
Abstract
Physical activity relies on muscular force. In adult skeletal muscle, force results from the contraction of postmitotic, multinucleated myofibres of different contractile and metabolic properties. Myofibres can adapt to (patho-)physiological conditions of altered functional demand by radial growth, longitudinal growth, and regulation of fibre type functional gene modules. The adaptation’s specificity depends on the distinct molecular and cellular events triggered by unique combinations of conditional cues. In order to derive effective and tailored exercise prescriptions, it must be determined (1) which mechano-biological condition leads to what molecular/cellular response, and (2) how this molecular/cellular response relates to the structural, contractile, and metabolic adaptation. It follows that a thorough mechano-biological description of the loading condition is imperative. Unfortunately, the definition of (resistance) exercise conditions in the past and present literature is insufficient. It is classically limited to load magnitude, number of repetitions and sets, rest in-between sets, number of interventions/week, and training period. In this review, we show why the current description is insufficient, and identify new determinants of quantitative and/or qualitative effects on skeletal muscle with respect to resistance exercise in healthy, adult humans. These new mandatory determinants comprise the fractional and temporal distribution of the contraction modes per repetition, duration of one repetition, rest in-between repetitions, time under tension, muscular failure, range of motion, recovery time, and anatomical definition. We strongly recommend to standardise the design and description of all future resistance exercise investigations by using the herein proposed set of 13 mechano-biological determinants (classical and new ones).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
From stimulus to adaptational effect
Physiological conditions such as resistance exercise perturb the skeletal muscle’s tensional integrity (Ingber 2003a, b). These perturbations are mechano-chemically transduced into a molecular and cellular response within and between myofibres and satellite cells (i.e. muscle stem cells) (Tidball 2005). The mechano-chemical transduction is based on the genetic background, age, gender, and several other factors. Finally, this molecular and cellular response leads to specific structural adaptations that result in task-specific functional enhancement (i.e. the adaptational effect) (Fig. 1). However, a causal connection only exists between the molecular/cellular response (i.e. signal transduction) and the adaptation (Fig. 1). “More strength”, i.e. the adaptational effect, is not necessarily the result of more muscle mass, since several distinct adaptations can lead to the same effect (at least in the short term). Conversely, training in the “6–12-repetition-maximum zone with multiple sets for 2–3 days week−1”, the so-called “hypertrophy training” (Kraemer and Ratamess 2004), doesn’t necessarily mean that muscle hypertrophy (i.e. an increase in muscle mass due to the increase in the size, as opposed to the number, of preexisting skeletal muscle fibres) will result. This is due to the fact that the ability to exercise is distinct from the ability to adapt. It has recently been shown for a large cohort of men and women that significant variability in muscle size and strength gain exists after unilateral resistance exercise of the elbow flexors (Hubal et al. 2005). While some subjects showed little to no gain, other showed profound changes. Also, sex differences were apparent. Men had a slight advantage in relative size gains compared to women, whereas women outpaced men considerably in relative gains in strength. Age differences exhibit a profound impact on the molecular response to resistance exercise, too (Hameed et al. 2003). Following a single bout of high load knee extensor resistance exercise, mechanogrowth factor (MGF) response was attenuated in older subjects. This is indicative of age-related desensitivity to mechanical loading. Also, various human polymorphisms at genetic loci have a quantitative effect on muscle phenotype (Thompson et al. 2004). Such polymorphisms, also known as muscle quantitative trait loci, drive the genetic variation that underlies muscle size and strength.

Simplified model for the transduction of exercise-related skeletal muscle perturbations into structural adaptations with associated adaptational effect. An exercise stimulus with defined mechano-biological characteristics (described in this paper) (a) is mechano-chemically transduced (c) into a quantitative and/or qualitative adaptation (d) of the muscle phenotype, based on the respective response matrix (b). The adaptation (d) is associated with the corresponding adaptational effect (e). A causal connection exists between (c) and (d) (red shading)
Therefore, several prerequisites for the identification of effective and specific exercise perturbations with desired functional enhancement are to be met. First, causal relationships between changes at the molecular and cellular level and the resulting adaptation need to be identified. These relationships need to be established on the basis of several factors such as genetics, age, sex, etc. Further, it must be ascertained whether the adaptation leads to functional enhancement. Finally, the various causal connections between signal transduction and adaptation can be interrogated by multiple exercise perturbations in order to identify the most effective and specific exercise perturbations. However, for the identification of effective and specific exercise perturbations it is imperative to unequivocally define and describe the exercise stimulus in mechano-biological relevant terms. These mechano-biological terms should be directly associated with the molecular response. Unfortunately, in current investigations into the molecular response to exercise, perturbation design and definition remain largely undefined or ignored. This review aims at identifying mechano-biological determinants of exercise conditions that have quantitative and/or qualitative effects on skeletal muscle phenotype. These mechano-biological determinants are proposed for standardised description of resistance exercise stimuli in both academic and recreational settings.
Fundamental mechanical stimuli decoded by skeletal muscle
Basically, muscles can adopt three strategies of quantitative or qualitative effect on muscular phenotype to adjust for altered functional demands (Goldspink 1985): (1) positive or negative longitudinal growth; (2) positive or negative radial growth and (3) contractile [myosin heavy chain (MyHC)] and metabolic tuning. These adaptational strategies can be adopted concurrently or separately, depending on the specificity of the (patho-)physiological condition. Generally, exercise-induced physiological conditions can be viewed as the perturbations of the muscle cells’ tensional integrity. Perturbation of tensional integrity occurs by increasing or decreasing myocellular active and/or passive tension, as well as energy production or absorption. Additionally, these tensional and energetic alterations can be sustained for different duration. Thus, every exercise condition is coded by a specific combination of changes in constant or intermittent active and/or passive tension of different duration. These temporal changes in active and/or passive tension, together with the inferred structural insults, are then decoded at the molecular level and transduced into an appropriate adaptational response. In the following sections, we provide a detailed description on how the three fundamental adaptations are regulated at the molecular and cellular levels. From this, we derive the relevant mechano-biological determinants for resistance exercise-induced muscular conditioning.
Molecular and cellular determinants of longitudinal muscle growth
Mechanical measurements of rabbit muscle strips before and after skinning indicate that total passive tension increases with increasing sarcomere length (second-order polynomial) (Prado et al. 2005), i.e. with increased strain. Total passive tension is believed to develop due to the lengthening of extramyofibrillar elements (especially the collagen content in the extracellular matrix) as well as to the lengthening of titin. Titin is a giant (~ 3–3.7 MDa) sarcomeric protein that contains a series of spring elements within its I-band region, which contribute to the elastic properties of myofibrils (Prado et al. 2005). External or internal forces applied to the myofibril lengthen or shorten the myofibril to above or below the slack length, respectively. The lengthening or shortening of the myofibril creates a titin force, which is directed to restore resting length (Miller et al. 2003). Myofibrils can be lengthened actively or passively. This means that myofibrils can lengthen while contracting (“lengthening contraction”, “eccentric contraction”, “active stretch”) or without concurrent contraction. In contrast, myofibrils can only actively shorten, i.e. while contracting (“shortening contraction”, “concentric contraction”). Consequently, passive tension in myofibrils can develop with or without concurrent lengthening contraction or with shortening contraction.
Molecular sensing of myofibrillar strain
In line with titin’s structural and elastic properties, mounting evidence indicates that myofibrillar strain mediates signalling pathways that involve titin’s Z-line region (Miller et al. 2003). Signalling pathways comprise (1) the titin-muscle LIM protein (MLP) pathway, (2) the N2A-muscle ankyrin repeat protein (MARP) pathway, and (3) the titin-muscle RING finger protein (MuRF) pathway:
-
(1)
is part of a stretch-dependent myocardial signalling pathway whose impairment contributes to the pathogenesis of a subset of dilated cardiomypathies in humans (Knoll et al. 2002) and is also induced by skeletal muscle injury due to eccentric exercise (Barash et al. 2004, 2005; Chen et al. 2002; Hentzen et al. 2006).
-
(2)
involves 3 homologous MARPs: CARP/MARP1, Ankrd2/Arpp/MARP2, and DARP/MARP3. MARPs show cytokine-like induction following cardiac injury, muscle denervation, and eccentric exercise in vivo (CARP/MARP1) (Aihara et al. 2000; Barash et al. 2004; Baumeister et al. 1997; Kuo et al. 1999); strain in culture, immobilisation at stretched muscle length, and eccentric exercise in vivo (Ankrd2/Arpp/MARP2) (Barash et al. 2004, 2005; Kemp et al. 2000; McKoy et al. 2005); or during recovery after metabolic challenge (DARP/MARP3) (Ikeda et al. 2003), respectively.
-
(3)
is likely to be involved in the regulation of ubiquitin-proteasome-mediated myofibrillar protein degradation (see “Atrophy signalling”). MuRF1 was found to bind the C-terminal immunoglobulin domains of titin (Centner et al. 2001), and in the nucleus MuRF1 can bind the transcription factor, glucocorticoid modulatory element binding protein-1 (McElhinny et al. 2002). Furthermore, mechanical tension and the titin catalytic domain have been shown to regulate the nuclear localisation of MuRF2 and serum response factor (SRF) (Lange et al. 2005).
Recently, it has also been demonstrated in an in vivo rat model that CARP and MLP are sensitive to both muscle tissue stress and contraction mode, while Ankrd2/Arpp is sensitive only to contraction mode (Barash et al. 2005). This raises the possibility that strain can be sensed independently of stress.
Further evidence for the molecular transduction of passive tension comes from the ~ 1.5- and ~ 2-fold increase in the serine/threonine kinase Akt [protein kinase B (PKB)] activity following 5 and 10–20 min of passive stretch, respectively, of the fast-twitch rat extensor digitorum longus muscle but not in the slow-twitch soleus muscle (Sakamoto et al. 2003). Once activated, Akt phosphorylates an array of substrates, including proteins that mediate protein synthesis, gene transcription, cell proliferation, and survival (Vivanco and Sawyers 2002). In mammals, there are three forms of Akt, (Akt1-3), encoded by distinct genes (Vivanco and Sawyers 2002). Expression of a constitutively active form of Akt1 in skeletal muscle cells, either in vitro (Takahashi et al. 2002) or in mice and rats (Bodine et al. 2001b; Lai et al. 2004; Pallafacchina et al. 2002), causes hypertrophy. Conversely, Akt1 inhibits atrophy in vitro and in mice (Sandri et al. 2004; Stitt et al. 2004). Based on their finding that passive stretch has no effect on Akt activity in rat slow-twitch soleus muscle (in contrast to rat extensor digitorum longus muscle), the authors suggested that susceptibility to mechanical stretch is fibre type-specific (Sakamoto et al. 2003). This notion is also supported by the recent finding that the relative importance of titin and the extracellular matrix for total passive tension can vary between different adult rabbit skeletal muscles (Prado et al. 2005). Slow-twitch rabbit muscles exhibit low titin-based passive tension but this tension is highly variable in fast-twitch muscles. Furthermore, titin-based passive tension, but not extramyofibrillar passive tension correlates with the muscle type (Prado et al. 2005).
Cellular sensing of mechanical stretch
Satellite cells are muscle precursor cells that lie between the basal lamina and sarcolemma of skeletal muscle fibres (Mauro 1961). In normal adult muscle, satellite cells are mitotically and metabolically quiescent (Schultz et al. 1978). With appropriate environmental signals, satellite cells enter into the cell cycle, i.e. are “activated”, to provide the precursors needed for new muscle formation in growth and repair (Charge and Rudnicki 2004; Hill et al. 2003; McKinnell et al. 2005). Results from in vitro stretch assays demonstrate that mechanical stretch can result in satellite cell activation (Anderson 2000; Anderson and Pilipowicz 2002; Tatsumi et al. 2001; Wozniak et al. 2003). This mechanical stretch induces hepatocyte growth factor (HGF) release from its tethering in the extracellular matrix in a nitric oxide-dependent manner (Tatsumi and Allen 2004; Tatsumi et al. 2002). Once released, HGF binds to the c-met receptor which is located on the plasma membrane of the satellite cells. This interaction initiates a cascade of signalling events that lead to DNA synthesis, and, thus, to satellite cell proliferation.
Structural adaptation to strain perturbation
It has long been known that muscles adapt to a new functional length by adding or removing sarcomeres in series at the ends of the existing myofibrils (Dix and Eisenberg 1990; Griffin et al. 1971; Tabary et al. 1972; Williams and Goldspink 1971). Immobilisation at long muscle length results in an increase in the number of sarcomeres in series. Conversely, immobilisation at short muscle length leads to a decrease in the number of sarcomeres in series. Furthermore, remodelling of the connective tissue following immobilisation has been demonstrated multiple times in mice, rats, rabbits, and cats (Goldspink 1985; Tabary et al. 1976; Tardieu et al. 1977, 1982; Williams and Goldspink 1984). However, both the occurrence and the extent of remodelling seem to depend on the connective tissue type (series elastic element and/or parallel elastic element), species, age, muscle length during immobilisation, and time period of immobilisation.
