
Abstract
Ovarian adenocarcinomas, like human ovarian surface epithelial cells, form functional tight junctions. Tight junction molecules claudin-3 and claudin-4, which are the receptors of Clostridium perfringens enterotoxin (CPE), are abnormally upregulated in epithelial ovarian cancers of all subtypes including, mucinous cystadenocarcinoma and serous cystadenocarcinoma. Clostridium perfringens enterotoxin may be a novel tumor-targeted therapy for ovarian cancers. In epithelial ovarian cancers, overexpression of epidermal growth factor receptor has been observed and the exogenous ligand EGF induces epithelial–mesenchymal transition in ovarian surface epithelium. Epidermal growth factor (EGF) signaling modulates expression of claudins with changes of fence and barrier functions in various cell types. However, the regulation of tight junctions by EGF in ovarian cancers remains unclear. In the present study, to investigate the mechanisms of the regulation of tight junctions in ovarian cancers, ovarian cancer cell lines mucinous cystadenocarcinoma (MCAS) and serous cystadenocarcinoma (HUOA) were treated with EGF. Epidermal growth factor downregulated claudin-3 in MCAS and claudin-4 in HUOA by inducing degradation of the proteins with changes in structures and functions of tight junctions via the MEK/ERK or PI3K/Akt signaling pathway. In addition, in HUOA but not MCAS, EGF downregulated the cytotoxic effect of CPE via claudin-4. Thus, there were different mechanisms for regulation of claudins by EGF between subtypes of epithelial ovarian cancer cells in vitro. These results indicate that EGF may affect claudins and tight junctional functions in ovarian cancer cells during cancer progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ovarian carcinomas comprise a heterogeneous group of neoplasms, the four most common subtypes being serous, endometrioid, clear cell and mucinous (McCluggage 2011). Ovarian surface epithelium is the most likely cell origin of epithelial ovarian carcinomas (Feeley and Wells 2001). Normal human ovarian surface epithelial cells express and form functional tight junctions both in vivo and vitro (Zhu et al. 2004). It is well known that epithelial ovarian adenocarcinomas form functional tight junctions in vitro (Zhu et al. 2006; Zhu and Sundfeldt 2007). Modulation of the tight junctional barrier in ovarian cancers may be a potential target in the treatment of platinum-resistant tumors (Yoshida et al. 2011).
Tight junctions are the most apical components of intercellular junctional complexes. They inhibit solute and water flow through the paracellular space (termed the “barrier” function) (Gumbiner 1993; Schneeberger and Lynch 1992). They also separate the apical from the basolateral cell surface domains to establish cell polarity (termed the “fence” function) (van Meer et al. 1986; Cereijido et al. 1998). Recent evidence suggests that tight junctions also participate in signal transduction mechanisms that regulate epithelial cell proliferation, gene expression, differentiation and morphogenesis (Matter and Balda 2003). The tight junction is formed by not only the integral membrane proteins claudins, occludin, and JAMs, but also many peripheral membrane proteins (Tsukita et al. 2001; Sawada et al. 2003; Schneeberger and Lynch 2004). Recently, tricellulin was identified at tricellular contacts where there are three epithelial cells and shown to have a barrier function (Ikenouchi et al. 2005). These tight junction proteins are regulated by various cytokines and growth factors via distinct signal transduction pathways (González-Mariscal et al. 2008; Kojima et al. 2009).
The claudin family, which consists of at least 27 members, is solely responsible for forming tight junction strands and these strands are directly involved in both the ‘barrier’ and ‘fence’ functions in epithelial and endothelial cell sheets (Mineta et al. 2011; Tsukita et al. 2001). The claudins have four transmembrane domains and two extracellular loops and the second extracellular loop of claudin-3 and claudin-4 is the receptor of Clostridium perfringens enterotoxin (CPE) (Fujita et al. 2000). This 35-kDa polypeptide CPE binds to its receptor, and then the CPE causes changes in the membrane permeability via a complex formation on the plasma membrane followed by the induction of apoptosis (McClane and Chakrabarti 2004). In ovarian cancers, claudin-4 is frequently overexpressed (Boylan et al. 2011) and is a high-affinity receptor of CPE (Katahira et al. 1997; Fujita et al. 2000). It is anticipated that it may be possible to develop a novel tumor-targeted therapy for ovarian cancers using a claudin-4-targeting molecule (Walther et al. 2011; Cocco et al. 2010).
In some human cancers, including ovarian, breast, pancreatic and prostate cancers, tight junction protein claudins are abnormally regulated and thus promising molecular targets for diagnosis and therapy (Morin 2005; Tsukita et al. 2008). Claudin-3 and claudin-4 are highly upregulated in epithelial ovarian cancers of all subtypes, including mucinous cystadenocarcinoma and serous cystadenocarcinoma (Rangel et al. 2003; Boylan et al. 2011). Expression of claudin-3 and claudin-4 in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity and angiogenic effects (Agarwal et al. 2005; Li et al. 2009).
In epithelial ovarian cancers, overexpression of various epidermal growth factor receptor (EGFR) family members, including EGFR, Her2, and ErbB3, has been reported and anti-EGFR-targeted therapy shows limited clinical activity in ovarian cancers (Sheng and Liu 2011). The exogenous ligand epidermal growth factor (EGF) is one of important growth factors that promote biological responses including cell proliferation, differentiation and migration in normal cells, whereas EGF is linked to malignant transformation in epithelial cancer cells (Price et al. 1996; Tobita et al. 2003; Zuo et al. 2011). Epidermal growth factor induces epithelial–mesenchymal transition (EMT) in ovarian surface epithelium via distinct signaling transduction pathways (Ahmed et al. 2006). Furthermore, EGF signaling modulates expression of claudins with changes of fence and barrier functions in various cell types (Kojima et al. 2004; Singh and Harris 2004; Chen et al. 2005; Flores-Benítez et al. 2007; Singh et al. 2007; Ikari et al. 2009; Ikari et al. 2011). However, the regulation of tight junctions by EGF in ovarian cancers remains unclear.
In the present study, to investigate the detailed mechanisms of regulation of tight junctions in ovarian cancers, ovarian cancer cell lines MCAS (a model of mucinous cystadenocarcinoma) and HUOA (a model of serous cystadenocarcinoma) were treated with EGF and the changes in expression of tight junction proteins, including claudin-3 and claudin-4, the structures and the functions of the fence and barrier, and the cytotoxic effects of CPE via signal transduction pathways were examined.
Materials and methods
Antibodies, activators and inhibitors
Recombinant human EGF and transforming growth factor-β1 (TGF-β1) were purchased from PeproTech EC (London, UK). Rabbit polyclonal anti-claudin-1, -3, and -4, anti-occludin, anti-tricellulin (c-term), and anti-phospho-threonine antibodies and mouse monoclonal anti-claudin-1 and -4 antibodies were obtained from Zymed Laboratories (San Francisco, CA). Rabbit polyclonal anti-phospho-MAPK, anti-Akt, anti-Snail and anti-EGFR antibodies and a mouse monoclonal phospho-Akt antibody were purchased from Cell Signaling (Beverly, MA). A rabbit polyclonal anti-ERK 1/2 antibody was purchased from Promega Corporation (Madison, WI). A rabbit polyclonal anti-actin antibody and cycloheximide were purchased from Sigma-Aldrich Inc. (St. Louis, MO). Alexa 488 (green)-conjugated anti-rabbit IgG and Alexa594 (red)-conjugated anti-mouse IgG antibodies were purchased from Molecular Probes, Inc. (Eugene, OR). Inhibitors of EGF receptor (AG1478), TGF-β receptor I kinase, MAPK (U0126), PI3K (LY294002), p38 MAPK (SB203580), JNK (SP600125), pan-PKC (GF109203X), COX-1 (FR122047) and COX-2, as well as prostaglandin E2 (PGE2) were purchased from Calbiochem-Novabiochem Corporation (San Diego, CA).
