyperventilation protocol (with PETCO2 maintained at ~20 mmHg), is evidence in support of the effectiveness of the prolonged hyperventilation maneuver in achieving equilibration of CO2 (and its effect on acid-base balance) among the tissue compartments.
In the study of Hayashi et al. (1999), hyperventilation was started only 2 min before the exercise transition and PETCO2 was allowed to increase during the exercise transition (by ~10 mmHg), whereas in the study of Ward et al. (1983), hyperventilation began 9 min before and was stopped ~20 s before the start of the exercise. As it takes ~15 min for 90% of the body’s CO2 stores to be removed (Brandi and Clode 1969) and at least 7 min for increases in cardiac output and heart rate to return to control levels (Richardson et al. 1972) after the start of the hyperventilation maneuver, comparisons with these studies should be made with caution.
Hayashi et al. (1999) attributed the slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics that was observed in HYPO to impaired O2 off-loading from hemoglobin, a consequence of a leftward-shift of the oxy-hemoglobin dissociation curve. Indeed in this and in a previous study (Chin et al. 2007), we observed that immediately after the onset of hyperventilation, all subjects demonstrated an increase in the oxy-hemoglobin (Δ[O2Hb]) signal and decrease in the deoxy-hemoglobin (Δ[HHb]) signal (without a change in Δ[HbTOT]) (Fig. 5), consistent with an increase in hemoglobin affinity for O2 assuming little or no change in microvascular PO2. However, this response was only transient, lasting ~8 min, such that for the remainder of the 20 min accommodation period there was no difference in the Δ[O2Hb] and Δ[HHb] signals between HYPO and CON, suggesting that factors in addition to an increased affinity of hemoglobin for O2 are responsible for slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in the HYPO condition.
During baseline cycling (at 20 W) prior to MOD1, baseline Δ[O2Hb] was lower and Δ[HHb] tended to be higher in HYPO than CON, a trend that continued throughout MOD1. As \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) was not different between conditions at baseline or end-exercise, these data suggest that local muscle perfusion (and O2 delivery) were lower in HYPO, thereby requiring a greater O2 extraction to support muscle mitochondrial O2 utilization. Previous studies have reported a lower skeletal muscle blood flow in animals at rest during hypocapnic alkalosis (Brice and Welch 1985; Gustafsson et al. 1993; Karlsson et al. 1994) which is accompanied by unchanged perfusion pressure (Gustafsson et al. 1993) and increased vascular resistance (Kontos et al. 1972). In humans at rest, Kontos et al. (1972) reported decreased forearm blood flow and increased forearm vascular resistance following 6 min voluntary hyperventilation, while Straub and Bühlmann (1970) reported a decrease in blood volume following 20 min of hyperventilation. More recently, Chin et al. (2007) using NIRS, reported a lower Δ[HbTOT] (and presumably muscle blood volume) during moderate-intensity exercise in HYPO compared to CON. This may reflect lower O2 diffusive conductance (DO2) and impaired O2 flux between the red blood cell (RBC) and muscle mitochondrial cytochrome c oxidase in HYPO, as lower amounts of hemoglobin would reflect increased RBC spacing in capillaries and decreased functional capillary surface area for O2 diffusion.
In the present study, Δ[HbTOT] was not different between conditions, although baseline, end-exercise and amplitude (Table 2) tended to be lower in HYPO. Also, the greater Δ[HHb] amplitude in HYPO and similar time course for the Δ[HHb] increase (τΔ[HHb]; MRT Δ[HHb]) between conditions, despite similar increase in \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) amplitude but slower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in HYPO compared to CON (reflecting the kinetics of muscle O2 utilization), suggests that microvascular blood flow was adapting at a slower rate in HYPO requiring a faster and greater rate of O2 extraction to meet the muscle O2 requirements.