In exercise physiology, serial sarcomere number modulation has been a neglected topic so far (Morgan and Proske 2004). Only recently has serial sarcomere number modulation been investigated in the context of exercise. Direct evidence for exercise-induced modulation of serial sarcomere number has come from treadmill-trained rat vastus intermedius muscles, the postural knee extensors (Lynn and Morgan 1994; Lynn et al. 1998). Rats were trained by running on a climbing or descending treadmill for approximately 10 min day−1 for 5 days. The latter had previously been shown to cause muscle damage. Subsequently, serial sarcomere analysis for single fibres was performed by laser diffraction. As a result, the descending-trained rats had the largest sarcomere count, the climbing-trained rats had the smallest count, and sedentary rats had intermediate counts, although closer to the climbing group. In another series of experiments, rat vastus intermedius muscles were tested mechanically while still in situ, i.e. attached to the bones, but with all other muscles about the knee joint removed. As a result, in descending-trained rats, the knee angle for optimum torque generation corresponded to longer muscle lengths than in climbing-trained rats. It follows from these results that eccentric exercise (lengthening contractions) leads to accretion of serial sarcomeres. Conversely, exercise comprising only shortening contractions leads to a decrease in the number of serial sarcomeres. Due to the finding that 38% of the difference in sarcomere numbers between decline- and incline-trained rats does not appear as a difference in optimum angle, the authors suggest that it has been taken up by shortened tendons (Lynn et al. 1998).
Recently, the contraction type-dependent differential serial sarcomere number adaptation has been confirmed by measuring vastus intermedius and vastus lateralis muscle fibre dynamics of up- and downhill-running rats in vivo (Butterfield et al. 2005). It was shown in that study that vastus intermedius and vastus lateralis muscles of uphill-walking rats undergo repeated concentric contractions, and therefore they suffer no contraction-induced injury. Conversely, the vastus intermedius and vastus lateralis muscles during downhill walking undergo repeated eccentric contractions (Butterfield et al. 2005). Accordingly, short muscle lengths for uphill concentric-biased contractions result in a loss of serial sarcomeres, while long muscle lengths for downhill eccentric-biased contractions result in a gain of serial sarcomeres (Butterfield et al. 2005).
In humans, the optimum angle for torque generation can be measured reliably, e.g. by isokinetic dynamometry. By that means, an angle-torque curve is measured during maximum voluntary contraction with constant velocity shortening. As determined by this measure, a series of eccentric contractions (“hamstring lowers”) of human hamstring muscles produced a significant shift of approximately 7° in optimum knee angle for torque generation to longer muscle lengths (Brockett et al. 2001). The shift in optimum knee angle for torque generation was parallelled by delayed-onset muscle soreness (DOMS) in the hamstrings. The shift occurred immediately after exercise and persisted 8 days postexercise, consistent with a training effect. The mechanism by which eccentric exercise produces muscle damage, DOMS, and increased optimum length for torque generation has been postulated in the “popping sarcomere hypothesis” (Morgan 1990). The popping sarcomere hypothesis states that stretch-induced muscle damage results from nonuniform lengthening of sarcomeres, when active muscle is stretched beyond optimum length. If sarcomeres are beyond optimum length, then the longest sarcomeres will be the weakest and, so, will be stretched more rapidly than the others. Thus, they will become weaker, until rising passive tension compensates for falling active tension. For at least some muscles this corresponds to lengths beyond filament overlap. The term “popping” is used to describe the uncontrolled, virtually instantaneous lengthening of a sarcomere from a length commensurate with its passive length to a length where passive structures primarily support the tension. Because the weakest sarcomeres are not at the same point along each myofibril, this nonuniform lengthening leads to a shearing of myofibrils, exposing membranes, especially T-tubules, to large deformations. This is thought to lead to loss of Ca2+ homeostasis, and, hence, damage, either through tearing of membranes or opening of stretch-activated channels (Allen et al. 2005). Support for the nonuniform lengthening of sarcomeres comes from a recent study of myofibrils from rabbit psoas muscle and left ventricles of guinea pig during activation and relaxation (Telley et al. 2006a). However, these authors also show that albeit half-sarcomeres of contracting single rabbit psoas myofibrils lengthen to different extents during a stretch, rapid elongation of individual sarcomeres beyond filament overlap (popping) does not occur. Moreover, in contrast to predictions of the popping sarcomere hypothesis, they postulate that a stretch rather stabilises the uniformity of half-sarcomere lengths and sarcomere symmetry (Telley et al. 2006b).
With respect to eccentric exercise, it is postulated that the structural adaptation consists of an increase in the number of sarcomeres in series so that a given muscle length corresponds to a shorter sarcomere length (Morgan and Talbot 2002). Whether and how serial sarcomere adaptation in humans following eccentric exercise is parallelled by changes in tendinous and/or muscle belly connective tissue remains to be established.
A consequence of the popping sarcomere hypothesis is that the unloaded shortening velocity of muscle fibres should increase with eccentric training. The reason for this is that the unloaded shortening velocity of a fibre is the sum of the velocities of its sarcomeres. Thus, the more sarcomeres in series, the faster the unloaded shortening velocity, provided that no alterations in MyHC composition occur. However, this will have to be demonstrated in future experiments, especially with respect to the MyHC isoform gene switching associated with stretch and force production (Goldspink et al. 1991). Another consequence of the popping sarcomere hypothesis is that significant muscle damage also can occur with endurance exercise, provided that the duration (marathon running) or mode (downhill running) of exercise is extreme. Therefore, it is predicted that under certain circumstances, endurance exercise can lead to serial sarcomere accretion with concurrent increase in unloaded fibre shortening velocity. On the contrary, “conventional” endurance exercise, which is associated with a bias towards shortening contractions at short muscle lengths, will lead to a decrease in the serial sarcomere number and, thus, to shorter muscle lengths. Short muscle lengths come with a reduction in functional range of motion (ROM). In general, a reduction in ROM is not desirable in health-based settings that aim at increasing musculoskeletal and cardiovascular function. Hence, in order to increase or preserve a functional ROM by serial sarcomere number modulation, eccentric resistance exercise covering the functional articular range might be the method of choice.
In conclusion, there is a substantial body of evidence that muscle fibres and satellite cells can sense changes in length. Accordingly, active and passive excursions from resting length are transduced into a molecular and cellular response with subsequent structural adaptation. However, with respect to active excursions from resting length, the response at the molecular, cellular, and structural level is dependent on the contraction mode. It follows directly that muscle length change as well as contraction mode are two mechano-biological determinants of exercise-induced skeletal muscle length adaptation. Therefore, these two mechano-biological determinants, among others (described below), need to be specified in reports coming from investigations into the plasticity of skeletal muscle following (resistance) exercise. As a measure of muscle length change we suggest to specify ROM (x 11, Table 1) during exercise and the number of length excursions [i.e. the number of repetitions (x 2, Table 1)]. It must be pointed out, however, that ROM might not always be indicative of fascicle length excursion. The reason for this is that length changes of the muscle-tendon units do not necessarily correspond to the length changes in the muscle fascicles (Hoyt et al. 2005). This means that the muscle-tendon-unit may lengthen, while the contracting muscle is shortening or isometric. As a measure of contraction mode we propose to report the fractional distribution of the three contraction types [shortening (concentric), isometric, lengthening (eccentric)] per repetition in terms of occurrence and temporal requirement (x 7, Table 1). Also, the number of contractions should be reported (x 2 and x 3, Table 1). For example, did the exercise comprise one set of several, only lengthening contractions or was one repetition composed of one shortening, one isometric, and one lengthening contraction? How much time did it take to perform one repetition and how was this time distributed over the respective contraction modes? The importance of the latter point for inducing muscle hypertrophy and gains in strength has been demonstrated in studies where the effect of fast lengthening contractions versus slow lengthening or slow shortening contractions has been investigated (Farthing and Chilibeck 2003; Shepstone et al. 2005).
Molecular and cellular determinants of radial muscle hypertrophy
Previous work showed that if the tibialis anterior in the mature rabbit was electrically stimulated while held in the stretched position by plaster cast immobilisation, it increased in mass by 35% within 7 days (Goldspink et al. 1992). Thus, if the lengthened (stretched) rodent muscle is additionally subjected to electrical stimulation, it increases in girth as well as length. The distinct role of active tension in generating radial growth is evidenced by this finding. Conversely, when muscle contractile activity is reduced by means of immobilisation (e.g. casting) or unloading (bed rest, space flight), rapid muscle loss (atrophy) occurs (Booth and Kelso 1973; Thomason and Booth 1990). Muscle loss is accentuated when immobilisation occurs at short muscle length and attenuated when immobilisation occurs at long muscle length, i.e. in a stretched position (Dupont Salter et al. 2003). Therefore, muscle growth and muscle atrophy are two opposing phenomena that are mechanistically linked. Either the activity or inactivity of a common set of molecules controlling a few cellular pathways determines whether the skeletal muscle tissue will respond to defined stimuli with increased protein synthesis and stimulation of cell growth or with increased protein breakdown and reduced cell proliferation (Glass 2003a, b, 2005; Nader 2005; Rennie et al. 2004; Sartorelli and Fulco 2004). In essence, the maintenance of skeletal muscle mass is the result of the dynamic balance between muscle protein synthesis and muscle protein degradation. Thus, these two opposite processes are believed to hold the key to the understanding of the mechanisms involved in the regulation of skeletal muscle mass.
Resistance exercise in humans and relevant animal models such as functional overload via synergist ablation can produce a significant increase in the mass of the overloaded muscles. In contrast to endurance exercise, resistance exercise is associated with high-intensity-short-duration workloads. The high-intensity-short-duration workloads placed on skeletal muscle during resistance exercise are at or near maximal capacity, and as such produce significant perturbations to the skeletal muscle fibres and the associated extracellular matrix. These perturbations can lead to significant muscle damage, especially if lengthening contractions (eccentric exercise) with supramaximal loads are performed. However, while both resistance and endurance exercise can result in muscle injury, resistance exercise is more likely to be associated with increases in fibre cross-sectional area and mass. The reasons for the response’s specificity point to differences in the integration of hormonal, metabolic, mechanical, neuronal, and immune responses, which are all likely involved in the distinct transcriptional responses that characterise endurance and resistance training.
Molecular determinants of skeletal muscle hypertrophy and atrophy
Exercise-induced hypertrophy mediators upstream of Akt
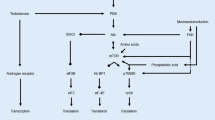
Resistance exercise-induced muscle hypertrophy results when muscle protein synthesis exceeds muscle protein degradation. In contrast, muscle atrophy is the result from increased muscle protein degradation over muscle protein synthesis. The integration of both anabolic and catabolic signals that lead to the increase or decrease in skeletal muscle mass (Fig. 2) is believed to occur at the molecular nodal point Akt (Nader 2005). Thus, activated (phosphorylated) Akt is both an effector of anabolic signals and a dominant inhibitor of catabolic signals. Activation of Akt is mediated by the insulin-like growth factor 1 (IGF-1)/phosphatidylinositol-3 kinase (PI3K) pathway. The IGF-1/PI3K pathway is triggered by increased muscle loading and subsequent expression of the gene encoding IGF-1 in both animal models (DeVol et al. 1990) and humans (Bamman et al. 2001). On the basis of their mRNA transcripts, three human muscle IGF-1 isoforms have been identified so far: IGF-1Ea (“liver type” isoform), IGF-1Eb, and IGF-1Ec (MGF) (Goldspink 2005; Hameed et al. 2003). In overloaded rodent muscle there are two clearly identified transcripts, IGF-1Ea and IGF-1Eb, of which IGF-1Eb has been termed MGF (Goldspink 2005). Rodent MGF differs slightly from the human MGF sequence as it contains a 52 base pair insert in exon 5 (Goldspink 2005). Other terms such as “mIGF-1”, which corresponds to the IGF-1Ea isoform, have also been used to describe the different isoforms (Musaro et al. 2001). However, only MGF appears to be activated by mechanical signals (Yang et al. 1996).

Simplified model for the relationship between muscle fibre size and the balance between anabolic and catabolic stimuli. Muscle size (girth and/or length) is set by the balance between activity-induced hypertrophy (anabolic) (blue) and counteracting atrophy (catabolic) (red) signals. In normal muscle, subjected to some amount of tear and wear, hypertrophy and atrophy signals are in balance (a). Resistance exercise perturbs the balance by inducing hypertrophy signals over atrophy signals (b), or by inhibiting atrophy signals (c), or both (d), thus driving hypertrophy. This model does not take into account changes in the contractile and metabolic profile that may occur following resistance exercise
These muscle-specific isoforms of IGF-1 are believed to be sufficient to induce hypertrophy through either autocrine or paracrine mechanisms (DeVol et al. 1990). Transgenic mice engineered to overexpress systemic or liver type IGF-1 under the control of a muscle-specific promoter have skeletal muscles that are twofold greater in mass than those seen in normal mice (Coleman et al. 1995; Musaro et al. 2001). Binding of the cytokine IGF-1 induces a conformational change in the muscle IGF-1 receptor (IGFR) tyrosine kinase, resulting in its trans-phosphorylation and the subsequent phosphorylation of insulin receptor substrate 1 (IRS-1). In turn, this results in the activation of PI3K. Finally, activation of PI3K results in the production of phosphatidylinositol-3,4,5-triphosphate and activation of Akt via 3′-phosphoinositide-dependent protein kinase 1 (PDK1). However, whether IGF-1 acts as an extracellular cue in muscle biology depends on its availability for muscle IGFR. Indeed, the availability of IGF-1 for muscle IGFR is controlled by IGF-1-binding proteins (IGFBPs). Binding of IGF-1 to IGFBPs can lead either to stimulation or inhibition of IGF-1 effects.