Immunohistochemical analysis
Immunohistochemical analysis was performed to evaluate the expression and distribution of claudin-1, -3 and -4 in a total of 26 ovarian cancer tissues, 13 cases of searous cystadenocarcinoma and 13 cases of mucinous cystadenocarcinoma. Deparaffinized tissue sections were immersed in 10 mmol/L sodium citrate buffer (pH 6.0) and autoclaved for antigen retrieval. Endogenous peroxidase activity was blocked using methanol containing 0.03 % H2O2. After incubation with blocking buffer (0.01 mol/L PBS containing 5 % bovine serous albumin, Sigma Co., Tokyo, Japan), the sections were incubated with rabbit polyclonal claudin-1, -3 and -4 antibodies (1:100 dilution) at 4 °C. After gentle rinsing with 0.05 mol/L of Tris–HCl, the sections were incubated with a biotinylated secondary antibody (LSAB2 Kit, Dako A/S, Copenhagen, Denmark) for 30 min. Next, the sections were incubated with horseradish peroxidase-conjugated streptavidin reagent (LSAB2 Kit, Dako A/S) for 30 min. Chromogenic fixation was performed for 5 min in a solution of 3-amino-9-ethyl carbazole substrate chromogen (Dako). The sections were counterstained with Mayer’s hematoxylin. Staining was considered positive only when localized to the cell membrane. Staining extent was scored on a scale of 1 to 3, as follows: 0 no staining; 1 staining of 25–50 %; 2 staining of 51–75 %; 3 staining of 76–100 % of cancer cells, and the tumors were divided into negative (0) and positive (1–3) groups.
Cultures of cell lines and treatment
Human ovarian cancer cell lines MCAS and HUOA, were kindly obtained from Dr. Yoshiro Kidera (Saga University, Saga, Japan) (Kidera et al. 1985) and Dr. Isamu Ishiwata (Obstetrics and Gynecologic Hospital, Ibaraki, Japan) (Ishiwata et al. 1987), respectively, were maintained with RPMI-1640 (Sigma-Aldrich) supplemented with 10 % dialyzed fetal bovine serum (FBS, Invitrogen; Carlsbad, CA). The medium contained 100 U/ml penicillin and 100 μg/ml streptomycin and these cells were plated on 35- and 60-mm culture dishes (Corning Glass Works, Corning, NY) that were coated with rat tail collagen (500 μg of dried tendon/ml in 0.1 % acetic acid), and incubated in a humidified 5 % CO2 incubator at 37 °C.
These cells grown to subconfluence were treated with 100 ng/ml EGF and 100 ng/ml TGF-β1 for 24 h after pretreatment with 10 μM EGFR inhibitor AG1478 and 10 μM TGF-βR inhibitor for 30 min. The cells were also treated with 100 ng/ml EGF for 24 h after being pretreated with 10 μM U0126, 10 μM LY294002, 10 μM SB203580, 10 μM SP600125, 10 μM GF109203X, 10 μM PGE2, 10 μM COX-1 inhibitor and 10 μM COX-2 inhibitor II for 30 min. Furthermore, in the presence of 20 or 50 μM cycloheximide, a translation inhibitor, the protein levels of claudin-1, -3, and -4, were measured in a time-dependent manner.
RNA isolation, reverse transcription polymerase chain reaction (RT-PCR) analysis and real-time PCR analysis
Total RNA was extracted and purified using TRIzol (Invitrogen, Carlsbad, CA). One microgram of total RNA was reverse-transcribed into cDNA using a mixture of oligo (dT) and Superscript II reverse transcriptase according to the manufacturer’s recommendations (Invitrogen). Synthesis of each cDNA was performed in a total volume of 20 μl for 50 min at 42 °C and terminated by incubation for 15 min at 70 °C. PCR was performed in a 20 μl total mixture containing 100 pM primer pairs, 1.0 μl of the 20 μl total RT product, PCR buffer, dNTPs and Taq DNA polymerase according to the manufacturer’s recommendations (Takara, Kyoto, Japan). Amplifications were for 25–40 cycles depending on the PCR primer pair with cycle times of 15 s at 96 °C, 30 s at 55 °C and 60 s at 72 °C. Final elongation time was 7 min at 72 °C. Seven microliters of the total 20 μl PCR product was analyzed by 1 % agarose gel electrophoresis with ethidium bromide staining and standardized using a GeneRuler ™ 100 bp DNA ladder (Fermentas, ON, Canada). The PCR primers used for claudin-1, 3, 4, 7, 8, 9, 12, 15, 18, tricellulin, occludin, JAM, ZO-1, ZO-2 and glutaraldehyde-3-phosphate dehydrogenase (G3PDH) are indicated in Table 1.
Real-time PCR detection was performed using a TaqMan Gene Expression Assay kit with a StepOnePlus™ real-time PCR system (Applied Biosystems, Foster City, CA). The amount of 18S ribosomal RNA (rRNA) (Hs99999901) mRNA in each sample was used to standardize the quantity of the following mRNAs: claudin-1 (Hs00221623), -3 (Hs00265816), and -4 (Hs00533616). The relative mRNA-expression levels between the control and treated samples were calculated by the difference of the threshold cycle [comparative C T (∆∆C T) method] and presented as the average of triplicate experiments with a 95 % confidence interval.
Immunocytostaining
The cells were grown on 35-mm glass-base dishes (Iwaki, Chiba, Japan) coated with rat tail collagen and incubated with 10 % FBS. Then they were fixed with cold acetone and ethanol (1:1) at 20 °C for 10 min. After rinsing in PBS, the sections and the cells were incubated with a polyclonal anti-claudin-3 antibody (1:100) or monoclonal anti-claudin-1, and -4 antibodies (1:100) at room temperature (RT) for 1 h and then with Alexa 488 (green)-conjugated anti-rabbit IgG or Alexa 584 (red)-conjugated anti-mouse IgG antibodies (1:200) at RT for 1 h. DAPI (Sigma-Aldrich) was used for counterstaining of nuclei in the cells. The specimens were examined using an epifluorescence microscope (Olympus, Tokyo, Japan) and a confocal laser scanning microscope (LSM510; Carl Zeiss, Jena, Germany).