HYPO with prior heavy-intensity exercise (MOD2)
A novel finding in the present study was that prior heavy-intensity exercise (HVY) resulted in a speeding of the slowed \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in HYPO (i.e., \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) reduced by ~20 s), and in CON (i.e., \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) reduced by ~8 s) along with a greater Δ[HHb] amplitude relative to \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) amplitude \( \left( {\Updelta {\text{HHb}}/\Updelta \dot{V}{\text{O}}_{{ 2 {\text{p}}}} } \right) \) following heavy-intensity exercise, which likely is related to the lower \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) amplitude and higher end-exercise \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) observed in MOD2 compared to MOD1. However, in several studies where HVY priming exercise was used prior to the start of a bout of MOD, a measurable speeding of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics was not observed (Burnley et al. 2000; DeLorey et al. 2004; Gerbino et al. 1996; Scheuermann et al. 2002). In these studies the \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) reported in the unprimed, control state was relatively short (e.g., 15–25 s; Burnley et al. 2000; DeLorey et al. 2004; Scheuermann et al. 2002). However, as demonstrated recently, the reduction in \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) after prior HVY priming exercise was related directly to how “slow” the \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response was to the “unprimed” state (Gurd et al. 2005, 2006), and thus failure to observe a measurable reduction in \( \tau \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) in these studies was not unexpected.
A prior bout of HVY was shown previously to increase heart rate (DeLorey et al. 2004; Gurd et al. 2005; Scheuermann et al. 2002) and cardiac output (Faisal et al. 2009); to increase muscle perfusion, as shown by elevated bulk muscle blood flow (determined using Doppler ultrasonography) (DeLorey et al. 2007; Endo et al. 2005; Fukuba et al. 2004; Hughson et al. 2003; MacDonald et al. 2001; Paterson et al. 2005) and elevated local muscle oxy- and total hemoglobin concentrations (measured using NIRS) (DeLorey et al. 2004, 2007; Gurd et al. 2006; Gurd et al. 2005; Jones et al. 2006); and to increase mitochondrial PDH activity (Gurd et al. 2006). In the present study, the speeding of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in MOD2 in HYPO (and CON), without a change in deoxygenation kinetics, along with a greater Δ[HbTOT] and Δ[O2Hb], suggest that greater muscle perfusion and distribution prior to and during MOD2 resulted in a higher muscle blood flow-to-O2 utilization ratio during the exercise transition which would maintain a higher microvascular PO2 and greater diffusive delivery of O2 into the muscle. Also, the higher Δ[HbTOT] during MOD2 might reflect a higher microvascular hematocrit and reduced red blood cell spacing which would contribute to a greater functional capillary surface area, which contributes to an improved muscle O2 diffusive conductance and promotes greater O2 diffusion into the muscle (for a given ΔPO2).
Although the approximate doubling of ventilation during HYPO would be expected to be associated with higher \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) [as demonstrated in our previous study (Chin et al. 2007)], there was no observable difference of end-exercise \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) between HYPO and CON in the present study. Similarly, Hayashi et al. (1999) also showed no increase in \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) when CON was compared to a hyperventilation-induced hypocapnic alkalosis condition. Perhaps the lower work rate within moderate-intensity exercise employed by this study and Hayashi et al. (1999) (~80% \( \hat{\theta }_{\text{L}} \) vs. 90% \( \hat{\theta }_{\text{L}} \) in the study by Chin et al. (2007)) was not enough to result in significant increases in the cost of breathing and measured \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \).
HYPO and metabolic activation
A focus of this present study was to extend the findings of Chin et al. (2007) showing that slowed \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics in HYPO were attributed to a limitation imposed by microvascular O2 delivery. However, activation of the mitochondrial PDH enzyme complex has been discussed as a limitation to the provision of acetyl CoA and reducing equivalents to the TCA cycle and ETC, respectively, at exercise onset (i.e., sometimes referred to as a “metabolic inertia”), thereby delaying the full activation of mitochondrial oxidative phosphorylation (Grassi 2003; Spriet and Heigenhauser 2002; Tschakovsky and Hughson 1999). Of importance to the present study, LeBlanc et al. (2002) observed that hyperventilation-induced respiratory alkalosis was associated with a delayed activation of PDH during the first minute of exercise at ~55% \( \dot{V}{\text{O}}_{ 2} \) max. Given that the hyperventilation protocol used by LeBlanc et al. (2002) was similar to that used in the present study [and that of Chin et al. (2007)], a similar attenuated activation of PDH would be expected in the present study, which could contribute to the slowed MOD1 \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics seen during HYPO in this study.