Hypertrophy mediators downstream of PI3K and Akt
Two pathways downstream of PI3K and Akt are believed to mediate hypertrophy (Glass 2005; Nader 2005): the Akt/mammalian target of rapamycin (mTOR) pathway, and the Akt/glycogen synthase kinase 3 beta (GSK3β) pathway. Both pathways lead to marked hypertrophy through activation of the protein synthetic machinery. Activation of mTOR by phosphorylated Akt results in an increase in protein translation by two mechanisms: first, mTOR activates 70 kDa ribosomal S6 protein kinase (S6K1/p70S6k), a positive regulator of protein translation; second, mTOR inhibits the activity of PHAS-1 (also known as 4E-BP1), a negative regulator of the protein initiation factor eIF-4E. Conversely, phosphorylation of Akt results in the inactivation of GSK3β. GSK3β blocks protein translation initiated by the eIF2B protein. Therefore, GSK3β inhibition may induce hypertrophy by stimulating protein synthesis independent of the mTOR pathway.
Other growth-signalling pathways in skeletal muscle
Other signal transduction pathways shown to be activated in response to various forms of muscle contraction include those involving the mitogen-activated protein kinase (MAPK) signalling pathways (Long et al. 2004). The MAPK-signalling pathways constitute a network of phosphorylation cascades that link cellular stress to changes in transcriptional activity. Relevant to the present review is the observation that exercise leads to the activation of at least three MAPK-signalling pathways, i.e. extracellular signal-regulated kinases (ERK)1/2, p38 MAPK, c-JUN NH2-terminal kinase (JNK), in skeletal muscle (Aronson et al. 1998; Boppart et al. 1999; Widegren et al. 2000). Further, AMP-activated protein kinase (AMPK) activity has been shown to be increased during contractions and exercise both in rodents and humans (Winder 2001). However, the relevance of the AMPK-signalling pathway has recently been questioned (Brooks 2005; Wadley et al. 2006). With respect to calcineurin signalling in working skeletal muscle, the reader is referred to chapter “Molecular and cellular determinants of contractile and metabolic tuning”.
Atrophy signalling
As with protein synthesis, degradation of cellular proteins is an essential process for the maintenance of myocellular homeostasis. However, in some specific situations, when protein degradation exceeds protein synthesis, skeletal muscle mass loss occurs. This process of mass loss is termed atrophy. Skeletal muscle atrophy is a serious consequence of various conditions such as microgravity, hindlimb suspension, immobilisation, and numerous diseases, including cancer and AIDS (Baracos 2001; Booth and Kelso 1973; Miro et al. 1997; Thomason and Booth 1990). Muscle loss is parallelled by profound transcriptomic (Bey et al. 2003; Cros et al. 2001; St-Amand et al. 2001; Stein et al. 2002; Stevenson et al. 2003; Wittwer et al. 2002) and proteomic (Isfort et al. 2000, 2002a, b; Toigo et al. 2005) changes. Over the years, several studies have identified at least five different systems involved in the degradation of proteins during muscle atrophy (Jackman and Kandarian 2004; Kandarian and Jackman 2006). These are the lysosomal system, the calpain system, the caspase or apoptotic protease system, the ubiquitin proteasome system, and the nuclear factor kappa B (NF-κB) system. At present, it remains unclear what the relative contribution of these systems to the atrophy process are, and which specific roles they may play during each particular disease state or context in which muscle atrophy develops. However, among the various systems involved in muscle proteolysis during atrophy, the ubiquitin-proteasome system is thought to play a major role (Jagoe et al. 2002). In addition to ubiquitin, three distinct enzymatic components are required, an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin-ligating enzyme (Glickman and Ciechanover 2002). The E3 ubiquitin ligases are the components which confer substrate specificity. In multiple models of skeletal muscle atrophy, the expression levels of two genes increased significantly: Muscle Ring Finger 1 (MuRF1) (Bodine et al. 2001a) and Muscle Atrophy F-box (MAFbx) (Bodine et al. 2001a) [also called Atrogin-1 (Gomes et al. 2001)]. Both MuRF1 and MAFbx/Atrogin were shown to encode E3 ubiquitin ligases and to be specifically expressed in skeletal muscle (Bodine et al. 2001a). However, the upregulation of MuRF1 and MAFbx/Atrogin requires the nuclear translocation and activity of a family of transcription factors termed Forkhead box O (FOXO). Indeed, in the context of skeletal muscle atrophy, an increase in FOXO1 mRNA in addition to several other atrophy-related genes was reported (Lecker et al. 2004). Also, FOXO3 activation was demonstrated to be sufficient to induce atrophy (Sandri et al. 2004). However, recent evidence shows that FOXO transcription factors are excluded from the nucleus when phosphorylated by Akt, and translocate to the nucleus upon dephosphorylation. Thus, muscle atrophy is prevented by FOXO inhibition through nuclear exclusion by phosphorylation through Akt. This finding highlights Akt’s role as a molecular checkpoint for the integration of both anabolic and catabolic signals that lead either to the increase or decrease in skeletal muscle mass. However, although there is a distinct set of genes which are inversely regulated by hypertrophy and atrophy (Latres et al. 2005), distinct transcriptional pathways are activated in skeletal muscle atrophy. These distinct transcriptional pathways are not necessarily the converse of those seen during hypertrophy. Thus, it seems that atrophy is not simply the converse of hypertrophy.
Muscle mass enhancement by inhibition of negative regulators
Myostatin, also known as growth and differentiation factor 8 (GDF-8), is a transforming growth factor–β (TGF-β) family member. It inhibits the progression of myoblasts from G1- to S-phase of the cell cycle through upregulation of p21, the only cyclin-dependent kinase 2 (Cdk2) inhibitor (McCroskery et al. 2003). Myostatin also inhibits myoblast differentiation by downregulation of MyoD/Myogenin expression (Langley et al. 2002). Consequently, myostatin acts as a negative regulator of skeletal muscle mass in (1) cattle, (2) mice, and (3) humans. (1) Naturally occurring mutations in the myostatin gene are responsible for the “double-muscling” phenotype, which is characterised by a dramatic increase in muscle mass of certain breeds of cattle (McPherron and Lee 1997). (2) Myostatin-null mice show an increase in muscle mass due to muscle hyperplasia and hypertrophy (McPherron et al. 1997). (3) Recently, a child with muscle hypertrophy was found to have a loss-of-function mutation in the myostatin gene (Schuelke et al. 2004). This individual showed a quadriceps cross-sectional area 7.2 standard deviations above the mean for age- and sex-matched controls and the ability to hold two 3 kg dumbbells in “horizontal suspension with arms extended” at the age of 4.3 years (Schuelke et al. 2004). As suggested, other less dramatic changes in the myostatin gene (or heterozygosity for the splice site mutation) may confer enhanced athletic prowess in a less conspicuous manner (McNally 2004). However, the child’s mutation has not been found in any other individual, and is therefore not a polymorphism-driving normal human variation (Gordon et al. 2005). Furthermore, genetic association studies with myostatin polymorphisms have consistently failed to demonstrate any statistically significant relationship with any human muscle trait (Ferrell et al. 1999; Ivey et al. 2000; Thomis et al. 2004).
In summary, skeletal muscle mass depends on the dynamic balance of protein synthesis versus protein breakdown. Whether muscle fibre protein synthesis outweighs protein degradation depends on the activity of intracellular hypertrophy- and atrophy-inducing mediators (Fig. 2). The activity of intracellular hypertropy- and atrophy-inducing mediators is coordinated at molecular checkpoints within the myofibre. These molecular checkpoints integrate anabolic and catabolic signals that are triggered by (patho-)physiological conditions. Resistance exercise is a physiological condition that aims at inducing hypertrophy signalling while repressing atrophy signalling (Fig. 2), finally leading to myofibre hypertrophy. Resistance exercise is associated with high active tension that is imposed on skeletal muscle. As shown, active tension through muscular contraction is per se a potent anabolic stimulus for myofibre hypertrophy. However, the levels of active tension required to induce graded hypertrophic effects or to prevent atrophy are most likely to differ. Therefore, such graded tensional effects must be investigated at the molecular and cellular level, if specific exercise regimens, e.g. for the prevention or treatment of sarcopenia, are to be developed. Consequently, the level of active tension that is imposed on skeletal muscle during resistance exercise is a further significant mechano-biological determinant of skeletal muscle size adaptation (x 1, Table 1). As such, it should be quantified in resistance exercise reports. However, the quantification of the load magnitude poses some problems, since usually, load magnitude is reported in terms of the one-repetition-maximum (1RM), e.g. 75% 1RM. It is beyond the scope of this review to discuss issues related to the 1RM. Suffice it to say that in a scientific setting we do not consider the 1RM an appropriate measure to determine the magnitude of the tensional load for exercise. In a scientific setting, we suggest to construct maximal voluntary torque (MVT)-angle curves, whenever possible. Based on these MVT-angle curves, the respective choices with respect to tension magnitude can be legitimated. Moreover, MVT intramuscular imbalances (joint angles of disproportionate torque) can be detected and pre-/post-MVT-angle curves can be compared with respect to MVT as well as optimum angle for torque generation (see “Structural adaptation to strain perturbation”). In most other settings it might be more practical to report the load magnitude in terms of 1RM [e.g. % 1RM (x 1, Table 1)]. Importantly, information about the 1RM should always be combined with information about the time under tension (TUT) (x 9, Table 1) until failure. That is, how many seconds the exercise can maximally be sustained prior to volitional failure (x 10, Table 1). This will additionally give important information about the metabolic changes occurring with training (see “Molecular and cellular determinants of contractile and metabolic tuning”). However, load magnitude per se is not a measure of muscular loading. Only an anatomically perfect technique will allow the efficient “delivery” of the load to the muscle under investigation. It follows that a sound anatomical definition of the exercise in terms of joint positions, movement velocity (movement control), etc. should be an integral part of the exercise stimulus descriptions (x 13, Table 1). It is imperative to know if the muscle was under permanent tension and how much of the load effectively “reached” the target muscle.
Cellular determinants of muscle hypertrophy and atrophy
As mentioned above, satellite cells are lineage-committed adult muscle stem cells, located between the basal lamina and the sarcolemma of myofibres. Satellite cells contribute to postnatal muscle growth and muscle regeneration after injury (Charge and Rudnicki 2004; Dhawan and Rando 2005; McKinnell et al. 2005; Wagers and Conboy 2005). Upon myotrauma, quiescent satellite cells become activated, proliferate, and ultimately fuse to existing damaged muscle fibres or among themselves to form new myofibres. Satellite cells are activated in response to hypertrophic stimuli, such as those occurring during muscle mechanical overload (Darr and Schultz 1987; Moss and Leblond 1971; Schiaffino et al. 1976). In several animal models of compensatory hypertrophy (Hanzlikova et al. 1975; Snow 1990) or after resistance training in humans (Kadi et al. 1999a, b, 2004; Roth et al. 2001), the total number of activated satellite cells is substantially increased. The mechanisms leading to satellite cell activation during muscle hypertrophy are not entirely understood. It is postulated that extensive physical activity, such as resistance training or muscle overloading (chronic stretch, agonist muscle ablation, tenotomy), inflicts muscle injury (Allen et al. 2005; Armstrong et al. 1991; Faulkner et al. 1993; Gibala et al. 1995). Consequently, muscle injury, similar to more severe muscle damage, may initiate a process of regeneration. An indirect proof of muscle damage after mechanical stress is given by an increase of serum markers such as muscle creatine kinase, an enzyme that is usually restricted to the myofibre cytosol. Muscle injury initiates an inflammatory response with the attraction of nonmuscle mononucleated cells, such as neutrophils and macrophages, into the damaged zone (Fielding et al. 1993). Subsequently, several growth factors are released either by the infiltrating cells or by the damaged myofibres themselves. These growth factors may ultimately regulate satellite cell proliferation and differentiation. Indeed, several cytokines have been described that modulate proliferation and differentiation of satellite cells in vitro or during regeneration after (exercise-induced) muscle injury (Charge and Rudnicki 2004; Vierck et al. 2000). As mentioned above, HGF is considered to be a key regulator of satellite cell activity during muscle regeneration (Allen et al. 1995; Bischoff 1997). HGF is secreted by damaged tissue during the early phase of muscle regeneration in amounts proportional to the extent of muscle injury (Sheehan and Allen 1999; Tatsumi et al. 1998). It seems that HGF directly regulates satellite cell activation. As described, a large body of evidence supports the importance of IGF-1 in the genesis of skeletal muscle hypertrophy. IGF-1 can promote both proliferation and differentiation of cultured satellite cells, and these findings have been confirmed in animal models (Charge and Rudnicki 2004). Experiments showed that muscle-localised expression of IGF-1Ea (“mIGF-1”) prevented, through an increase of the regenerative potential of satellite cells, the age-related loss of muscle mass (Musaro et al. 2001). Also, satellite cells derived from mice overexpressing IGF-1Ea display an increased proliferative potential (Chakravarthy et al. 2000b). Increased proliferative potential seems to be mediated by activation of the IGF-1/PI3K/Akt pathway, which results in the inactivation (phosphorylation) of FOXO1 (Machida et al. 2003). Inactivation of FOXO1 downregulates the activation of the p27Kip1 promoter (Chakravarthy et al. 2000a). Therefore, the molecular pathways activated by IGF-1 in the muscle fibres to promote increased protein translation appear also to be activated in satellite cells. However, IGF-1 action on satellite cells seems to be IGF-1 isoform-specific with apparently different expression kinetics (Goldspink 2005). After exercise and/or damage, the IGF-1 gene is first spliced towards MGF but after a day or so becomes completely spliced towards the systemic IGF-1 isoforms, which in human muscle are IGF-1Ea and IGF1-Eb (Goldspink 2005; Haddad and Adams 2002; Hill and Goldspink 2003; Hill et al. 2003; Yang and Goldspink 2002).