Western blot analysis
For Western blotting of total cell lysates, the dishes were washed with PBS and 400 μl of sample buffer (1 mM NaHCO3 and 2 mM phenylmethylsulfonylfluoride) was added to 60 mm culture dishes. The cells were scraped and collected in microcentrifuge tubes and then sonicated for 10 s. The protein concentrations of samples were determined using a BCA Protein Assay Reagent Kit (Pierce Chemical Co., Rockford, IL). Aliquots of 15 μg of protein/lane for each sample were separated by electrophoresis in 4–20 % SDS polyacrylamide gels (Cosmo Bio Co., Tokyo, Japan). After electrophoretic transfer to nitrocellulose membranes (Immobilon; Millipore, Billerica, MA), the membranes were saturated with blocking buffer [Tris-buffered saline (TBS) with 0.1 % Tween 20 and 4 % skim milk] for 30 min at room temperature and incubated with polyclonal anti-claudin-1, -3, and -4, anti-tricellulin, anti-occludin, anti-phospho-MAPK, anti-ERK 1/2, anti-Akt, anti-EGFR and anti-Snail antibodies, and a monoclonal phospho-Akt antibody for 1 h at room temperature. Then the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Dako A/S) at room temperature for 1 h. The immunoreactive bands were detected using an ECL Western blotting analysis system (GE Healthcare, Little Chalfont, UK).
Immunoprecipitation
The dishes were washed with PBS twice, and 300 μl of NP-40 lysis buffer (50 mM Tris–HCl, 2 % NP-40, 0.25 mM Na-deoxycholate, 150 mM NaCl, 2 mM EGTA, 0.1 mM Na3VO4, 10 mM NaF, and 2 mM PMSF) was added to 60 mm dishes. The cells were scraped and collected in microcentrifuge tubes and then sonicated for 10 s. Cell lysates were incubated with protein A-Sepharose CL-4B (Pharmacia LKB Biotechnology, Inc., Uppsala, Sweden) for 1 h at 4 °C and then clarified by centrifugation at 15,000×g for 10 min. The supernatants were incubated with polyclonal anti-claudin-1, -3, and -4 antibodies bound to protein A-Sepharose CL-4B overnight at 4 °C. After incubation, immunoprecipitates were washed extensively with the same lysis buffer and then subjected to Western blot analysis using anti-claudin-1, -3 and -4, and anti-phospho-threonine antibodies.
Freeze-fracture analysis
The cells grown on 60 mm dishes were centrifuged into pellets and then immersed in 40 % glycerin solution after fixation in 2.5 % glutaraldehyde/0.1 M PBS, pH 7.3. The specimens mounted on a copper stage were frozen in liquid nitrogen, fractured at −150 to 160 °C and replicated by platinum/carbon from an electron beam gun positioned at a 45° angle followed by carbon applied from overhead in a JFD-7000 freeze-fracture device (JEOL Ltd., Tokyo, Japan). After the replicas were thawed, they were floated on filtered 10 % sodium hypochlorite solution for 30 min in Teflon dishes. Then they were washed in distilled water for 30 min, mounted on copper grids, and examined at an acceleration voltage of 100 kV with a JEOL-1200EX transmission electron microscope (JEOL Ltd., Tokyo, Japan).
Diffusion of bodipy-sphingomyelin
For measurement of the tight junctional fence function, we used diffusion of BODIPY-sphingomyelin (Balda et al. 1996) with some modification. Sphingomyelin/BSA complexes (5 nM) were prepared in P buffer (10 nM HEPES, pH 7.4, 1 mM sodium pyruvate, 10 mM glucose, 3 mM CaCl2, and 145 mM NaCl) using BODIPY-FL-sphingomyelin (Molecular Probes) and defatted BSA. Cells plated on glass-bottom microwell plates (Mat Tek Corp., Ashland, MA) were loaded with BODIPY-sphingomyelin/BSA complex for 1 min on ice, after which they were rinsed with cold DMEM and mounted in DMEM on a glass slide. The samples were analyzed by confocal laser scanning microscopy (LSM510; Carl Zeiss, Jena, Germany). All pictures shown were generated within the first 5 min of analysis.
Mesurement of transepithelial electrical resistance (TER)
The cells were cultured to confluence on inner chambers of 12 mm Transwell with 0.4 μm pore-size filters (Corning Life Science). TER was measured using an EVOM voltmeter with an ENDOHM-12 (World Precision Instruments, Sarasota, FL) on a heating plate (Fine, Tokyo, Japan) adjusted to 37 °C. The values were expressed in standard units of ohms per square centimeter and presented as the mean ± SD of triplicate experiments. For calculation, the resistance of blank filters was subtracted from that of filters covered with cells.
CPE treatment and MTT assay
For purification of CPE, the bacterial DNA fragment encoding the full-length CPE gene was PCR amplified (primer 1, 5′-AGA TGT TAA TCA TAT GAT GCT TAG TAA CAA TTT AAA TCC-3′; primer 2, 5′-AGG ATC CTT AAA ATT TTT GAA ATA ATA TTG AAT AAG GG-3′). The PCR products were digested with the restriction enzymes NdeI/BamHI and cloned into an NdeI/BamHI-digested pet 16 (Novagen) expression vector to generate an in-frame NH2-terminus His-tagged CPE expression plasmid, pet 16-(His)5-CPE. His-tagged CPE toxin was prepared from pet 16-(His)5-CPE-transformed Escherichia coli BL-21 (DE 3). Transformed bacteria were grown overnight at 37 °C, after which CPE protein expression was induced for 3 h with 1 mM isopropyl β-d-thio-galactoside. The cells were harvested, resuspended in 20 mM Tris–HCl, pH 7.9, binding buffer, and lysed by sonication. The fusion protein was isolated from the supernatant on an His-Bind column (Novagen). After treatment with ten column volumes of binding buffer and six column volumes of wash buffer, His-tagged CPE was eluted with 0.5 M NaCl, 20 mM Tris–HCl, pH 7.9, and dialyzed (M r 3,500 cutoff dialysis tubing) against PBS overnight. The cell survival was evaluated with the colorimetric assay using an MTT Cell Growth Assay Kit (Millipore, Billerica, MA) according to the manufacturer’s recommendations. The ratio of absorbance was calculated and presented as the mean ± SD of triplicate experiments.
Data analysis
Signals were quantified using Scion Image Beta 4.02 Win (Scion Co.; Frederick, MA). Each set of results shown is representative of at least three separate experiments. Results are given as means ± SE. Differences between groups were tested by ANOVA followed by a post hoc test and an unpaired two-tailed Student’s t test and considered to be significant when p < 0.05.
Results
Expression and distribution of claudin-1, claudin-3 and claudin-4 in human ovarian cancers
Claudin-3 and claudin-4 are highly expressed in ovarian cancers, including mucinous cystadenocarcinoma and serous cystadenocarcinoma (Rangel et al. 2003; Boylan et al. 2011). In the present study, we examined the expression of claudin-3 and claudin-4 compared to claudin-1 in mucinous cystadenocarcinoma and serous cystadenocarcinoma by immunohistochemistry, using formalin-fixed ovarian cancer tissues. Claudin-4 was strongly expressed in 13 of 13 (100 %) cases of both mucinous cystadenocarcinoma and serous cystadenocarcinoma, whereas claudin-3 was expressed in 6 of 13 (46 %) cases of mucinous cystadenocarcinoma and 13 of 13 (100 %) cases of serous cystadenocarcinoma (Fig. 1). Claudin-3 and claudin-4 in both tumors were often heterogeneous and occasionally plaque-like and were expressed at the lateral membranes, with some specimens showing cytoplasmic expression (Fig. 1). Claudin-1 was in part detected in cancer cells of both types (Fig. 1).