Activation of PDH alone by dichloroacetate infusion has not contributed to measurably faster muscle O2 utilization (Grassi et al. 2002) or pulmonary \( \dot{V}{\text{O}}_{ 2} \) kinetics (Koppo et al. 2004) during MOD. However, Gurd et al. (2006) suggested that elevated PDH activity (and/or other mitochondrial or cytosolic rate-limiting enzymes) combined with greater muscle perfusion may contribute to the faster \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics through enhanced delivery of all substrates needed in oxidative phosphorylation. This would assume even greater importance when one or more substrates are limiting (as might occur in MOD1 of HYPO). Hence, the observed speeding of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) kinetics during MOD2 in the present study also could be related to an enhanced provision of acetyl CoA, reducing equivalents (NADH, FADH2), O2, ADP, and Pi to the mitochondria, through the combined effects of increased muscle perfusion and elevated or accelerated activation of PDH (and other mitochondrial rate-limiting enzymes) consequent to the prior bout of HVY. Prior exercise also was shown to speed the fall in intracellular PO2 in isolated Xenopus single muscle fibers (Hogan 2001) and in microvasculature PO2 of female rat spinotrapezius muscle (Behnke et al. 2002), both responses consistent with a higher PDH activity and speeding of muscle O2 utilization subsequent to the “priming” bout of exercise. Whether this mechanism contributes to the faster adjustment of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) in MOD2 in the present study in HYPO remains speculative.
Summary and conclusion
This study demonstrated that acute hypocapnic alkalosis (HYPO) induced by voluntary hyperventilation slowed the adjustment of \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) during the transition to moderate-intensity exercise, in part, as a consequence of an attenuated muscle perfusion-to-muscle O2 utilization ratio (with subsequent greater O2 extraction, thus lower microvascular PO2). However, a priming bout of heavy-intensity exercise which resulted in an elevated Δ[HbTOT], Δ[O2Hb] and HR prior to and throughout a subsequent bout of moderate-intensity exercise (MOD2) was associated with a speeding of the \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) response, with no change in Δ[HHb] kinetics, despite continued HYPO. These findings suggest that the slowed \( \dot{V}{\text{O}}_{{ 2 {\text{p}}}} \) in HYPO may be related, in part, to an attenuated muscle blood flow response and reduced local muscle blood flow-to-O2 utilization which was relieved by prior heavy-intensity exercise. To date, however, the role of HYPO on conduit artery or microvascular blood flow kinetics has not been established.
References
Beaver WL, Lamarra N, Wasserman K (1981) Breath-by-breath measurement of true alveolar gas exchange. J Appl Physiol 51:1662–1675
Beaver WL, Wasserman K, Whipp BJ (1986) A new method for detecting anaerobic threshold by gas exchange. J Appl Physiol 60:2020–2027
Behnke BJ, Kindig CA, Musch TI, Sexton WL, Poole DC (2002) Effects of prior contractions on muscle microvascular oxygen pressure at onset of subsequent contractions. J Physiol 539:927–934
Brandi G, Clode M (1969) CO2 washout during hyperventilation in man. Respir Physiol 7:163–172
Brice AG, Welch HG (1985) Effect of respiratory alkalosis on skeletal muscle metabolism in the dog. J Appl Physiol 58:658–664
Burnley M, Jones AM, Carter H, Doust JH (2000) Effects of prior heavy exercise on phase II pulmonary oxygen uptake kinetics during heavy exercise. J Appl Physiol 89:1387–1396