Concepts of myocellular enlargement
Muscle fibres, i.e. multinucleated muscle cells, develop during embryonic differentiation, when mononucleated myoblasts first proliferate and then fuse to form myotubes that become innervated. Following myoblast fusion, no further mitotic divisions occur within the myotubes or muscle fibres. Thus, under normal biological conditions, adult skeletal muscle is an extremely stable tissue with little turnover of nuclei (Decary et al. 1997; Schmalbruch and Lewis 2000). These findings about the postmitotic and multinucleated nature of muscle fibres have led to the concept of a DNA unit or myonuclear domain (Allen et al. 1999; Cheek 1985; Hall and Ralston 1989). The myonuclear domain is the theoretical amount of cytoplasm supported by a single myonucleus. However, the concept of a myonuclear domain is a theoretical one since regulation of the expression and distribution of individual proteins within the muscle fibre is dependent on a number of different variables related to the nature of each protein. Nonetheless, since each muscle fibre is made up of many myonuclear domains, muscle fibre radial or longitudinal hypertrophy could conceivably result from either an increase in the number of domains (by increasing myonuclear number) or by an increase in the size of existing domains (Edgerton and Roy 1991) (Fig. 3). Research to date has strongly supported the former concept by showing that satellite cell activation is required for muscle hypertrophy. The requirement of satellite cell activation was first demonstrated by an approach in which mild γ-irradiation was employed to block satellite cell proliferation. In response to functional overload, myonuclear number or muscle size was not increased in irradiated rat and mice muscles (Adams et al. 2002; Rosenblatt and Parry 1992). However, recent reports indicate that the size of myofibres can increase without the addition of new myonuclei (Kadi et al. 2004; Wada et al. 2003; Zhong et al. 2005). It was found that following 30 and 90 days of resistance exercise, the fibre area controlled by each myonucleus gradually increased throughout the training period and returned to pretraining values during detraining (Kadi et al. 2004). No alterations in the number of myonuclei were detected. Moreover, it has been shown that under normal physiological conditions myonuclear domain size might vary throughout mouse lifespan (Wada et al. 2003) and that myonuclear domain size is not constant during rat soleus muscle atrophy (Zhong et al. 2005).

Hypothetical model of skeletal muscle fibre cytoplasmic enlargement. Schematically depicted are satellite cells (red/orange) lying beneath the basal lamina and sarcolemma of multinucleated (dark blue) skeletal muscle fibres (cross and longitudinal sections). According to the concept of the myonuclear domain (see text for details), muscle fibre hypertrophy could conceivably result from either an increase in the size of existing domains (dark blue shading) (a→b and a→b′ for radial and longitudinal hypertrophy, respectively) or by an increase in the number of domains by addition of new myonuclei (dark blue) provided by the satellite cells (red) (b→c / a→c and b′→c′ / a′→c′ for radial and longitudinal hypertrophy, respectively)
Whether the increase in mass during hypertrophy results from an increase in the size of each fibre (hypertrophy) or by an increase in fibre number (hyperplasia), has been under debate. Although the evidence has been somewhat contradictory, there has been some suggestion that an increase in fibre number may occur in some animals under certain experimental conditions. Indeed, a review of several investigations into skeletal muscle growth concluded that in several animal species certain forms of mechanical overload increases muscle fibre number (Kelley 1996). However, it has been suggested that some reports have misinterpreted the intricate arrangements of elongating fibres as increases in fibre number (Paul and Rosenthal 2002). Indeed, studies reporting an increase in the number of muscle fibres used avian or cat muscles (Kelley 1996). Both avian and cat muscles have multiple endplate bands and fibres that do not insert into both tendons but terminate intrafascicularly (Paul 2001). Thus, it remains to be determined whether the radial growth of muscles with intrafascicularly terminating fibres in larger mammals arises from new fibre formation as assumed previously, or from elongation of existing fibres as recently proposed (Paul and Rosenthal 2002). However, most human muscle fascicles, despite their great length, consist of fibres that extend continuously from one tendon to the other with a single nerve endplate band. Therefore, muscle hypertrophy in the adult human apparently can be accounted for predominantly by hypertrophy of existing fibres via addition of newly constructed myofibrils to the contractile apparatus. Accordingly, it has been suggested that hyperplasia does not occur in humans following resistance exercise (MacDougall et al. 1984; McCall et al. 1996). However, hyperplasia still remains a thinkable mechanism of muscle enlargement. Thus, more conclusive evidence might come from future investigations into the plasticity of human muscle fibre number. Fibre splitting or branching is also a characteristic feature of muscle regeneration (Charge and Rudnicki 2004). Fibre splitting is commonly observed in muscles from patients suffering neuromuscular diseases, in hypertrophied muscles, and in ageing mouse muscles, all of which are associated with abnormal regenerative capacity (Bockhold et al. 1998; Charge et al. 2002; Schiaffino et al. 1979). It has been hypothesised that fibre splitting occurs due to the incomplete fusion of fibres regenerating within the same basal lamina (Blaivas and Carlson 1991; Bourke and Ontell 1984).
Summarising, mechanical stress through high-intensity resistance exercise (especially, but not exclusively supramaximal eccentric exercise) inflicts myotrauma. Upon myotrauma, quiescent satellite cells become activated: (1) through anabolic cytokines that are released by the perturbed extracellular matrix; (2) by infiltrating cells involved in the inflammatory response; (3) by the damaged myofibres; (4) in an autocrine manner by the satellite cells themselves. Following activation, the satellite cells proliferate, and ultimately fuse to existing muscle fibres or among themselves for tissue repair/regeneration. Thus, besides myofibre hypertrophy due to increased protein synthesis/decreased protein degradation (see “Cellular determinants of muscle hypertrophy and atrophy”), satellite cell-based myoplasmic enlargement is a further mechanism in adult skeletal muscle hypertrophy. It is consistent with the concept of the myonuclear domain, where the satellite cells provide the additional DNA for the establishment of additional myonuclear domains during myoplasmic enlargement. It follows that muscle damage is a further significant mechano-biological determinant of skeletal muscle size adaptation. Thus, load magnitude (as a measure of extracellular matrix and satellite cell perturbation) as well as the number of lengthening contractions (as a measure or eccentric damage) need to be specified in resistance exercise reports (x 1, x 2, x 7, Table 1). Additionally, successful recovery (repair) from muscle injury depends on the balance between the degenerative and regenerative processes. Importantly, degenerative and regenerative processes are time-dependent. Thus, if resistance exercise perturbations are delivered at very short time intervals, the degenerative processes prevail. If the degenerative processes prevail, muscle mass is lost. As a consequence, studies in which subjects are resistance-trained over a determinate period of time (x 6, Table 1) should report recovery times in between the exercise sessions (x 12, Table 1) and number of exercise interventions per week (x 5, Table 1), as a function of exercise-induced muscle damage (exercise intensity). The importance of these three factors (x 6, x 12, x 5, Table 1) has been demonstrated in studies, in which the duration of elevated protein turnover following resistance exercise sessions was investigated (Chesley et al. 1992; MacDougall et al. 1995; Phillips et al. 1997).
Molecular and cellular determinants of contractile and metabolic tuning
Muscle fibre has multiple, complex functional gene groupings adapting independently to environmental stimuli (Spangenburg and Booth 2003). The molecular regulation of these functional gene groupings is fibre type-specific and results in the fibre type’s phenotypical characteristics. Such characteristics comprise, e.g. contractile protein isoforms, mitochondrial volume, myoglobin levels, capillary density, and oxidative enzyme capacity. Each of these characteristics could be considered a functional gene domain within the respective fibre type (Spangenburg and Booth 2003). Not only can these functional modules be regulated within different myofibres but also MyHC protein expression can be heterogeneous within a single fibre (Pette and Staron 2000; Talmadge et al. 1996), resulting in “hybrid” (Baldwin and Haddad 2001) or “polymorphic” (Caiozzo et al. 2003) fibres. It is believed that contractile activity following neural activation induces changes in common regulatory factors within a subpopulation of genes (i.e. gene “modules”) to modify the muscle fibre phenotype (Spangenburg and Booth 2003). More specifically, neural activation of skeletal muscle results in the release of acetylcholine from the neuromuscular junction and depolarisation of the plasma membrane, which activates force production by a process known as excitation–contraction coupling. The frequency and duration of stimulation determine the amplitude and duration of the Ca2+ transients and, as a result, the level of force output by the muscle. Thus, both the amplitude and duration of the Ca2+ transient in skeletal muscle are determined by the motor unit (MU) firing frequency. The increases in amplitude, as well as the duration for which these amplitudes are achieved, are thought to encode signals that will be recognised by different downstream Ca2+-dependent pathways. The key signalling pathways downstream of the elevation in intracellular Ca2+ that translate this signal into a transcriptional response include the Ca2+/calmodulin(CaM)-dependent phosphatase calcineurin (Cn), Ca2+/calmodulin-dependent kinase II (CaMKII), Ca2+/calmodulin-dependent kinase IV (CaMKIV), and Ca2+-dependent protein kinase C (PKC) (Chin 2005). In turn, these Ca2+-dependent key signalling pathways will determine the set of genes expressed, thus providing a mechanism for tightly coupling the extent of muscle excitation to regulation of transcription (i.e. excitation–transcription coupling) (Chin 2005). Many Ca2+-sensitive target genes have been identified in skeletal muscle. Downstream Ca2+-sensitive target genes of varied expression levels between fibres include the nicotinic acetylcholine receptor (nAChR), glucose transporter 4 (GLUT4), sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA1), MyHC isoforms, oxidative enzymes, as well as genes that regulate mitochondrial biogenesis. Apart from the transcriptional regulation, it has been suggested that muscle fibre contractile characteristics might also be regulated by posttranslational modification of contractile proteins (Canepari et al. 2005). In addition to the role of Cn signalling in the determination of muscle fibre type characteristics, this phosphatase is known to play an important role in muscle hypertrophy (Dunn et al. 1999; Michel et al. 2004). Cn dephosphorylates the transcription factor nuclear factor of activated T cells (NFAT), enabling its nuclear translocation and DNA binding. The Cn–NFAT pathway has been linked to Ca2+-induced skeletal muscle hypertrophy, at least in cultured skeletal muscle (Semsarian et al. 1999). With regard to skeletal muscle hypertrophy in animal models, the role of Cn remains controversial. Current thought suggests that hyperactivation of Cn alone is not sufficient to induce skeletal muscle hypertrophy but that the activation of accessory parallel signalling pathways for growth is required (Michel et al. 2004).
The size principle of motor recruitment—consequences for resistance exercise
As mentioned above, amplitude and duration of Ca2+-transients in skeletal muscle as a function of MU firing frequency are decoded at the molecular level, resulting in expression changes of fibre type-specific functional gene modules. For mammals, it has been shown that for many activities there is a graded level of muscle recruitment that is driven by the different thresholds of the motor neurones: “the size principle of motor recruitment” (Denny-Brown and Pennybacker 1938; Henneman et al. 1965, 1974). The size principle predicts that MU recruitment is determined by force requirements. The majority of units are, in fact, recruited voluntarily in the order of increasing size (Monster and Chan 1977; Tanji and Kato 1973). Typically, small MUs are type I units, and unit size increases with progression through the fibre types: I < IIA < IIX (according to the MyHC isoform classification system in humans). Therefore, when low force is required, only type I MUs will be active. Only when force is high will recruitment demand involvement of the larger MUs. It follows that slow muscle fibres are activated for low-force contractions and fast muscle fibres are additionally activated to supply greater force demands. Several researchers have shown that MU recruitment is completed by ~ 50% of maximum voluntary contraction (MVC) in small muscles [adductor pollicis (Kukulka and Clamann 1981), and first dorsal interosseus (De Luca et al. 1982a, b, 1996; Milner-Brown et al. 1973)], and 70–80% of MVC in large muscles [biceps (Kukulka and Clamann 1981), deltoid (De Luca et al. 1982a, b), and tibialis anterior (De Luca et al. 1996; Erim et al. 1996)]. The size principle applies when either slow-ramp force is exerted or constant low forces are compared with constant higher forces. The size principle of orderly recruitment is also preserved during exercise (Gollnick et al. 1974a, b; Vollestad and Blom 1985; Vollestad et al. 1984, 1992), whether the comparison is of sustained contractions at different intensities, or at different times during exercise at the same submaximal force sustained until exhaustion (Adam and De Luca 2003).