Expression and distribution of claudin-1, -3 and -4 in human ovarian cancer tissues. Hematoxylin–eosin staining and immunohistochemical staining for claudin-1, -3 and -4 in ovarian cancer tissues, serous cystadenocarcinoma and mucinous cystadenocarcinoma. Bar 30 μm, mucinous mucinous cystadenocarcinoma, serous serous cystadenocarcinoma, H&E hematoxylin–eosin, CLDN claudin
Expression patterns of claudin-1, claudin-3 and claudin-4 in human ovarian cancer cell lines
To study the regulation of claudin-3 and claudin-4 in ovarian cancers in detail, we first investigated the expression patterns of tight junction molecules in ovarian cancer cell lines MCAS (a model of mucinous cystadenocarcinoma) and HUOA (a model of serous cystadenocarcinoma). In both cancer cell lines, claudin-1, -3, -4, -7, -8, -9, -12, -15 and -18, occludin, tricellulin, JAM-A, ZO-1 and -2 were detected by RT-PCR (Supplemental data 1).
We compared the expression and distribution of claudin-1, claudin-3, and claudin-4 between the cell lines. In Western blotting, the protein level of claudin-3 was higher in HUOA than in MCAS, whereas that of claudin-1 was higher in MCAS than in HUOA. For claudin-4 there was almost no difference between the cell lines (Fig. 2a). In real-time PCR, the difference between the mRNA levels of claudin-1, claudin-3, and claudin-4 was the same as in the results of Western blotting (Fig. 2b). Immunostaining showed that, in both cancer cell lines, claudin-1, claudin-3 and claudin-4 were localized at the apicalmost and basolateral membranes (Fig. 2c).

Expression patterns of claudin-1, -3 and -4 in human ovarian cancer cell lines. Western blotting (a), real-time PCR (b) and immunocytostaining (c) for claudin-1, -3 and -4 in MCAS and HUOA. CLDN claudin, M MCAS, H HUOA, *p < 0.05, **p < 0.01, Bar 40 μm
EGF decreases proteins but not mRNAs of claudin-3 and claudin-4 in ovarian cancer cell lines
Epidermal growth factor and TGF-β affect the expression of some claudins in various cell types (Grände et al. 2002; Flores-Benítez et al. 2007; Samak et al. 2011; Kojima and Sawada 2011). To investigate whether EGF and TGF-β1 affected the expression and distribution of claudin-1, claudin-3 and claudin-4 in human ovarian cancers, MCAS and HUOA were treated with 100 ng/ml EGF or 100 ng/ml TGF-β1 for 24 h. In Western blotting, EGF decreased the proteins of claudin-3 and claudin-4 in MCAS and HUOA, respectively, whereas TGF-β1 decreased claudin-3 protein in MCAS (Fig. 3a). Interestingly, EGF remarkably increased claudin-1 protein in HUOA (Fig. 3a). These changes induced by EGF or TGF-β1 in the cancer cell lines were prevented by pretreatment with 10 μM EGF receptor inhibitor AG1478 and 10 μM TGF-βR inhibitor, respectively (Fig. 3a). In real-time PCR, no change of the mRNAs of claudin-3 and claudin-4 was observed in either cancer cell line treated with EGF, whereas EGF increased claudin-1 mRNA in HUOA (Fig. 3b).

EGF decreases proteins but not mRNAs of claudin-3 and -4 by inducing degradation in ovarian cancer cell lines. a Western blotting for claudin-1, -3, and -4 in MCAS and HUOA treated with 100 ng/ml EGF and 100 ng/ml TGF-β1 for 24 h after pretreatment with or without 10 μM EGFR inhibitor AG1478 or 10 μM TGF-βR inhibitor for 30 min. b Real-time PCR for claudin-1, -3 and -4 in MCAS and HUOA treated with 100 ng/ml EGF for 24 h. **p < 0.01 c Western blotting for claudin-1, -3, and -4 in MCAS in the presence of 20 μM cycloheximide and in HUOA in the presence of 50 μM cycloheximide after treatment with 100 ng/ml EGF for 24 h with or without 10 μM AG1478. CLDN claudin, C control, E EGF, T TGF-β, AG AG1478, Ri TGF-β receptor inhibitor, CHX cycloheximide
EGF induces degradation of claudin-3 and claudin-4 in ovarian cancer cell lines
To elucidate the mechanism of the decrease of proteins but not mRNAs of claudin-3 and claudin-4 caused by the treatment with EGF in MCAS and HUOA, we examined the protein levels of claudin-3 and claudin-4 in a time-dependent manner after treatment with 20 or 50 μM cycloheximide (a translation inhibitor) 24 h after pretreatment with 100 ng/ml EGF. In the presence of cycloheximide, claudin-3 in MCAS and claudin-4 in HUOA treated with EGF, were decreased in a time-dependent manner (Fig. 3c). Epidermal growth factor receptor inhibitor AG1478 prevented the decreases of claudin-3 and claudin-4 of MCAS and HUOA by EGF in the presence of cycloheximide (Fig. 3c). These results indicated that EGF induced degradation of claudin-3 and claudin-4 in MCAS and HUOA.
EGF affects distribution of claudin-3 and claudin-4 and tight junction strands in ovarian cancer cell lines
We investigated the distribution of claudin-3 and claudin-4 in MCAS and HUOA at 24 h after treatment with 100 ng/ml EGF. In MCAS treated with EGF, claudin-3 was localized at the apicalmost regions, whereas it was localized at the apicalmost and basolateral membranes in the control and in cells pretreated with EGF receptor inhibitor AG1478 (Fig. 4a). In HUOA treated with EGF, claudin-4 was decreased at the apicalmost and basolateral membranes compared to the control, and pretreatment with AG1478 prevented the decrease of claudin-4 by EGF (Fig. 4a). In HUOA treated with EGF, claudin-1 was increased at the membranes compared to the control (supplemental data 2).

EGF affects localization of claudin-3 and -4, and tight junction strands in ovarian cancer cell lines. a immunocytostaining for claudin-3 in MCAS and for claudin-4 in HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM EGFR inhibitor AG1478 for 30 min. Bar 40 μm, XY XY-section, Z Z-section. b Freeze-fracture replicas of MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM AG1478 for 30 min. Black bar 500 nm, white bar 200 nm, CLDN claudin, AG AG1478
We performed freeze-fracture analysis to investigate changes of tight junction strands in MCAS and HUOA at 24 h after treatment with 100 ng/ml EGF. In the controls of both cancer cell lines, tight junction strands formed anastomosing networks (Fig. 4b). In both cancer cell lines treated with EGF, the strands were decreased and the change was returned to the control level by pretreatment with AG1478 (Fig. 4b). Interestingly, in HUOA treated with EGF, some tight junction-like strands were also observed in the vesicles (Fig. 4b). It was thought that these vesicles indicated endocytosis of tight junctions as previously reported (Matsuda et al. 2004; Tang 2006).