Chin LM, Leigh RJ, Heigenhauser GJ, Rossiter HB, Paterson DH, Kowalchuk JM (2007) Hyperventilation-induced hypocapnic alkalosis slows the adaptation of pulmonary O2 uptake during the transition to moderate-intensity exercise. J Physiol 583:351–364
DeLorey DS, Kowalchuk JM, Paterson DH (2004) Effects of prior heavy-intensity exercise on pulmonary O2 uptake and muscle deoxygenation kinetics in young and older adult humans. J Appl Physiol 97:998–1005
DeLorey DS, Kowalchuk JM, Heenan AP, duManoir GR, Paterson DH (2007) Prior exercise speeds pulmonary O2 uptake kinetics by increases in both local muscle O2 availability and O2 utilization. J Appl Physiol 103:771–778
Elwell CE (1995) A practical user’s guide to near infrared spectroscopy. Hamamatsu Photonics KK, London
Endo M, Okada Y, Rossiter HB, Ooue A, Miura A, Koga S, Fukuba Y (2005) Kinetics of pulmonary VO2 and femoral artery blood flow and their relationship during repeated bouts of heavy exercise. Eur J Appl Physiol 95:418–430
Faisal A, Beavers KR, Robertson AD, Hughson RL (2009) Prior moderate and heavy exercise accelerate oxygen uptake and cardiac output kinetics in endurance athletes. J Appl Physiol 106:1553–1563
Forbes SC, Kowalchuk JM, Thompson RT, Marsh GD (2007) Effects of hyperventilation on phosphocreatine kinetics and muscle deoxygenation during moderate-intensity plantar flexion exercise. J Appl Physiol 102:1565–1573
Fukuba Y, Ohe Y, Miura A, Kitano A, Endo M, Sato H, Miyachi M, Koga S, Fukuda O (2004) Dissociation between the time courses of femoral artery blood flow and pulmonary VO2 during repeated bouts of heavy knee extension exercise in humans. Exp Physiol 89:243–253
Gerbino A, Ward SA, Whipp BJ (1996) Effects of prior exercise on pulmonary gas-exchange kinetics during high-intensity exercise in humans. J Appl Physiol 80:99–107
Grassi B (2003) Oxygen uptake kinetics: old and recent lessons from experiments on isolated muscle in situ. Eur J Appl Physiol 90:242–249
Grassi B, Poole DC, Richardson RS, Knight DR, Erickson BK, Wagner PD (1996) Muscle O2 uptake kinetics in humans: implications for metabolic control. J Appl Physiol 80:988–998
Grassi B, Hogan MC, Greenhaff PL, Hamann JJ, Kelley KM, Aschenbach WG, Constantin-Teodosiu D, Gladden LB (2002) Oxygen uptake on-kinetics in dog gastrocnemius in situ following activation of pyruvate dehydrogenase by dichloroacetate. J Physiol 538:195–207
Gurd BJ, Scheuermann BW, Paterson DH, Kowalchuk JM (2005) Prior heavy-intensity exercise speeds VO2 kinetics during moderate-intensity exercise in young adults. J Appl Physiol 98:1371–1378
Gurd B, Peters SJ, Heigenhauser GJ, LeBlanc PJ, Doherty TJ, Paterson DH, Kowalchuk JM (2006) Prior heavy exercise elevates pyruvate dehydrogenase activity and speeds O2 uptake kinetics during subsequent moderate-intensity exercise in healthy young adults. J Physiol 577:985–996
Gustafsson U, Sjoberg F, Lewis DH, Thorborg P (1993) The effect of hypocapnia on skeletal muscle microcirculatory blood flow, oxygenation and pH. Int J Microcirc Clin Exp 12:131–141
Hayashi N, Ishihara M, Tanaka A, Yoshida T (1999) Impeding O2 unloading in muscle delays oxygen uptake response to exercise onset in humans. Am J Physiol 277:R1274–R1281
Hogan MC (2001) Fall in intracellular PO2 at the onset of contractions in Xenopus single skeletal muscle fibers. J Appl Physiol 90:1871–1876
Hughson RL, Schijvens H, Burrows S, Devitt D, Betik AC, Hopman MTE (2003) Blood flow and metabolic control at the onset of heavy exercise. Int J Sport Health Sci 1:9–18
Jones AM, Berger NJ, Wilkerson DP, Roberts CL (2006) Effects of “priming” exercise on pulmonary O2 uptake and muscle deoxygenation kinetics during heavy-intensity cycle exercise in the supine and upright positions. J Appl Physiol 101:1432–1441