However, during some forms of voluntary or reflex contraction, motor neurone size may not be the sole factor determining excitation threshold: (1) Specialisation of the synaptic input among the motor neurones, and joint position have been shown to affect recruitment order in some cases; (2) MU rotation strategy has been demonstrated for sustained isometric contraction of biceps brachii at 10 but not at 40% of maximum; (3) In certain very rapid or sudden corrective movements—such as accelerations or sudden changes in direction—high-threshold units that do not participate in walking or even in running might be selectively recruited, as shown for the human short extensor muscle of the toes (Grimby 1984). With respect to ballistic movements, there is some controversy over the extent to which selective recruitment may occur (if at all) (Zehr and Sale 1994); (4) It has been suggested that selective recruitment of large MUs occurs during lengthening contractions (Nardone and Schieppati 1988; Nardone et al. 1989); (5) In certain cases of altered motor tasks, recruitment order might vary. Based on these results, it has been suggested that differential recruitment simply represents the existence of distinct, task-related subpopulations of motor units, rather than “violations” of the size principle (Burke 2002; Cope and Sokoloff 1999). Hence, when the same motor task is undertaken in exactly the same way, the order in which MUs are recruited remains fixed. This is of special importance, since it supports the notion that recruitment order is maintained during muscular fatigue. In particular, it has been shown that submaximal fatiguing contractions in the vastus lateralis muscle of humans lead to the monotonic decline in the recruitment threshold of all MUs and the progressive recruitment of new MUs, without change in the recruitment order. Thus, as the force capacity of continuously active muscle fibres declines progressively, increased excitation is required to keep the muscle output constant. The increased excitation produces the recruitment of additional MUs. The recruited MUs thus become active at a lower torque level than their initial threshold, and the recruitment threshold continues to decrease in subsequent contractions as the force production of the active MUs continues to decrease (Adam and De Luca 2003). Indeed, it could be shown that the reduction in peak tetanic torque is linearly correlated with the decrease in the mean MU recruitment threshold at corresponding endurance times (beginning, middle, and end of fatiguing contractions) (Adam and De Luca 2003). However, these data do not prove a causal relationship between changes in muscle force output and changes in MU recruitment. Nonetheless, it can reasonably be assumed that the drop in peak tetanic torque that comes with muscular fatique corresponds to an increase in the relative force requirement to sustain the target torque level.
Based on these findings, we propose a theoretical model of muscle fatigue and MU recruitment during resistance exercise progression (Fig. 4). Provided that exercise is performed to volitional muscular failure, the three different load magnitudes in Fig. 4a–c will lead to similar, i.e. “complete” MU recruitment (Fig. 4d), and, thus, to a similar stimulation of protein synthesis. Indeed, preliminary studies aimed at delineating the dose–response relationship between the intensity of exercise and the rates of muscle protein synthesis have shown that when the same total amount of ATP is turned over and recruitment is complete, exercise at 60, 75, and 90% of the 1RM results in exactly the same stimulation of muscle protein synthesis (Bowtell et al. 2003). Additionally, increases in tension above 65% cause no further stimulation in muscle protein synthesis (Bowtell et al. 2003). However, the different TUT until muscular failure imply different MU recruitment dynamics. As a consequence, distinct metabolic loads are inflicted on the exercising muscle in Fig. 4a–c. In Fig. 4a, progression through the fibre types I → IIA → IIX occurs fast (30 s). In contrast, in Fig. 4c, progression through the fibre types occurs relatively slowly (180 s). Consequently, in Fig. 4c the fibres’ capability in providing ATP through oxidative metabolism is more pronouncedly challenged than in Fig. 4a. This is due to the fact that with low resistive loads in % of the maximum voluntary torque (MVT), the threshold for complete recruitment is reached later. Thus, if a biceps exercise is assumed for the examples in Fig. 4, only in Fig. 4a the recruitment is predicted to be complete at start. In Fig. 4b, c, complete recruitment will be reached later. It follows directly, that the two strategies of MU recruitment and MU firing rate-coding for the increase in force output will vary to different extents for Fig. 4a–c. Consequently, the distinct pattern of MU recruitment and rate-coding in Fig. 4a–c will have a different impact on the previously described excitation–transcription coupling (Chin 2005).

Hypothetical model of muscle fatigue and its consequences for motor unit recruitment and, thus, for the metabolic load imposed on a skeletal muscle. In this simplistic model (e.g. biceps brachii performing 1 set of resistance exercise to volitional muscular failure), it is assumed that the drop in % of the maximum voluntary torque (MVT) in-between the repetitions (i.e. the “fatigue inroad”) occurs with constant percentage. The magnitude of fatigue inroad per repetition is assumed to be 6, 3.5, and 2.5% for high (magenta bars), intermediate (cyan bars), and low (yellow bars) resistive loads, respectively. The primary ordinates in a–c indicate the load magnitude expressed in % of the MVT achieved at a determinate joint angle. The abscissae in a–d indicate the total time (s) under tension until muscular failure (TUTF) or the corresponding number of repetitions when 10 s movement time per repetition is assumed. The secondary ordinates in a–c indicate the resistive load (i.e. the “training load”) in % of MVT at t = t 0. The ordinate in d indicates the resistive load in % of MVT through exercise progression, which corresponds to the relative resistive MVT. A high (magenta bars), intermediate (cyan bars), and low (yellow bars) resistive load is assumed in a–c, respectively. These high, intermediate, and low resistive loads correspond to high, intermediate, and low % of MVT. Moreover, these resistive loads (“training loads”) remain unchanged during the same exercise set. However, the theoretical MVT (black bars) declines with exercise progression due to muscular fatigue, with a concurrent increase in motor unit (MU) recruitment and MU firing rate (see text for details). The time point of muscular failure corresponds to the point at which the theoretical MVT is below the MVT required to overcome the resistive load. Muscular failure occurs after 30 s (3 repetitions), 90 s (9 repetitions), and 180 s (18 repetitions) in examples a–c, respectively. In d, the hypothetical increase in relative resistive load (% MVT) for high (magenta), intermediate (cyan), and low (yellow) resistive loads [% MVT (t = t 0)] is plotted against the TUTF. The relative load [% MVT (t = t 0)] as exercise starts and the steepness with which it increases through exercise progression until voluntary failure, as a function of the muscle’s fibre type distribution, determines MU recruitment. The grey shaded area corresponds to the predicted resistive load in % of MVT at which MU recruitment for biceps brachii muscle is completed (see text for details)
In conclusion, we deduce that the TUT (x 9, Table 1), volitional muscular failure (x 10, Table 1), number of sets (x 3, Table 1), rest in-between sets (x 4, Table 1), and rest in-between repetitions (x 8, Table 1) are further mechano-biological determinants that affect MU recruitment and MU firing rate-coding, and, thus, excitation–transcription coupling. In addition, the number of exercise interventions per week (x 5, Table 1) and the duration of the experimental period (x 6, Table 1) should be reported for an estimation of the training’s stimulatory effect on muscle protein synthesis.
Molecular sensing of oxygen
Intramyocellular oxygen partial pressure has been suggested to drop with exercise, leading to “local hypoxia” (Gayeski et al. 1985). Local hypoxia, as determined by measurement of oxygen saturation via assessment of myoglobin desaturation, has been shown to occur within 20 s of onset of exercise in human quadriceps muscle (Richardson et al. 1995). Additionally, oxygen saturation is reduced to a plateau with the onset of exercise (Richardson et al. 2001). Thus, it has been hypothesised that local hypoxic conditions may prevail in muscle even during low intensity exercise in normoxia (Hoppeler et al. 2003).
The partial pressure of cellular oxygen is sensed by a family of prolyl hydroxylases (Wenger et al. 2005). This enzyme family contains three members: prolyl-4-hydroxylase domain 1 (PHD1), PHD2, and PHD3, also known as hypoxia-inducible factor (HIF) prolyl hydroxylase 3 (HPH3), HPH2, and HPH1, respectively. The prolyl hydroxylases covalently modify HIFα subunits. HIF is a heterodimeric transcription factor consisting of one of three different oxygen-sensitive HIFα subunits and a common constitutive HIFβ subunit. Under normoxic conditions, HIFα is hydroxylated. HIFα hydroxylation promotes von Hippel–Lindau (VHL) tumour suppressor protein binding to HIFα. Upon binding of VHL, HIFα is targeted for proteasomal destruction. Under hypoxic conditions, however, PHD activity (and thus HIFα hydroxylation) decreases. Thus, under hypoxic conditions, the high turnover rate of HIFα subunits enables the very rapid accumulation of HIFα. Following a further decrease in oxygen availability, the asparagine hydroxylase function of another enzyme, the factor inhibiting HIF (FIH), also becomes impaired, resulting in a decrease in HIFα C-terminal hydroxylation. This decrease in C-terminal HIFα hydroxylation enables the increased recruitment of the p300 and CREB-binding protein (p300/CBP) transcriptional coactivators, leading to the enhanced transcriptional activation of at least 70 different HIF effector genes (Wenger et al. 2005).
HIF effector genes regulate, e.g., oxygen supply, cellular metabolism, cell growth, and apoptosis (Wenger et al. 2005). Indeed, it has recently been shown that HIF1α protein abundance increases in response to the acute exercise-induced increase in oxygen demand in human tissue (Ameln et al. 2005). Concurrently, no changes in HIF1α mRNA abundance were detected in response to exercise. Thus, it was suggested that the observed increased HIF1α protein abundance mainly depends on posttranscriptional mechanisms. In parallel with the exercise-induced stabilisation of HIF1α protein, nuclear translocation of the protein was observed, together with an induced expression of the HIF regulated target genes. In particular, vascular endothelial factor (VEGF) and erythropoietin (EPO) mRNA abundance was increased with exercise (Ameln et al. 2005).
The role of HIF1α in regulating skeletal muscle function has recently also been investigated with HIF1α knockout mice (HIF1α KOs) (Mason et al. 2004). It was found that a significant exercise-induced increase in mRNA abundance of VEGF, GLUT4, muscle-specific phosphofructokinase (PFK-M), phosphoglycerate kinase (PGK), and lactate dehydrogenase-A (LDH-A) is missing in HIF1α KOs compared to wild-type (WT) mice. Also, changes in enzymatic activity in a number of key glycolytic enzymes are affected by deletion of HIF1α. In particular, the activity of one of the key rate-limiting enzymes, PFK, is significantly lower following exercise in HIF1α KOs compared to WT mice. In HIF1α KOs, overall, a metabolic shift away from glycolysis towards oxidation was observed. This metabolic shift away from glycolysis towards oxidation increases swimming and uphill running exercise times in the HIF1α KOs. Conversely, repeated bouts of downhill running (eccentric exercise) results in greatly reduced exercise times and increased muscle damage in HIF1α KOs relative to WT mice. The authors hypothesised that the reduced exercise times with eccentric exercise is due to the impaired glycolytic capacity in HIF1α KOs (Mason et al. 2004). This assumption was based on the previous findings that primarily fast-twitch glycolytic fibres are activated for eccentric contraction (Nardone and Schieppati 1988; Nardone et al. 1989).
Further evidence for the molecular regulation of skeletal muscle metabolic properties through regulation of fibre type functional modules has recently come from the engineering of a mouse capable of continuous running of up to twice the distance of a WT littermate (Wang et al. 2004). These authors uncovered peroxisome proliferator-activated receptor (PPAR)δ as the first transcription factor able to drive a metabolic fibre reprogramming following the targeted expression of an activated form of PPARδ. PPARδ produces profound and coordinated increases in oxidation enzymes, mitochondrial biogenesis, and production of specialised type I fibre contractile proteins—the three hallmarks for muscle fibre type switching. The muscle phenotypes described in that study are remarkably similar to those of transgenic mice expressing either Cn, CaM kinase, or peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) (Lin et al. 2002; Naya et al. 2000; Wu et al. 2002). Thus, it was suggested that PPARδ could be one of the hypothetical downstream transcription factors of these pathways (Wang et al. 2004). However, according to ligand and gain-of-function transgenic studies, PPARδ needs to be activated in order to direct the muscle fibre switch. Indeed, in a recent report, simple overexpression of WT PPARδ in muscle was found not to be sufficient to promote a fibre switch or obesity resistance, although certain oxidation enzymes are increased (Luquet et al. 2003). This supports the notion that the activating signal or ligand, but not the receptor, is limiting (Wang et al. 2004). Thus, PPARδ activation, rather than merely an increase of PPARδ levels, is an essential element for fibre switching and its associated functional manifestations. Two models have been proposed for the exercise-induced activation of endogenous PPARδ (Wang et al. 2004). First, it is possible that exercise generates or increases endogenous ligands for PPARδ as tissues are undergoing substantial increases in fatty-acid internalisation and oxidation. Fatty acids and their metabolites can activate PPARδ. A second model is that exercise may induce the expression of PGC-1α (Goto et al. 2000) and thereby activate PPARδ. This is consistent with previous work in which it was shown that PGC-1α physically associates with PPARδ in muscle tissue and can powerfully activate it even in the absence of ligands (Wang et al. 2003). In line with these findings in rodent muscles, two recent studies show that one-legged knee extensor exercise of different duration and blood supply conditions induce transcriptional activation of the PGC-1α gene in human skeletal muscle (Norrbom et al. 2004; Pilegaard et al. 2003). Impairment of blood supply following low-intensity exercise with superimposed vascular occlusion has also been shown to stimulate hypertrophy to a greater degree than exercise without occlusion (Abe et al. 2005; Moore et al. 2004; Takarada et al. 2002, 2004).