EGF decreases claudin-3 and claudin-4 via activation of MEK/ERK and PI3K/Akt signaling pathways in ovarian cancer cell lines
To investigate which signal transduction pathways were associated with the decreases of claudin-3 and claudin-4 caused by EGF in MCAS and HUOA, the cells were pretreated with various inhibitors before treatment with 100 ng/ml EGF (supplemental data 3). In MCAS, pretreatment with inhibitors of EGFR (AG1478), MEK/ERK (U0126) and PI3K/Akt (LY294002) prevented the decrease of claudin-3 by EGF (Fig. 5a). In HUOA, pretreatment with inhibitors of EGFR, MEK/ERK, but not PI3K/Akt, prevented the decrease of claudin-4 by EGF (Fig. 5a). In MCAS, upregulation of pMAPK and pAkt was induced by treatment with EGF and prevented by pretreatment with the inhibitors of EGFR and MEK/ERK or the inhibitors of EGFR and PI3K/Akt, respectively (Fig. 5b). In HUOA, upregulation of pMAPK was induced by treatment with EGF and was prevented by pretreatment with the inhibitors of EGFR and MEK/ERK, whereas pAkt was not detected (Fig. 5b).

EGF decreases claudin-3 and -4 via MEK/ERK and/or PI3K/Akt signaling pathways in ovarian cancer cell lines. a Western blotting for claudin-1, -3 and -4 in MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM EGFR inhibitor AG1478, 10 μM MEK/ERK inhibitor U0126 or 10 μM PI3K/Akt inhibitor LY294002 for 30 min. b Western blotting for pMAPK, MAPK, pAkt and Akt in MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM AG1478, 10 μM U0126 or 10 μM LY294002 for 30 min. CLDN claudin, C control, AG AG1478, LY LY294002, pMAPK phospho-MAPK, pAkt phosphor-Akt
EGF modulates phosphorylation of claudin-3 and claudin-4 in ovarian cancer cell lines
The immunoprecipitates of claudin-3 and claudin-4 were analyzed by immunoblotting with antibodies to phosphothreonine and the corresponding claudins. In MCAS treated with 100 ng/ml EGF, threonine-phosphorylated claudin-3 was detected, whereas it was not detected in the control or in the cells pretreated with the inhibitors of EGFR (AG1478), MEK/ERK (U0126) and PI3K/Akt (LY294002) before treatment with EGF (Fig. 6). Threonine-phosphorylated claudin-4 was detected in the control and with all treatments of MCAS (Fig. 6). In HUOA treated with 100 ng/ml EGF, threonine-phosphorylated claudin-4 was not detected, whereas it was detected in the control and in the cells pretreated with the inhibitors of EGFR, MEK/ERK and PI3K/Akt before treatment with EGF (Fig. 6). Threonine-phosphorylated claudin-3 was detected in the control and with all treatments of HUOA (Fig. 6).

EGF modulates phosphorylation of claudin-3 and -4 in ovarian cancer cell lines. Western blotting for phosphothreonine in the immunoprecipitates; use of claudin-3 and -4 antibodies in MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM EGFR inhibitor AG1478, 10 μM MEK/ERK inhibitor U0126 or PI3K/Akt inhibitor LY294002 for 30 min. IP immunoprecipitation, CLDN claudin, C control, AG AG1478, LY LY294002, p-thre phosphothreonine
EGF affects fence function and barrier function
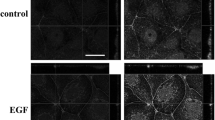
To investigate whether EGF affected the fence function of tight junctions, BODIPY-sphingomyelin diffusion in the membrane was measured in MCAS and HUOA at 24 h after treatment with 100 ng/ml EGF. BODIPY-sphingomyelin was effectively retained in the apical domain in control MCAS and HUOA (Fig. 7a). The BODIPY-sphingomyelin of MCAS after treatment with EGF was diffused through the tight junctions, labeled the basolateral surfaces and appeared to penetrate the cells, and the diffusion was prevented by pretreatment with EGF receptor inhibitor AG1478 (Fig. 7a). In HUOA treated with EGF and pretreated with AG1478 before treatment with EGF, the probe was well retained in the apical domain as in the control (Fig. 7a).

EGF affects fence and barrier functions and the cytotoxic effects of CPE in ovarian cancer cell lines. a Fence function examined by diffusion of labeled BODIPY-sphingomyelin into MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM EGFR inhibitor AG1478 for 30 min. Bar 20 μm. b Barrier function examined by TER values in MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM AG1478 for 30 min. **p < 0.01 versus control at 24 h, ## p < 0.01 versus EGF at 24 h. c The cell viability was measured by MTT assay after treatment with 2 mg/ml CPE for 2 h at 24 h after treatment with 100 ng/ml EGF after pretreatment with or without 10 μM AG1478 for 30 min. **p < 0.01 versus control with CPE, ## p < 0.01 versus EGF. AG AG1478, CPE Clostridium perfringens enterotoxin
The barrier function of tight junctions was measured by TER values in MCAS and HUOA at 24 h after treatment with 100 ng/ml EGF. The TER value of MCAS after treatment with EGF was decreased compared to the control, but was maintained at the control level by pretreatment with AG1478 (Fig. 7b). In HUOA after treatment with EGF, the TER value was conversely increased compared to the control, but was maintained at control level by pretreatment with AG1478 (Fig. 7b).
EGF downregulates the cytotoxic effect of CPE in HUOA not but MCAS
To investigate whether EGF affected the cytotoxic effect of CPE via claudin-3 and claudin-4 in MCAS and HUOA, the cell viability was measured by MTT assay after treatment with 2 mg/ml CPE for 2 h at 24 h after treatment with 100 ng/ml EGF. In control MCAS and HUOA, the cell viability was decreased by treatment with CPE (Fig. 7c). In HUOA, but not MCAS, the cytotoxic effect of CPE was downregulated by treatment with EGF but was maintained at the control level by pretreatment with EGF receptor inhibitor AG1478 (Fig. 7c).
Discussion
In the present study, we demonstrated that EGF downregulated claudin-3 in MCAS (mucinous cystadenocarcinoma) and claudin-4 in HUOA (serous cystadenocarcinoma) by inducing degradation of the proteins with changes in the structures and functions of tight junctions. In addition, in HUOA but not MCAS, EGF downregulated the cytotoxic effect of CPE via claudin-4. The mechanisms of the regulation of claudins by EGF differed between the subtypes of epithelial ovarian cancer cells in vitro.
Epidermal growth factor/EGFR signaling modulates expression of claudins in various cell types (Kojima et al. 2004; Singh and Harris 2004; Chen et al. 2005; Flores-Benítez et al. 2007; Singh et al. 2007; Ikari et al. 2009; Ikari et al. 2011). In polarized Madin-Darby canine kidney (MDCK) II cells, EGF-induced EGFR activation significantly inhibits claudin-2 expression, while simultaneously inducing cellular redistribution and increased expression of claudin-1, claudin-3, and claudin-4 (Singh and Harris 2004). In addition, EGF accelerates clathrin-dependent endocytosis and lysosomal degradation of claudin-2 protein in MDCK-II cells (Ikari et al. 2011). In present study, EGF decreased the protein but not the mRNA of claudin-3 in MCAS and claudin-4 in HUOA and enhanced the decrease in the protein level in the presence of cycloheximide. In HUOA, upregulation of claudin-1 by EGF was also observed at the mRNA level. EGF decreased the numbers of tight junction strands in both cell lines. Furthermore, in freeze-fracture replicas, we also found images of endocytosed vesicles of tight junctions in HUOA treated with EGF (Fig. 4b, arrows). The images of endocytosed vesicles of tight junctions can be observed by transmission electron microscopy during remodeling of tight junctions (Matsuda et al. 2004; Tang 2006). All changes in both cell lines induced by treatment with EGF were prevented by an EGFR inhibitor. These findings suggested that EGF downregulated claudin-3 in MCAS and claudin-4 in HUOA by enhancing degradation of the proteins at the post-translational level.