Karlsson T, Stjernstrom EL, Stjernstrom H, Norlen K, Wiklund L (1994) Central and regional blood flow during hyperventilation. An experimental study in the pig. Acta Anaesthesiol Scand 38:180–186
Kontos HA, Richardson DW, Raper AJ, Zubair-ul-Hassan, Patterson JL Jr (1972) Mechanisms of action of hypocapnic alkalosis on limb blood vessels in man and dog. Am J Physiol 223:1296–1307
Koppo K, Wilkerson DP, Bouckaert J, Wilmshurst S, Campbell IT, Jones AM (2004) Influence of DCA on pulmonary VO2 kinetics during moderate-intensity cycle exercise. Med Sci Sports Exerc 36:1159–1164
Krustrup P, Jones AM, Wilkerson DP, Calbet JA, Bangsbo J (2009) Muscular and pulmonary O2 uptake kinetics during moderate and high-intensity sub-maximal knee-extensor exercise in humans. J Physiol 587:1843–1856
Lamarra N, Whipp BJ, Ward SA, Wasserman K (1987) Effect of interbreath fluctuations on characterizing exercise gas exchange kinetics. J Appl Physiol 62:2003–2012
LeBlanc PJ, Parolin ML, Jones NL, Heigenhauser GJ (2002) Effects of respiratory alkalosis on human skeletal muscle metabolism at the onset of submaximal exercise. J Physiol 544:303–313
MacDonald MJ, Naylor HL, Tschakovsky ME, Hughson RL (2001) Peripheral circulatory factors limit rate of increase in muscle O2 uptake at onset of heavy exercise. J Appl Physiol 90:83–89
Paterson ND, Kowalchuk JM, Paterson DH (2005) Effects of prior heavy-intensity exercise during single-leg knee extension on VO2 kinetics and limb blood flow. J Appl Physiol 99:1462–1470
Poole DC, Barstow TJ, McDonough P, Jones AM (2008) Control of oxygen uptake during exercise. Med Sci Sports Exerc 40:462–474
Richardson DW, Kontos HA, Raper AJ, Patterson JL Jr (1972) Systemic circulatory responses to hypocapnia in man. Am J Physiol 223:1308–1312
Rossiter HB, Ward SA, Doyle VL, Howe FA, Griffiths JR, Whipp BJ (1999) Inferences from pulmonary O2 uptake with respect to intramuscular [phosphocreatine] kinetics during moderate exercise in humans. J Physiol 518:921–932
Scheuermann BW, Bell C, Paterson DH, Barstow TJ, Kowalchuk JM (2002) Oxygen uptake kinetics for moderate exercise are speeded in older humans by prior heavy exercise. J Appl Physiol 92:609–616
Spriet LL, Heigenhauser GJ (2002) Regulation of pyruvate dehydrogenase (PDH) activity in human skeletal muscle during exercise. Exerc Sport Sci Rev 30:91–95
Straub PW, Bühlmann AA (1970) Reduction of blood volume by voluntary hyperventilation. J Appl Physiol 29:816–817
Tschakovsky ME, Hughson RL (1999) Interaction of factors determining oxygen uptake at the onset of exercise. J Appl Physiol 86:1101–1113
Ward SA, Whipp BJ, Koyal S, Wasserman K (1983) Influence of body CO2 stores on ventilatory dynamics during exercise. J Appl Physiol 55:742–749
Whipp BJ, Ward SA, Wasserman K (1986) Respiratory markers of the anaerobic threshold. Adv Cardiol 35:47–64
Acknowledgments
This study was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) grants. Additional support was provided by Western’s Graduate Thesis Research Fund. The authors would like to thank Tim J.H. Chaplin, Natasha R. Rowling and Ryan J. Leigh for assistance with data collection, Brad Hansen for his technical support, and all the subjects who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Susan Ward.
Rights and permissions
About this article
Cite this article
Chin, L.M.K., Heigenhauser, G.J.F., Paterson, D.H. et al. Effect of hyperventilation and prior heavy exercise on O2 uptake and muscle deoxygenation kinetics during transitions to moderate exercise. Eur J Appl Physiol 108, 913–925 (2010). https://doi.org/10.1007/s00421-009-1293-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-009-1293-1