In summary, it can be derived that decreased oxygen supply to skeletal muscle during resistance exercise affects myocellular oxygen homeostasis. Oxygen supply to skeletal muscle is influenced by the magnitude of active and/or passive tension that is generated during exercise as well as by the modality of exercise (Vedsted et al. 2006). Magnitude of active and/or passive tension dictates to which extent blood flow is reduced. Modality of exercise, i.e. constant versus intermittent tension delivery, is a measure of how long and how many times blood flow is reduced. For example, during “eccentric only” resistance exercise (such as “negative chins” or “negative dips”), only lengthening contractions are performed. Negative chins/dips are performed by climbing into the top position using the legs and by lowering the body back down by the lengthening contractions of the respective target muscles. Consequently, while climbing into the top position, target muscle blood flow is less challenged than during the lowering phase. Thus, we conclude that load magnitude (x 1, Table 1), TUT (x 9, Table 1), contraction mode and fractional distribution per repetition (x 7, Table 1), rest in-between repetitions (x 8, Table 1), as well as ROM (x 11, Table 1), are significant mechano-biological determinants of myocellular oxygen homeostasis. Therefore, they need to be specified in resistance exercise protocols. It must also be emphasised that the total TUT does not necessarily just correspond to the number of repetitions times the time requirement for one repetition. If the exercise is performed to volitional muscular failure, the TUT corresponds to the TUT until failure (TUTF). TUTF includes an isometric contraction of maximal time duration at the end of exercise. Thus, TUTF may exceed the product of the number of repetitions times the time requirement for one repetition.
Recommendations for optimal exercise stimulus design and description
Skeletal muscle is a biological tissue. As such, it adapts to mechano-biological conditions that are present or missing. As described in this paper, these mechano-biological conditions lead to the induction of molecular and cellular determinants for the regulation of skeletal muscle girth, length, and fibre type. Thus, we propose to view (resistance) “exercise” as physiological tissue conditioning, i.e., the targeted delivery of muscular stimuli of quantitative and/or qualitative effect on the muscular phenotype. Whether and to which extent an adaptation occurs is subordinate to the “response matrix” of the respective subject (Fig. 1). Targeted delivery of adaptational stimuli precludes an accurate mechano-biological description of the condition. Unfortunately, most of the past and present resistance exercise prescriptions (Kraemer and Ratamess 2004) still use ambiguous stimulus descriptors. This shortcoming makes it a priori virtually impossible to identify which stimuli lead to what kind of adaptation. Consequently, no exercise prescriptions with specific effect, e.g. for the prevention and/or treatment of sarcopenia and metabolic diseases, exist. In order to allow for a dissection of the relative contributions of exercise stimuli and response matrix to muscular adaptation, we have identified and described a basic set of resistance exercise-related muscular conditions and termed them “mechano-biological determinants of muscular adaptation” (Table 1). While most of these determinants are scientifically tested, some are hypothetical (e.g. rest in-between repetitions). Understanding the relative contribution of theses determinants in modulating the skeletal muscle phenotype should provide the basis for the design and prescription of training stimuli with specific quantitative and/or qualitative effect. We are aware of the fact that the use of 13 descriptors might raise some concerns with respect to practicability. However, we consider these determinants mandatory information for scientific experiments that aim at identifying causal connections between stimulus and response. Consequently, we strongly recommend to standardise the design and description of all future resistance exercise investigations by using the herein proposed basic set of 13 mechano-biological determinants (classical and new ones). Finally, although some of the newly proposed descriptors (e.g. TUT) are already employed by some exercise specialists, the identification of graded quantitative and qualitative effects of the most important descriptors should improve the effectiveness of future exercise recommendations.
References
Abe T, Kearns CF, Sato Y (2006) Muscle size and strength are increased following walk training with restricted venous blood flow from the leg muscle, Kaatsu-walk training. J Appl Physiol 100:1460–1466
Adam A, De Luca CJ (2003) Recruitment order of motor units in human vastus lateralis muscle is maintained during fatiguing contractions. J Neurophysiol 90:2919–2927
Adams GR, Caiozzo VJ, Haddad F, Baldwin KM (2002) Cellular and molecular responses to increased skeletal muscle loading after irradiation. Am J Physiol Cell Physiol 283:C1182–C1195
Aihara Y, Kurabayashi M, Saito Y, Ohyama Y, Tanaka T, Takeda S, Tomaru K, Sekiguchi K, Arai M, Nakamura T, Nagai R (2000) Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: role of M-CAT element within the promoter. Hypertension 36:48–53
Allen RE, Sheehan SM, Taylor RG, Kendall TL, Rice GM (1995) Hepatocyte growth factor activates quiescent skeletal muscle satellite cells in vitro. J Cell Physiol 165:307–312
Allen DL, Roy RR, Edgerton VR (1999) Myonuclear domains in muscle adaptation and disease. Muscle Nerve 22:1350–1360
Allen DG, Whitehead NP, Yeung EW (2005) Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: role of ionic changes. J Physiol (Lond) 567:723–735
Ameln H, Gustafsson T, Sundberg CJ, Okamoto K, Jansson E, Poellinger L, Makino Y (2005) Physiological activation of hypoxia inducible factor-1 in human skeletal muscle. Faseb J 19:1009–1011
Anderson JE (2000) A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell 11:1859–1874
Anderson J, Pilipowicz O (2002) Activation of muscle satellite cells in single-fiber cultures. Nitric Oxide 7:36–41
Armstrong RB, Warren GL, Warren JA (1991) Mechanisms of exercise-induced muscle fibre injury. Sports Med 12:184–207
Aronson D, Boppart MD, Dufresne SD, Fielding RA, Goodyear LJ (1998) Exercise stimulates c-Jun NH2 kinase activity and c-Jun transcriptional activity in human skeletal muscle. Biochem Biophys Res Commun 251:106–110
Baldwin KM, Haddad F (2001) Effects of different activity and inactivity paradigms on myosin heavy chain gene expression in striated muscle. J Appl Physiol 90:345–357
Bamman MM, Shipp JR, Jiang J, Gower BA, Hunter GR, Goodman A, McLafferty CL Jr, Urban RJ (2001) Mechanical load increases muscle IGF-I and androgen receptor mRNA concentrations in humans. Am J Physiol Endocrinol Metab 280:E383–E390
Baracos VE (2001) Management of muscle wasting in cancer-associated cachexia: understanding gained from experimental studies. Cancer 92:1669–1677
Barash IA, Mathew L, Ryan AF, Chen J, Lieber RL (2004) Rapid muscle-specific gene expression changes after a single bout of eccentric contractions in the mouse. Am J Physiol Cell Physiol 286:C355–C364
Barash IA, Mathew L, Lahey M, Greaser ML, Lieber RL (2005) Muscle LIM protein plays both structural and functional roles in skeletal muscle. Am J Physiol Cell Physiol 289:C1312–C1320
Baumeister A, Arber S, Caroni P (1997) Accumulation of muscle ankyrin repeat protein transcript reveals local activation of primary myotube endcompartments during muscle morphogenesis. J Cell Biol 139:1231–1242
Bey L, Akunuri N, Zhao P, Hoffman EP, Hamilton DG, Hamilton MT (2003) Patterns of global gene expression in rat skeletal muscle during unloading and low-intensity ambulatory activity. Physiol Genomics 13:157–167
Bischoff R (1997) Chemotaxis of skeletal muscle satellite cells. Dev Dyn 208:505–515
Blaivas M, Carlson BM (1991) Muscle fiber branching—difference between grafts in old and young rats. Mech Ageing Dev 60:43–53
Bockhold KJ, Rosenblatt JD, Partridge TA (1998) Aging normal and dystrophic mouse muscle: analysis of myogenicity in cultures of living single fibers. Muscle Nerve 21:173–183
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ (2001a) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294:1704–1708
Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD (2001b) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3:1014–1019
Booth FW, Kelso JR (1973) Production of rat muscle atrophy by cast fixation. J Appl Physiol 34:404–406
Boppart MD, Aronson D, Gibson L, Roubenoff R, Abad LW, Bean J, Goodyear LJ, Fielding RA (1999) Eccentric exercise markedly increases c-Jun NH2-terminal kinase activity in human skeletal muscle. J Appl Physiol 87:1668–1673
Bourke DL, Ontell M (1984) Branched myofibers in long-term whole muscle transplants: a quantitative study. Anat Rec 209:281–288
Bowtell J, Park DM, Smith K, Cuthbertson DJR, Waddell T, Rennie MJ (2003) Stimulation of human quadriceps protein synthesis after strenous exercise: no effects of varying intensity between 60 and 90% of one repetition maximum (1RM). J Physiol (Lond) 547P:P16
Brockett CL, Morgan DL, Proske U (2001) Human hamstring muscles adapt to eccentric exercise by changing optimum length. Med Sci Sports Exerc 33:783–790
Brooks GA (2005) Governor recalled! Now what? J Physiol (Lond) 568:355
Burke RE (2002) Some unresolved issues in motor unit research. Adv Exp Med Biol 508:171–178
Butterfield TA, Leonard TR, Herzog W (2005) Differential serial sarcomere number adaptations in knee extensor muscles of rats is contraction type dependent. J Appl Physiol 99:1352–1358
Caiozzo VJ, Baker MJ, Huang K, Chou H, Wu YZ, Baldwin KM (2003) Single-fiber myosin heavy chain polymorphism: how many patterns and what proportions? Am J Physiol Regul Integr Comp Physiol 285:R570–R580
Canepari M, Rossi R, Pellegrino MA, Orrell RW, Cobbold M, Harridge S, Bottinelli R (2005) Effects of resistance training on myosin function studied by the in vitro motility assay in young and older men. J Appl Physiol 98:2390–2395
Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S (2001) Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 306:717–726
Chakravarthy MV, Abraha TW, Schwartz RJ, Fiorotto ML, Booth FW (2000a) Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol 3’-kinase/Akt signaling pathway. J Biol Chem 275:35942–35952
Chakravarthy MV, Davis BS, Booth FW (2000b) IGF-I restores satellite cell proliferative potential in immobilized old skeletal muscle. J Appl Physiol 89:1365–1379
Charge SB, Rudnicki MA (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84:209–238
Charge SB, Brack AS, Hughes SM (2002) Aging-related satellite cell differentiation defect occurs prematurely after Ski-induced muscle hypertrophy. Am J Physiol Cell Physiol 283:C1228–C1241
Cheek DB (1985) The control of cell mass and replication. The DNA unit - a personal 20-year study. Early Hum Dev 12:211–239
Chen YW, Nader GA, Baar KR, Fedele MJ, Hoffman EP, Esser KA (2002) Response of rat muscle to acute resistance exercise defined by transcriptional and translational profiling. J Physiol (Lond) 545:27–41
Chesley A, MacDougall JD, Tarnopolsky MA, Atkinson SA, Smith K (1992) Changes in human muscle protein synthesis after resistance exercise. J Appl Physiol 73:1383–1388
Chin ER (2005) Role of Ca2+/calmodulin-dependent kinases in skeletal muscle plasticity. J Appl Physiol 99:414–423
Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ (1995) Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem 270:12109–12116
Cope TC, Sokoloff AJ (1999) Orderly recruitment among motoneurons supplying different muscles. J Physiol (Paris) 93:81–85
Cros N, Tkatchenko AV, Pisani DF, Leclerc L, Leger JJ, Marini JF, Dechesne CA (2001) Analysis of altered gene expression in rat soleus muscle atrophied by disuse. J Cell Biochem 83:508–519
Darr KC, Schultz E (1987) Exercise-induced satellite cell activation in growing and mature skeletal muscle. J Appl Physiol 63:1816–1821
De Luca CJ, LeFever RS, McCue MP, Xenakis AP (1982a) Behaviour of human motor units in different muscles during linearly varying contractions. J Physiol (Lond) 329:113–128
De Luca CJ, LeFever RS, McCue MP, Xenakis AP (1982b) Control scheme governing concurrently active human motor units during voluntary contractions. J Physiol (Lond) 329:129–142
De Luca CJ, Foley PJ, Erim Z (1996) Motor unit control properties in constant-force isometric contractions. J Neurophysiol 76:1503–1516
DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ (1990) Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol 259:E89–E95
Decary S, Mouly V, Hamida CB, Sautet A, Barbet JP, Butler-Browne GS (1997) Replicative potential and telomere length in human skeletal muscle: implications for satellite cell-mediated gene therapy. Hum Gene Ther 8:1429–1438
Denny-Brown D, Pennybacker JB (1938) Fibrillation and fasciculation in voluntary muscle. Brain 61:311–334
Dhawan J, Rando TA (2005) Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol 15:666–673
Dix DJ, Eisenberg BR (1990) Myosin mRNA accumulation and myofibrillogenesis at the myotendinous junction of stretched muscle fibers. J Cell Biol 111:1885–1894
Dunn SE, Burns JL, Michel RN (1999) Calcineurin is required for skeletal muscle hypertrophy. J Biol Chem 274:21908–21912