Epidermal growth factor-activated growth factor receptors induce the activation of signal transduction pathways involving MEK/ERK, p38 MAPK, and PI3K/Akt, and play fundamental roles in the development, proliferation, and differentiation of epithelial cells (Scaltriti and Baselga 2006). Epidermal growth factor decreases claudin-2 and increases claudin-4 via MEK/ERK or PI3K/Akt pathways in MDCK cells (Singh et al. 2007; Ikari et al. 2009). Inhibitors of COX-1 and -2 enhance TER induced by EGF, and PGE2 prevents upregulation of TER and claudin-4 expression by EGF in MDCK cells (Flores-Benítez et al. 2009). Furthermore, it is also known that, in epithelal ovarian cancers, abnormal activity of MEK/ERK and PI3K/Akt pathways due to somatic mutation of the signal molecules may play a crucial role in malignant progression (Kurman and Shih 2011). In the present study, the cells were pretreated with inhibitors of EGFR, MEK/ERK, PI3K/Akt, p38 MAPK, JNK, pan-PKC, COX-1 and COX-2 as well as PGE2 before treatment with EGF. In MCAS, pretreatment with the inhibitors of EGFR, MEK/ERK and PI3K/Akt prevented the decrease of claudin-3 by EGF, and in HUOA, pretreatment with the inhibitors of EGFR, MEK/ERK, but not PI3K/Akt, prevented the decrease of claudin-4 by EGF. In both cell lines, activity of pMAPK was upregulated by EGF, whereas activity of pAkt was upregulated by EGF in MCAS but not HUOA. These findings indicated that EGF modulated expression of claudins in ovarian cancer cells via MEK/ERK or PI3K/Akt pathways and that there were different signal transduction pathways in the subtypes of the cancer cells treated with EGF.
Epidermal growth factor/EGFR signaling modulates fence and barrier functions of tight junctions via changes of claudins (Kojima et al. 2004; Singh and Harris 2004; Flores-Benítez et al. 2007; Singh et al. 2007; Ikari et al. 2009). In primary cultured rat hepatocytes, EGF downregulates the fence function measured by BODIPY-sphingomyelin diffusion with a decrease of claudin-1 (Kojima et al. 2004). In MDCK cells, EGF upregulates the barrier function measured by TER with a decrease of claudin-2 and an increase of claudin-4 (Flores-Benítez et al. 2007; Ikari et al. 2009). In the present study, in MCAS treated with EGF, fence and barrier functions were downregulated with a decrease of claudin-3. In HUOA treated with EGF, only the barrier function was upregulated, with an increase of claudin-1 and a decrease of claudin-4. All changes caused by treatment with EGF were prevented by an EGFR inhibitor. These results indicated that in MCAS, claudin-3 expression might be mainly associated with fence and barrier functions, and in HUOA, claudin-1 might be important for both functions.
On the other hand, claudin phosphorylation has been linked to the assembly and function of tight junction (D’Souza et al. 2007; Aono and Hirai 2008; Banan et al. 2005). Threonine phosphorylation of claudin-3 by cAMP-dependent protein kinase regulates the tight junction barrier function in ovarian cancer cells (D’Souza et al. 2005). In the present study, we examined changes of threonine phosphorylation of claudin-3 and claudin-4 in MCAS and HUOA treated with EGF. In MCAS, but not HUOA, threonine-phosphorylation of claudin-3 was upregulated by treatment with EGF and the change was prevented by inhibitors of EGFR, MEK/ERK and PI3K/Akt, whereas in HUOA, but not MCAS, threonine phosphorylation of claudin-4 was downregulated by treatment with EGF and the change was prevented by inhibitors of EGFR and MEK/ERK. These findings suggested that upregulation of threonine phosphorylation of claudin-3 by treatment with EGF might also associated with the barrier function in MCAS.
Claudin-3 and claudin-4, highly expressed in ovarian cancers, are receptors of CPE and the cytotoxic effects of CPE are thought to be useful as a novel therapeutic tool for ovarian cancers (Walther et al. 2011; Cocco et al. 2010). In the present study, as EGF downregulated claudin-3 in MCAS and claudin-4 in HUOA, we examined whether EGF affected the cytotoxic effects of CPE via claudin-3 and claudin-4. In HUOA, but not MCAS, EGF downregulated the cytotoxic effects of CPE and the change was prevented by an EGFR inhibitor. These results indicated that claudin-4 had higher affinity as a receptor for CPE than claudin-3 as previously reported (Katahira et al. 1997; Fujita et al. 2000), and it is possible that EGF may affect effective therapy targeting for claudin-4 using CPE in the subtype of serous cystadenocarcinoma.
In the present study, claudin-3 and claudin-4 were expressed not only in the apicalmost regions, where are tight junction areas, but also along basolateral membrane in both ovarian cancer cell lines (Fig. 2). It is known that some claudins express along basolateral membranes of some cancer cells (Dhawan et al. 2005; Ladwein et al. 2005; Lanigan et al. 2009; Yamaguchi et al. 2011). In addition, claudin-7 directly interacts with EpCAM at the basolateral membranes, and the EpCAM-claudin-7 complex strongly promotes tumorigenicity (Ladwein et al. 2005; Nübel et al. 2009). Furthermore, we recently reported that in pancreatic cancer cell lines, claudin-4 was expressed not only in the apicalmost regions but also at basolateral membranes, whereas in normal human pancreatic duct epithelail cells (HPDE cells), it was only localized in the apicalmost regions, and CPE led to an acute dose-dependent cytotoxic effect in pancreatic cell lines but not normal HPDE cells (Yamaguchi et al. 2011). Because it is thought that CPE binds to the free second extracellular loop of claudin-4 outside tight junctions and that, in normal HPDE cells, it cannot bind to that of claudin-4 in tight junction areas (Winkler et al. 2009).
In conclusion, in epithelial ovarian cancer cells, EGF modulated claudins and the tight junctional functions via MEK/ERK and PI3K/Akt signaling pathways. However, there were different mechanisms for regulation of claudins by EGF between the subtypes in vitro. Ovarian surface epithelium is the most likely cell origin of epithelial ovarian carcinomas (Feeley and Wells 2001). Normal human ovarian surface epithelial cells express and form functional tight junctions both in vivo and in vitro (Zhu et al. 2004). Epidermal growth factor induces epithelial–mesenchymal transition (EMT) in ovarian surface epithelium via distinct signaling transduction pathways (Ahmed et al. 2006). Continued investigation of the regulation of claudins by EGF using normal human ovarian surface epithelial cells is necessary to determin whether claudins can be used as molecular targets in the diagnosis and therapy for epithelial ovarian cancers.