Dupont Salter AC, Richmond FJ, Loeb GE (2003) Effects of muscle immobilization at different lengths on tetrodotoxin-induced disuse atrophy. IEEE Trans Neural Syst Rehabil Eng 11:209–217
Edgerton VR, Roy RR (1991) Regulation of skeletal muscle fiber size, shape and function. J Biomech 24(Suppl 1):123–133
Erim Z, De Luca CJ, Mineo K, Aoki T (1996) Rank-ordered regulation of motor units. Muscle Nerve 19:563–573
Farthing JP, Chilibeck PD (2003) The effects of eccentric and concentric training at different velocities on muscle hypertrophy. Eur J Appl Physiol 89:578–586
Faulkner JA, Brooks SV, Opiteck JA (1993) Injury to skeletal muscle fibers during contractions: conditions of occurrence and prevention. Phys Ther 73:911–921
Ferrell RE, Conte V, Lawrence EC, Roth SM, Hagberg JM, Hurley BF (1999) Frequent sequence variation in the human myostatin (GDF8) gene as a marker for analysis of muscle-related phenotypes. Genomics 62:203–207
Fielding RA, Manfredi TJ, Ding W, Fiatarone MA, Evans WJ, Cannon JG (1993) Acute phase response in exercise. III. Neutrophil and IL-1β accumulation in skeletal muscle. Am J Physiol 265:R166–R172
Gayeski TE, Connett RJ, Honig CR (1985) Oxygen transport in rest-work transition illustrates new functions for myoglobin. Am J Physiol 248:H914–H921
Gibala MJ, MacDougall JD, Tarnopolsky MA, Stauber WT, Elorriaga A (1995) Changes in human skeletal muscle ultrastructure and force production after acute resistance exercise. J Appl Physiol 78:702–708
Glass DJ (2003a) Molecular mechanisms modulating muscle mass. Trends Mol Med 9:344–350
Glass DJ (2003b) Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol 5:87–90
Glass DJ (2005) Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 37:1974–1984
Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82:373–428
Goldspink G (1985) Malleability of the motor system: a comparative approach. J Exp Biol 115:375–391
Goldspink G (2005) Mechanical signals, IGF-I gene splicing, and muscle adaptation. Physiology 20:232–238
Goldspink G, Scutt A, Martindale J, Jaenicke T, Turay L, Gerlach GF (1991) Stretch and force generation induce rapid hypertrophy and myosin isoform gene switching in adult skeletal muscle. Biochem Soc Trans 19:368–373
Goldspink G, Scutt A, Loughna PT, Wells DJ, Jaenicke T, Gerlach GF (1992) Gene expression in skeletal muscle in response to stretch and force generation. Am J Physiol 262:R356–R363
Gollnick PD, Karlsson J, Piehl K, Saltin B (1974a) Selective glycogen depletion in skeletal muscle fibres of man following sustained contractions. J Physiol (Lond) 241:59–67
Gollnick PD, Piehl K, Saltin B (1974b) Selective glycogen depletion pattern in human muscle fibres after exercise of varying intensity and at varying pedalling rates. J Physiol (Lond) 241:45–57
Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL (2001) Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98:14440–14445
Gordon ES, Gordish Dressman HA, Hoffman EP (2005) The genetics of muscle atrophy and growth: the impact and implications of polymorphisms in animals and humans. Int J Biochem Cell Biol 37:2064–2074
Goto M, Terada S, Kato M, Katoh M, Yokozeki T, Tabata I, Shimokawa T (2000) cDNA cloning and mRNA analysis of PGC-1 in epitrochlearis muscle in swimming-exercised rats. Biochem Biophys Res Commun 274:350–354
Griffin GE, Williams PE, Goldspink G (1971) Region of longitudinal growth in striated muscle fibres. Nat New Biol 232:28–29
Grimby L (1984) Firing properties of single human motor units during locomotion. J Physiol (Lond) 346:195–202
Haddad F, Adams GR (2002) Selected contribution: acute cellular and molecular responses to resistance exercise. J Appl Physiol 93:394–403
Hall ZW, Ralston E (1989) Nuclear domains in muscle cells. Cell 59:771–772
Hameed M, Orrell RW, Cobbold M, Goldspink G, Harridge SD (2003) Expression of IGF-I splice variants in young and old human skeletal muscle after high resistance exercise. J Physiol (Lond) 547:247–254
Hanzlikova V, Mackova EV, Hnik P (1975) Satellite cells of the rat soleus muscle in the process of compensatory hypertrophy combined with denervation. Cell Tissue Res 160:411–421
Henneman E, Somjen G, Carpenter DO (1965) Functional significance of cell size in spinal motoneurons. J Neurophysiol 28:560–580
Henneman E, Clamann HP, Gillies JD, Skinner RD (1974) Rank order of motoneurons within a pool: law of combination. J Neurophysiol 37:1338–1349
Hentzen ER, Lahey M, Peters D, Mathew L, Barash IA, Friden J, Lieber RL (2006) Stress-dependent and -independent expression of the myogenic regulatory factors and the MARP genes after eccentric contractions in rats. J Physiol (Lond) 570:157–167
Hill M, Goldspink G (2003) Expression and splicing of the insulin-like growth factor gene in rodent muscle is associated with muscle satellite (stem) cell activation following local tissue damage. J Physiol (Lond) 549:409–418
Hill M, Wernig A, Goldspink G (2003) Muscle satellite (stem) cell activation during local tissue injury and repair. J Anat 203:89–99
Hoppeler H, Vogt M, Weibel ER, Fluck M (2003) Response of skeletal muscle mitochondria to hypoxia. Exp Physiol 88:109–119
Hoyt DF, Wickler SJ, Biewener AA, Cogger EA, De La Paz KL (2005) In vivo muscle function vs speed. I. Muscle strain in relation to length change of the muscle-tendon unit. J Exp Biol 208:1175–1190
Hubal MJ, Gordish-Dressman H, Thompson PD, Price TB, Hoffman EP, Angelopoulos TJ, Gordon PM, Moyna NM, Pescatello LS, Visich PS, Zoeller RF, Seip RL, Clarkson PM (2005) Variability in muscle size and strength gain after unilateral resistance training. Med Sci Sports Exerc 37:964–972
Ikeda K, Emoto N, Matsuo M, Yokoyama M (2003) Molecular identification and characterization of a novel nuclear protein whose expression is up-regulated in insulin-resistant animals. J Biol Chem 278:3514–3520
Ingber DE (2003a) Tensegrity I. Cell structure and hierarchical systems biology. J Cell Sci 116:1157–1173
Ingber DE (2003b) Tensegrity II. How structural networks influence cellular information processing networks. J Cell Sci 116:1397–1408
Isfort RJ, Hinkle RT, Jones MB, Wang F, Greis KD, Sun Y, Keough TW, Anderson NL, Sheldon RJ (2000) Proteomic analysis of the atrophying rat soleus muscle following denervation. Electrophoresis 21:2228–2234
Isfort RJ, Wang F, Greis KD, Sun Y, Keough TW, Bodine SC, Anderson NL (2002a) Proteomic analysis of rat soleus and tibialis anterior muscle following immobilization. J Chromatogr B Analyt Technol Biomed Life Sci 769:323–332
Isfort RJ, Wang F, Greis KD, Sun Y, Keough TW, Farrar RP, Bodine SC, Anderson NL (2002b) Proteomic analysis of rat soleus muscle undergoing hindlimb suspension-induced atrophy and reweighting hypertrophy. Proteomics 2:543–550
Ivey FM, Roth SM, Ferrell RE, Tracy BL, Lemmer JT, Hurlbut DE, Martel GF, Siegel EL, Fozard JL, Jeffrey Metter E, Fleg JL, Hurley BF (2000) Effects of age, gender, and myostatin genotype on the hypertrophic response to heavy resistance strength training. J Gerontol A Biol Sci Med Sci 55:M641–M648
Jackman RW, Kandarian SC (2004) The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 287:C834–843
Jagoe RT, Lecker SH, Gomes M, Goldberg AL (2002) Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. Faseb J 16:1697–1712
Kadi F, Eriksson A, Holmner S, Butler-Browne GS, Thornell LE (1999a) Cellular adaptation of the trapezius muscle in strength-trained athletes. Histochem Cell Biol 111:189–195
Kadi F, Eriksson A, Holmner S, Thornell LE (1999b) Effects of anabolic steroids on the muscle cells of strength-trained athletes. Med Sci Sports Exerc 31:1528–1534
Kadi F, Schjerling P, Andersen LL, Charifi N, Madsen JL, Christensen LR, Andersen JL (2004) The effects of heavy resistance training and detraining on satellite cells in human skeletal muscles. J Physiol (Lond) 558:1005–1012
Kandarian SC, Jackman RW (2006) Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 33:155–165
Kelley G (1996) Mechanical overload and skeletal muscle fiber hyperplasia: a meta-analysis. J Appl Physiol 81:1584–1588
Kemp TJ, Sadusky TJ, Saltisi F, Carey N, Moss J, Yang SY, Sassoon DA, Goldspink G, Coulton GR (2000) Identification of Ankrd2, a novel skeletal muscle gene coding for a stretch-responsive ankyrin-repeat protein. Genomics 66:229–241
Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR (2002) The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111:943–955
Kraemer WJ, Ratamess NA (2004) Fundamentals of resistance training: progression and exercise prescription. Med Sci Sports Exerc 36:674–688
Kukulka CG, Clamann HP (1981) Comparison of the recruitment and discharge properties of motor units in human brachial biceps and adductor pollicis during isometric contractions. Brain Res 219:45–55
Kuo H, Chen J, Ruiz-Lozano P, Zou Y, Nemer M, Chien KR (1999) Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development 126:4223–4234
Lai KM, Gonzalez M, Poueymirou WT, Kline WO, Na E, Zlotchenko E, Stitt TN, Economides AN, Yancopoulos GD, Glass DJ (2004) Conditional activation of Akt in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol 24:9295–9304
Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, Gunnarsson LG, Hughes SM, Marchand S, Sejersen T, Richard I, Edstrom L, Ehler E, Udd B, Gautel M (2005) The kinase domain of titin controls muscle gene expression and protein turnover. Science 308:1599–1603
Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R (2002) Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem 277:49831–49840
Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ (2005) Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 280:2737–2744
Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. Faseb J 18:39–51
Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM (2002) Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 418:797–801
Long YC, Widegren U, Zierath JR (2004) Exercise-induced mitogen-activated protein kinase signalling in skeletal muscle. Proc Nutr Soc 63:227–232
Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA (2003) Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. Faseb J 17:2299–2301
Lynn R, Morgan DL (1994) Decline running produces more sarcomeres in rat vastus intermedius muscle fibers than does incline running. J Appl Physiol 77:1439–1444
Lynn R, Talbot JA, Morgan DL (1998) Differences in rat skeletal muscles after incline and decline running. J Appl Physiol 85:98–104
MacDougall JD, Sale DG, Alway SE, Sutton JR (1984) Muscle fiber number in biceps brachii in bodybuilders and control subjects. J Appl Physiol 57:1399–1403
MacDougall JD, Gibala MJ, Tarnopolsky MA, MacDonald JR, Interisano SA, Yarasheski KE (1995) The time course for elevated muscle protein synthesis following heavy resistance exercise. Can J Appl Physiol 20:480–486
Machida S, Spangenburg EE, Booth FW (2003) Forkhead transcription factor FoxO1 transduces insulin-like growth factor’s signal to p27Kip1 in primary skeletal muscle satellite cells. J Cell Physiol 196:523–531
Mason SD, Howlett RA, Kim MJ, Olfert IM, Hogan MC, McNulty W, Hickey RP, Wagner PD, Kahn CR, Giordano FJ, Johnson RS (2004) Loss of skeletal muscle HIF-1α results in altered exercise endurance. PLoS Biol 2:e288
Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495
McCall GE, Byrnes WC, Dickinson A, Pattany PM, Fleck SJ (1996) Muscle fiber hypertrophy, hyperplasia, and capillary density in college men after resistance training. J Appl Physiol 81:2004–2012
McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R (2003) Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol 162:1135–1147
McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC (2002) Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol 157:125–136
McKinnell IW, Parise G, Rudnicki MA (2005) Muscle stem cells and regenerative myogenesis. Curr Top Dev Biol 71:113–130
McKoy G, Hou Y, Yang SY, Vega Avelaira D, Degens H, Goldspink G, Coulton GR (2005) Expression of Ankrd2 in fast and slow muscles and its response to stretch are consistent with a role in slow muscle function. J Appl Physiol 98:2337–2343
McNally EM (2004) Powerful genes - myostatin regulation of human muscle mass. N Engl J Med 350:2642–2644
McPherron AC, Lee SJ (1997) Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA 94:12457–12461
McPherron AC, Lawler AM, Lee SJ (1997) Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387:83–90
Michel RN, Dunn SE, Chin ER (2004) Calcineurin and skeletal muscle growth. Proc Nutr Soc 63:341–349
Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S (2003) The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol 333:951–964
Milner-Brown HS, Stein RB, Yemm R (1973) The orderly recruitment of human motor units during voluntary isometric contractions. J Physiol (Lond) 230:359–370
Miro O, Pedrol E, Cebrian M, Masanes F, Casademont J, Mallolas J, Grau JM (1997) Skeletal muscle studies in patients with HIV-related wasting syndrome. J Neurol Sci 150:153–159
Monster AW, Chan H (1977) Isometric force production by motor units of extensor digitorum communis muscle in man. J Neurophysiol 40:1432–1443
Moore DR, Burgomaster KA, Schofield LM, Gibala MJ, Sale DG, Phillips SM (2004) Neuromuscular adaptations in human muscle following low intensity resistance training with vascular occlusion. Eur J Appl Physiol 92:399–406
Morgan DL (1990) New insights into the behavior of muscle during active lengthening. Biophys J 57:209–221