References
Agarwal R, D’Souza T, Morin PJ (2005) Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased metalloproteinase-2 activity. Cancer Res 65:7378–7385
Ahmed N, Maines-Bandiera S, Quinn MA, Unger WG, Dedhar S, Auersperg N (2006) Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. Am J Physiol Cell Physiol 290:C1532–C1542
Aono S, Hirai Y (2008) Phosphorylation of claudin-4 is required for tight junction formation in a human keratinocyte cell line. Exp Cell Res 314:3326–3339
Balda MS, Whitney JA, Flores C, González S, Cereijido M, Matter K (1996) Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol 134:1031–1049
Banan A, Zhang LJ, Shaikh M, Fields JZ, Choudhary S, Forsyth CB, Farhadi A, Keshavarzian A (2005) θ Isoform of protein kinase C alters barrier function in intestinal epithelium through modulation of distinct claudin isotypes: a novel mechanism for regulation of permeability. J Pharmacol Exp Ther 313:962–982
Boylan KL, Misemer B, Derycke MS, Andersen JD, Harringon KM, Kalloger SE, Gilks CB, Pambuccian SE, Skubitz AP (2011) Claudin 4 is differentially expressed between ovarian cancer subtypes and plays a role in spheroid formation. Int J Mol Sci 12:1334–1358
Cereijido M, Valdés J, Shoshani L, Contreras RG (1998) Role of tight junctions in establishing and maintaining cell polarity. Annu Rev Physiol 60:161–177
Chen SP, Zhou B, Willis BC, Sandoval AJ, Liebler JM, Kim KJ, Ann DK, Crandall ED, Borok Z (2005) Effects of transdifferentiation and EGF on claudin isoform expression in alveolar epithelial cells. J Appl Physiol 98:322–328
Cocco E, Casagrande F, Bellone S, Richter CE, Bellone M, Todeschini P, Holmberg JC, Fu HH, Montagna MK, Mor G, Schwartz PE, Arin-Silasi D, Azoudi M, Rutherford TJ, Abu-Khalaf M, Pecorelli S, Santin AD (2010) Clostridium perfringens enterotoxin carboxy-terminal fragment is a novel tumor-homing peptide for human ovarian cancer. BMC Cancer 10:349–359
D’Souza T, Agawal R, Morin PJ (2005) Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem 280:26233–26240
D’Souza T, Indig FE, Morin PJ (2007) Phosphorylation of claudin-4 by PKCε regulates tight junction barrier function in ovarian cancer cells. Exp Cell Res 313:3364–3375
Dhawan P, Singh AB, Deane NG, No Y, Shiou SR, Schmidt C, Neff J, Washington MK, Beauchamp RD (2005) Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest 115:1765–1776
Feeley KM, Wells M (2001) Precursor lesion of ovarian epithelial malignancy. Histopathology 38:87–95
Flores-Benítez D, Ruiz-Cabrera A, Flores-Maldonado C, Shoshani L, Cereijido M, Contreras RG (2007) Control of tight junctional sealing: role of epidermal growth factor. Am J Physiol Renal Physiol 292:F828–F836
Flores-Benítez D, Rincon-Heredia R, Razgado LF, Larre I, Cereijido M, Contreras RG (2009) Control of tight junctional sealing: role of epidermal growth factor and prostaglandin E2. Am J Physiol Cell Physiol 297:C611–C620
Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tsukita S (2000) Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett 476:258–261
González-Mariscal L, Tapia R, Chamorro D (2008) Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta 1778:729–756
Grände M, Franzen A, Karlsson JO, Ericson LE, Heldin NE, Nilsson M (2002) Transforming growth factor-β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J Cell Sci 115:4227–4236
Gumbiner BM (1993) Breaking through the tight junction barrier. J Cell Biol 123:1631–1633
Ikari A, Atomi K, Takiguchi A, Yamazaki Y, Miwa M, Sugatani J (2009) Epidermal growth factor increases claudin-4 expression mediated by Sp1 elevation in MDCK cells. Biochem Biophys Res Commun 384:306–310
Ikari A, Takiguchi A, Atomi K, Sugatani J (2011) Epidermal growth factor increases clathrin-dependent endocytosis and degradation of claudin-2 protein in MDCK cells. J Cell Physiol 226:2448–2456
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171:939–945
Ishiwata I, Ishiwata C, Kobayashi N, Ishikawa H (1987) Characterization of newly established cell line HUOA from a human ovarian serous cystadenocarcinoma. Act Obstet Gynaecol Jpn 39:831–836 [in Japanese]
Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N (1997) Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J Biol Chem 272:26652–26658
Kidera Y, Yoshimura T, Ohkuma Y, Iwasaka T, Sugimori H (1985) Establishment and characterization of a cell line derived from mucinous cystadenocarcinoma of human ovary. Acta Obstet Gynaecol Jpn 37:1820–1824 [in Japanese]
Kojima T, Sawada N (2011) Expression and function of claudins in hepatocytes. Methods Mol Biol 762:233–244
Kojima T, Yamamoto T, Lan M, Murata M, Takano K, Go M, Ichimiya S, Chiba H, Sawada N (2004) Inhibition of MAP kinase activity moderates changes in expression and function of Cx32 but not claudin-1 during DNA synthesis in primary cultures of rat hepatocytes. Med Electron Microsc 37:101–113
Kojima T, Murata M, Yamamoto T, Lan M, Imamura M, Son S, Takano K, Yamaguchi H, Ito T, Tanaka S, Chiba H, Hirata K, Sawada N (2009) Tight junction proteins and signal transduction pathways in hepatocytes. Histol Histopathol 24:1463–1472
Kurman RJ, Shih IeM (2011) Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm. Hum Pathol 42:918–931
Ladwein M, Pape UF, Schmidt DS, Schnölzer M, Fiedler S, Langbein L, Franke WW, Moldenhauer G, Zöller M (2005) The cell–cell adhesion molecule EpCAM interacts directly with the tight junction protein claudin-7. Exp Cell Res 309:345–357
Lanigan F, McKiernan E, Brennan DJ, Hegarty S, Millikan RC, McBryan J, Jistrom K, Landberg G, Martin F, Duffy MJ, Gallagher WM (2009) Increased claudin-4 expression is associated with poor prognosis and high tumour grade in breast cancer. Int J Cancer 124:2088–2097
Li J, Chigurupati S, Agarwal R, Mughal MR, Mattson MP, Becker KG, Wood WH 3rd, Zhang Y, Morin PJ (2009) Possible angiogenic roles for claudin-4 in ovarian cancer. Cancer Biol Ther 8:1806–1814
Matsuda M, Kubo A, Furuse M, Tsukita S (2004) A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J Cell Sci 117:1247–1257
Matter K, Balda MS (2003) Functional analysis of tight junctions. Methods 30:228–234
McClane BA, Chakrabarti G (2004) New insights into the cytotoxic mechanisms of Clostridium perfringens enterotoxin. Anaerobe 10:107–114
McCluggage WG (2011) Morphological subtypes of ovarian carcinoma: a review with emphasis on new developments and pathogenesis. Pathology 43:420–432
Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, Tamura A, Igarashi M, Endo T, Takeuchi K, Tsukita S (2011) Predicted expansion of the claudin multigene family. FEBS Lett 585:606–612
Morin PJ (2005) Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res 65:9603–9606
Nübel T, Preobraschenski J, Tuncay H, Weiss T, Kuhn S, Ladwein M, Langbein L, Zöller M (2009) Claudin-7 regulates EpCAM-mediated functions in tumor progression. Mol Cancer Res 7:285–299
Price JT, Wilson HM, Haites NE (1996) Epidermal growth factor (EGF) increases the in vitro invasion, motility and adhesion interactions of the primary renal carcinoma cell line, A704. Eur J Cancer 32A:1977–1982
Rangel LB, Agarwal R, D’Souza T, Pizer ES, Alò PL, Lancaster WD, Gregoire L, Schwartz DR, Cho KR, Morin PJ (2003) Tight junction proteins claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin Cancer Res 9:2567–2575
Samak G, Aggarwal S, Rao RK (2011) ERK is involved in EGF-mediated protection of tight junctions, but not adherens junctions, in acetaldehyde-treated Caco-2 cell monolayers. Am J Physiol Gastrointest Liver Physiol 301:G50–G59
Sawada N, Murata M, Kikuchi K, Osanai M, Tobioka H, Kojima T, Chiba H (2003) Tight junctions and human diseases. Med Electron Microsc 36:147–156
Scaltriti M, Baselga J (2006) The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 12:5268–5272
Schneeberger EE, Lynch RD (1992) Structure, function, and regulation of cellular tight junctions. Am J Physiol 262:L647–L661
Schneeberger EE, Lynch RD (2004) The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 286:C1213–C1228
Sheng Q, Liu J (2011) The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br J Cancer 104:1241–1245
Singh AB, Harris RC (2004) Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J Biol Chem 279:3543–3552
Singh AB, Sugimoto K, Dhawan P, Harris RC (2007) Juxtacrine activation of EGFR regulates claudin expression and increases transepithelial resistance. Am J Physiol Cell Physiol 293:C1660–C1668
Tang VW (2006) Proteomic and bioinformatic analysis of epithelial tight junction reveals an unexpected cluster of synaptic molecules. Biol Direct 1:37–66
Tobita K, Kijima H, Dowaki S, Kashiwagi H, Ohtani Y, Oida Y, Yamazaki H, Nakamura M, Ueyama Y, Tanaka M, Inokuchi S, Makuuchi H (2003) Epidermal growth factor receptor expression in human pancreatic cancer: significance for liver metastasis. Int J Mol Med 11:305–309
Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2:285–293
Tsukita S, Yamazaki Y, Katsuno T, Tamura A, Tsukita S (2008) Tight junction-based epithelial microenvironment and cell proliferation. Oncogene 27:6930–6938
van Meer G, Gumbiner B, Simons K (1986) The tight junction does not allow lipid molecules to diffuse from one epithelial cell to the next. Nature 322:639–641
Walther W, Petkov S, Kuvardina ON, Aumann J, Kobelt D, Fichtner I, Lemm M, Piontek J, Blasig IE, Stein U, Schlag PM (2011) Novel Clostridium perfringens enterotoxin suicide gene therapy for selective treatment of claudin-3- and -4-overexpressing tumors. Gene Ther 136:1–10
Winkler L, Gehring C, Wenzel A, Müller SL, Piehl C, Krause G, Blasig IE, Piontek J (2009) Molecular determinants of the interaction between Clostridium perfringens enterotoxin fragments and claudin-3. J Biol Chem 284:18863–18872
Yamaguchi H, Kojima T, Ito T, Kyuno D, Kimura Y, Imamura M, Hirata K, Sawada N (2011) Effects of Clostridium perfringens enterotoxin via claudin-4 on nomal human pancreatic duct epithelial cells and cancer cells. Cell Mol Biol Lett 16:385–395
Yoshida H, Sumi T, Zhi X, Yasui T, Honda K, Ishiko O (2011) Claudin-4: a potential therapeutic target in chemotherapy-resistant ovarian cancer. Anticancer Res 31:1271–1277
Zhu Y, Sundfeldt K (2007) Tight junction formation in epithelial ovarian adenocarcinoma. Acta Obstet Gynecol Scand 86:1011–1019
Zhu Y, Maric J, Nilsson M, Brännström M, Janson PO, Sundfeldt K (2004) Formation and barrier function of tight junctions in human ovarian surface epithelium. Biol Reprod 71:53–59
Zhu Y, Brännström M, Janson PO, Sundfeldt K (2006) Differences in expression patterns of the tight junction proteins, claudin 1, 3, 4 and 5, in human ovarian surface epithelium as compared to epithelia in inclusion cysts and epithelial ovarian tumours. Int J Cancer 118:1884–1891
Zuo JH, Zhu W, Li MY, Li XH, Yi H, Zeng GQ, Wan XX, He QY, Li JH, Qu JQ, Chen Y, Xiao ZQ (2011) Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem 112:2508–2517
Acknowledgments
This work was supported by the Suhara Memorial Foundation, the Pancreas Research Foundation of Japan, the Ministry of Education, Culture, Sports, Science, and Technology, and the Ministry of Health, Labour and Welfare of Japan, the Japan Science and Technology Agency, and Program for developing the supporting system for upgrading education and research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
418_2012_956_MOESM1_ESM.tif
Expression of mRNAs of tight junction molecules in ovarian cancer cell lines RT-PCR for claudin-1, -3, -4, -7, -8, -9, -12, -15 and -18, occludin, tricellulin, JAM-A, ZO-1 and -2 in MCAS and HUOA.CLDN: claudin, OCDN: occludin, TRIC: tricellulin. (TIFF 885 kb)
418_2012_956_MOESM2_ESM.tif
EGF affects localization of claudin-1, -3 and -4 in ovarian cancer cell lines Immunocytostaining for claudin-1, -3 and -4 in MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without 10 μM EGFR inhibitor AG1478 for 30 min. Bar: 40 μm, CLDN: claudin, AG: AG1478. (TIFF 5408 kb)
418_2012_956_MOESM3_ESM.tif
Effects of inhibitors of signal transduction pathways on downregulation of claudin-3 and -4 by EGF in ovarian cancer cell lines Western blotting for claudin-3 and -4 in MCAS and HUOA treated with 100 ng/ml EGF for 24 h after pretreatment with or without various inhibitors of 10 μM EGFR (AG1478), 10 μM MEK/ERK (U0126), 10 μM PI3K/Akt (LY294002), 10 μM p38 MAPK (SB203580), 10 μM JNK (SP600125) 10 μM pan-PKC (GF109203X), 10 μM COX-1 and 10 μM COX-2, and 10 μM prostaglandin E2 for 30 min. CLDN: claudin, C: control, AG: AG1478, LY: LY294002, SB: SB203580, SP: SP600125, GF: GF109203X, PGE2: prostaglandin E2, COX1i: COX-1 inhibitor, COX2i: COX-2 inhibitor. (TIFF 2710 kb)
Rights and permissions
About this article
Cite this article
Ogawa, M., Kojima, T., Someya, M. et al. Epidermal growth factor modulates claudins and tight junctional functions in ovarian cancer cell lines. Histochem Cell Biol 138, 323–338 (2012). https://doi.org/10.1007/s00418-012-0956-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-012-0956-x