Morgan DL, Proske U (2004) Popping sarcomere hypothesis explains stretch-induced muscle damage. Clin Exp Pharmacol Physiol 31:541–545
Morgan DL, Talbot JA (2002) The addition of sarcomeres in series is the main protective mechanism following eccentric exercise. J Mech Med Biol 2:421–431
Moss FP, Leblond CP (1971) Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec 170:421–435
Musaro A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N (2001) Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 27:195–200
Nader GA (2005) Molecular determinants of skeletal muscle mass: getting the “AKT” together. Int J Biochem Cell Biol 37:1985–1996
Nardone A, Schieppati M (1988) Shift of activity from slow to fast muscle during voluntary lengthening contractions of the triceps surae muscles in humans. J Physiol (Lond) 395:363–381
Nardone A, Romano C, Schieppati M (1989) Selective recruitment of high-threshold human motor units during voluntary isotonic lengthening of active muscles. J Physiol (Lond) 409:451–471
Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN (2000) Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem 275:4545–4548
Norrbom J, Sundberg CJ, Ameln H, Kraus WE, Jansson E, Gustafsson T (2004) PGC-1alpha mRNA expression is influenced by metabolic perturbation in exercising human skeletal muscle. J Appl Physiol 96:189–194
Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino S (2002) A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc Natl Acad Sci USA 99:9213–9218
Paul AC (2001) Muscle length affects the architecture and pattern of innervation differently in leg muscles of mouse, guinea pig, and rabbit compared to those of human and monkey muscles. Anat Rec 262:301–309
Paul AC, Rosenthal N (2002) Different modes of hypertrophy in skeletal muscle fibers. J Cell Biol 156:751–760
Pette D, Staron RS (2000) Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech 50:500–509
Phillips SM, Tipton KD, Aarsland A, Wolf SE, Wolfe RR (1997) Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am J Physiol 273:E99–E107
Pilegaard H, Saltin B, Neufer PD (2003) Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol (Lond) 546:851–858
Prado LG, Makarenko I, Andresen C, Kruger M, Opitz CA, Linke WA (2005) Isoform diversity of giant proteins in relation to passive and active contractile properties of rabbit skeletal muscles. J Gen Physiol 126:461–480
Rennie MJ, Wackerhage H, Spangenburg EE, Booth FW (2004) Control of the size of the human muscle mass. Annu Rev Physiol 66:799–828
Richardson RS, Noyszewski EA, Kendrick KF, Leigh JS, Wagner PD (1995) Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J Clin Invest 96:1916–1926
Richardson RS, Newcomer SC, Noyszewski EA (2001) Skeletal muscle intracellular PO2 assessed by myoglobin desaturation: response to graded exercise. J Appl Physiol 91:2679–2685
Rosenblatt JD, Parry DJ (1992) Gamma irradiation prevents compensatory hypertrophy of overloaded mouse extensor digitorum longus muscle. J Appl Physiol 73:2538–2543
Roth SM, Martel GF, Ivey FM, Lemmer JT, Tracy BL, Metter EJ, Hurley BF, Rogers MA (2001) Skeletal muscle satellite cell characteristics in young and older men and women after heavy resistance strength training. J Gerontol A Biol Sci Med Sci 56:B240–B247
Sakamoto K, Aschenbach WG, Hirshman MF, Goodyear LJ (2003) Akt signaling in skeletal muscle: regulation by exercise and passive stretch. Am J Physiol Endocrinol Metab 285:E1081–E1081
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117:399–412
Sartorelli V, Fulco M (2004) Molecular and cellular determinants of skeletal muscle atrophy and hypertrophy. Sci STKE 2004:re11
Schiaffino S, Bormioli SP, Aloisi M (1976) The fate of newly formed satellite cells during compensatory muscle hypertrophy. Virchows Arch B Cell Pathol 21:113–118
Schiaffino S, Bormioli SP, Aloisi M (1979) Fibre branching and formation of new fibres during compensatory muscle hypertrophy. In: Mauro A (ed) Muscle regeneration. Raven, New York, pp 177–188
Schmalbruch H, Lewis DM (2000) Dynamics of nuclei of muscle fibers and connective tissue cells in normal and denervated rat muscles. Muscle Nerve 23:617–626
Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ (2004) Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350:2682–2688
Schultz E, Gibson MC, Champion T (1978) Satellite cells are mitotically quiescent in mature mouse muscle: an EM and radioautographic study. J Exp Zool 206:451–456
Semsarian C, Wu MJ, Ju YK, Marciniec T, Yeoh T, Allen DG, Harvey RP, Graham RM (1999) Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signalling pathway. Nature 400:576–581
Sheehan SM, Allen RE (1999) Skeletal muscle satellite cell proliferation in response to members of the fibroblast growth factor family and hepatocyte growth factor. J Cell Physiol 181:499–506
Shepstone TN, Tang JE, Dallaire S, Schuenke MD, Staron RS, Phillips SM (2005) Short-term high- vs. low-velocity isokinetic lengthening training results in greater hypertrophy of the elbow flexors in young men. J Appl Physiol 98:1768–1776
Snow MH (1990) Satellite cell response in rat soleus muscle undergoing hypertrophy due to surgical ablation of synergists. Anat Rec 227:437–446
Spangenburg EE, Booth FW (2003) Molecular regulation of individual skeletal muscle fibre types. Acta Physiol Scand 178:413–424
St-Amand J, Okamura K, Matsumoto K, Shimizu S, Sogawa Y (2001) Characterization of control and immobilized skeletal muscle: an overview from genetic engineering. Faseb J 15:684–692
Stein T, Schluter M, Galante A, Soteropoulos P, Tolias P, Grindeland R, Moran M, Wang T, Polansky M, Wade C (2002) Energy metabolism pathways in rat muscle under conditions of simulated microgravity. J Nutr Biochem 13:471–478
Stevenson EJ, Giresi PG, Koncarevic A, Kandarian SC (2003) Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J Physiol (Lond) 551:33–48
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14:395–403
Tabary JC, Tabary C, Tardieu C, Tardieu G, Goldspink G (1972) Physiological and structural changes in the cat’s soleus muscle due to immobilization at different lengths by plaster casts. J Physiol 224:231–244
Tabary JC, Tardieu C, Tardieu G, Tabary C, Gagnard L (1976) Functional adaptation of sarcomere number of normal cat muscle. J Physiol (Paris) 72:277–291
Takahashi A, Kureishi Y, Yang J, Luo Z, Guo K, Mukhopadhyay D, Ivashchenko Y, Branellec D, Walsh K (2002) Myogenic Akt signaling regulates blood vessel recruitment during myofiber growth. Mol Cell Biol 22:4803–4814
Takarada Y, Sato Y, Ishii N (2002) Effects of resistance exercise combined with vascular occlusion on muscle function in athletes. Eur J Appl Physiol 86:308–314
Takarada Y, Tsuruta T, Ishii N (2004) Cooperative effects of exercise and occlusive stimuli on muscular function in low-intensity resistance exercise with moderate vascular occlusion. Jpn J Physiol 54:585–592
Talmadge RJ, Roy RR, Edgerton VR (1996) Distribution of myosin heavy chain isoforms in non-weight-bearing rat soleus muscle fibers. J Appl Physiol 81:2540–2546
Tanji J, Kato M (1973) Recruitment of motor units in voluntary contraction of a finger muscle in man. Exp Neurol 40:759–770
Tardieu C, Tabary JC, Tabary C, Huet de la Tour E (1977) Comparison of the sarcomere number adaptation in young and adult animals. Influence of tendon adaptation. J Physiol (Paris) 73:1045–1055
Tardieu C, Tabary JC, Tabary C, Tardieu G (1982) Adaptation of connective tissue length to immobilization in the lengthened and shortened positions in cat soleus muscle. J Physiol (Paris) 78:214–220
Tatsumi R, Allen RE (2004) Active hepatocyte growth factor is present in skeletal muscle extracellular matrix. Muscle Nerve 30:654–658
Tatsumi R, Anderson JE, Nevoret CJ, Halevy O, Allen RE (1998) HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev Biol 194:114–128
Tatsumi R, Sheehan SM, Iwasaki H, Hattori A, Allen RE (2001) Mechanical stretch induces activation of skeletal muscle satellite cells in vitro. Exp Cell Res 267:107–114
Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE (2002) Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol Biol Cell 13:2909–2918
Telley IA, Denoth J, Stussi E, Pfitzer G, Stehle R (2006a) Half-sarcomere dynamics in myofibrils during activation and relaxation studied by tracking fluorescent markers. Biophys J 90:514–530
Telley IA, Stehle R, Ranatunga KW, Pfitzer G, Stussi E, Denoth J (2006b) Dynamic behaviour of half-sarcomeres during and after stretch in activated psoas myofibrils: sarcomere asymmetry but no “sarcomere popping”. J Physiol (Lond) 573:173–185
Thomason DB, Booth FW (1990) Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol 68:1–12
Thomis MA, Huygens W, Heuninckx S, Chagnon M, Maes HH, Claessens AL, Vlietinck R, Bouchard C, Beunen GP (2004) Exploration of myostatin polymorphisms and the angiotensin-converting enzyme insertion/deletion genotype in responses of human muscle to strength training. Eur J Appl Physiol 92:267–274
Thompson PD, Moyna N, Seip R, Price T, Clarkson P, Angelopoulos T, Gordon P, Pescatello L, Visich P, Zoeller R, Devaney JM, Gordish H, Bilbie S, Hoffman EP (2004) Functional polymorphisms associated with human muscle size and strength. Med Sci Sports Exerc 36:1132–1139
Tidball JG (2005) Mechanical signal transduction in skeletal muscle growth and adaptation. J Appl Physiol 98:1900–1908
Toigo M, Donohoe S, Sperrazzo G, Jarrold B, Wang F, Hinkle R, Dolan E, Isfort RJ, Aebersold R (2005) ICAT-MS-MS time course analysis of atrophying mouse skeletal muscle cytosolic subproteome. Mol BioSyst 1:229–241
Vedsted P, Blangsted AK, Sogaard K, Orizio C, Sjogaard G (2006) Muscle tissue oxygenation, pressure, electrical, and mechanical responses during dynamic and static voluntary contractions. Eur J Appl Physiol 96:165–177
Vierck J, O’Reilly B, Hossner K, Antonio J, Byrne K, Bucci L, Dodson M (2000) Satellite cell regulation following myotrauma caused by resistance exercise. Cell Biol Int 24:263–272
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2:489–501
Vollestad NK, Blom PC (1985) Effect of varying exercise intensity on glycogen depletion in human muscle fibres. Acta Physiol Scand 125:395–405
Vollestad NK, Vaage O, Hermansen L (1984) Muscle glycogen depletion patterns in type I and subgroups of type II fibres during prolonged severe exercise in man. Acta Physiol Scand 122:433–441
Vollestad NK, Tabata I, Medbo JI (1992) Glycogen breakdown in different human muscle fibre types during exhaustive exercise of short duration. Acta Physiol Scand 144:135–141
Wada KI, Katsuta S, Soya H (2003) Natural occurrence of myofiber cytoplasmic enlargement accompanied by decrease in myonuclear number. Jpn J Physiol 53:145–150
Wadley GD, Lee-Young RS, Canny BJ, Wasuntarawat C, Chen ZP, Hargreaves M, Kemp BE, McConell GK (2006) Effect of exercise intensity and hypoxia on skeletal muscle AMPK signaling and substrate metabolism in humans. Am J Physiol Endocrinol Metab 290:E694–E702
Wagers AJ, Conboy IM (2005) Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 122:659–667
Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM (2003) Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 113:159–170
Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM (2004) Regulation of muscle fiber type and running endurance by PPARδ. PLoS Biol 2:e294
Wenger RH, Stiehl DP, Camenisch G (2005) Integration of oxygen signaling at the consensus HRE. Sci STKE 2005:re12
Widegren U, Wretman C, Lionikas A, Hedin G, Henriksson J (2000) Influence of exercise intensity on ERK/MAP kinase signalling in human skeletal muscle. Pflugers Arch 441:317–322
Williams PE, Goldspink G (1971) Longitudinal growth of striated muscle fibres. J Cell Sci 9:751–767
Williams PE, Goldspink G (1984) Connective tissue changes in immobilised muscle. J Anat 138(Pt 2):343–350
Winder WW (2001) Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. J Appl Physiol 91:1017–1028
Wittwer M, Fluck M, Hoppeler H, Muller S, Desplanches D, Billeter R (2002) Prolonged unloading of rat soleus muscle causes distinct adaptations of the gene profile. Faseb J 16:884–886
Wozniak AC, Pilipowicz O, Yablonka-Reuveni Z, Greenway S, Craven S, Scott E, Anderson JE (2003) C-Met expression and mechanical activation of satellite cells on cultured muscle fibers. J Histochem Cytochem 51:1437–1445
Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS (2002) Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296:349–352
Yang SY, Goldspink G (2002) Different roles of the IGF-I Ec peptide (MGF) and mature IGF-I in myoblast proliferation and differentiation. FEBS Lett 522:156–160
Yang S, Alnaqeeb M, Simpson H, Goldspink G (1996) Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J Muscle Res Cell Motil 17:487–495
Zehr EP, Sale DG (1994) Ballistic movement: muscle activation and neuromuscular adaptation. Can J Appl Physiol 19:363–378
Zhong H, Roy RR, Siengthai B, Edgerton VR (2005) Effects of inactivity on fiber size and myonuclear number in rat soleus muscle. J Appl Physiol 99:1494–1499
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Toigo, M., Boutellier, U. New fundamental resistance exercise determinants of molecular and cellular muscle adaptations. Eur J Appl Physiol 97, 643–663 (2006). https://doi.org/10.1007/s00421-006-0238-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-006-0238-1