
Abstract
It is well established that cAMP stabilizes endothelial barrier functions, in part by regulation of VE-cadherin via EPAC/Rap 1. The aim of the present study was to investigate whether cAMP activates Rac 1 in microvascular endothelium. In human dermal microvascular endothelial cells (HDMEC), treatment with forskolin/rolipram (F/R) to increase cAMP by as well as the Epac/Rap 1-stimulating cAMP analogue 8-pCPT-2′-O-methyl-cAMP (O-Me-cAMP) stabilized endothelial barrier properties as revealed by raised transendothelial electrical resistance (TER). Under these conditions, immunostaining of VE-cadherin and claudin 5 were increased and linearized. This was paralleled by activation of Rac 1 by 153 ± 16% (F/R) or 281 ± 65% (O-Me-cAMP) whereas activity of Rho A was unchanged. F/R and O-Me-cAMP increased the peripheral actin belt and recruited the Rac 1 effector cortactin to cell junctions, similar to direct activation of Rac 1 by CNF-1. Thrombin was used to further test the physiologic relevance of cAMP-mediated Rac 1 activation. Thrombin-induced drop of TER was paralleled by intercellular gap formation, inactivation of Rac 1 and activation of Rho A at 5 and 15 min whereas baseline conditions where re-established following 60 min. Both, F/R and O-Me-cAMP completely blocked the thrombin-induced barrier breakdown. F/R completely abolished thrombin-induced Rac 1 inactivation and Rho A activation whereas O-Me-cAMP only partially blocked Rac 1 inactivation. Taken together, these results indicate that Rac 1 activation likely contributes to the barrier-stabilizing effects of cAMP in microvascular endothelium and that these effects may in part be mediated by Epac/Rap 1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The vascular endothelium provides a barrier between blood and the surrounding tissues by limiting water and macromolecule exchange (Mehta and Malik 2006). Maintenance of endothelial barrier functions is crucial and edema formation in consequence to a loss of barrier integrity contributes to the pathogenesis of several severe disorders such as sepsis or allergic shock. Since it was reported in 1989 that adenylate cyclase-stimulating agents such as forskolin reduce endothelial permeability (Stelzner et al. 1989), it has been well established that increased intracellular cAMP strengthens endothelial barrier functions in vivo and in vitro (Lorenowicz et al. 2007; Michel and Curry 1999; van Nieuw Amerongen and van Hinsbergh 2002). The mechanisms involved include decreased actin-myosin-based contractility via a reduction of myosin light chain (MLC) phosphorylation by protein kinase A (PKA)-mediated inhibition of MLC kinase (Nishikawa et al. 1984) or by inhibition of the GTPase Rho A, which increases MLC phosphorylation by blocking MLC phosphatase (Qiao et al. 2003). Besides, cAMP via PKA phosphorylates actin-binding proteins such as vasodilator-stimulated phosphoprotein (VASP) (Blume et al. 2007; Rosenberger et al. 2007) or filamin (Hastie et al. 1997), which may regulate endothelial barrier functions by controlling dynamics of the actin cytoskeleton. Actin filaments are thought to stabilize endothelial adherens and tight junctions (Bazzoni and Dejana 2004) and accordingly dysbalanced actin polymerization was found to increase microvascular permeability in vivo (Waschke et al. 2005).
More recently, it has been demonstrated that cAMP also directly strengthens endothelial barrier functions on the level of endothelial cell junctions by redistribution of adherens and tight junction components (Cullere et al. 2005) as well as by augmenting VE-cadherin-mediated binding (Fukuhara et al. 2005; Kooistra et al. 2005). This regulation of VE-cadherin was found to be independent of PKA but rather to be mediated by activation of the GTPase Rap 1 via the GTP exchange factor (GEF) Epac. This has been shown by using the cAMP analogue 8-pCPT-2′-O-methyl-cAMP (O-Me-cAMP), which preferentially binds to Epac but only ineffectively activates PKA (Christensen et al. 2003; Holz et al. 2006). Accordingly, O-Me-cAMP was effective to block the reorganization of endothelial adherens junctions in response to platelet-activating factor (PAF) in vivo (Adamson et al. 2008).
The downstream effectors by which cAMP and Rap 1 stabilize endothelial barrier functions are largely unclear. Because the Rho family GTPases Rac 1 and Rho A are known to be effective regulators of endothelial barrier properties (Baumer et al. 2008; Waschke et al. 2004a; Wojciak-Stothard and Ridley 2002) and Rap 1 can activate Rac 1 in fibroblasts via the GEFs Vav 2 and Tiam 1 and also is capable to inhibit Rho A via the GTPase activating protein (GAP) Arap 3, it is reasonable to assume that Rho A and Rac 1 would be good candidates to mediate the protective effects of cAMP in endothelial cells (Kooistra et al. 2007; Lorenowicz et al. 2007). Indeed, it has been shown that O-Me-cAMP-mediated inhibition of thrombin-induced permeability was paralleled by blocking thrombin-induced activation of Rho A (Cullere et al. 2005), at least in macrovascular endothelium. However, the role of Rac 1 as a potential downstream effector of cAMP is less clear. We found that cAMP, in part via PKA-dependent mechanisms, can reduce Rac 1 inactivation by C. sordelii lethal toxin (LT) and thereby block LT-induced permeability in vivo suggesting that cAMP might stabilize endothelial barrier functions via regulation of Rac 1 (Waschke et al. 2004b). Therefore, a protective effect of cAMP on endothelial barrier functions via crosstalk of Rap 1 and Rac 1 is possible but has not been investigated so far in microvascular endothelium.
Therefore, the aim of the present study was to investigate whether cAMP activates Rac 1 in microvascular endothelium. We used forskolin and rolipram (F/R) to increase intracellular cAMP in human dermal microvascular endothelial cells (HDMEC). In addition, O-Me-cAMP was applied because it was shown that it primarily activates Rap 1 via Epac but only weakly activates PKA (Christensen et al. 2003). We found that both F/R and O-Me-cAMP stabilized endothelial barrier functions, activated Rac1 and caused Rac 1-dependent cytoskeletal reorganization which might be involved in the well-established barrier-stabilizing effects of cAMP.
Materials and methods
Cell culture
The primary human dermal microvascular endothelial cells (HDMEC) obtained from breast explants (PromoCell, Heidelberg, Germany) were grown in endothelial growth medium MV containing supplement mix and were passaged using Detach kit-30 (both PromoCell). HDMEC were used from passages 2–5.
Test reagents
We used forskolin/rolipram (F/R) (both Sigma–Aldrich, Taufkirchen, Germany) at 5 and 10 μM for 1 h and O-Me-cAMP (Biolog, Bremen, Germany) at 200 μM for 1 h. Cytotoxic necrotizing factor 1 (CNF-1) from E. Coli (300 ng/ml, 120 min) was used to activate Rac 1. CNF-1 was supplied by Gudula Schmidt (Department of Pharmacology and Toxicology, University of Freiburg). Human thrombin (Sigma–Aldrich, Taufkirchen, Germany) was used at 10 U/ml for 5, 15 and 60 min. To inhibit the effects of thrombin, F/R and O-Me-cAMP were used as pretreatment for 1 h as described above before thrombin was added.
Measurement of transendothelial resistance (TER)
ECIS 1600R (Applied BioPhysics Inc., USA) was used to measure the transendothelial resistance (TER) of HDMEC monolayers to assess endothelial barrier integrity as described in the literature (Garcia et al. 2001; Hartmann et al. 2007; Tiruppathi et al. 1992). The sensing area within a single well of the 8-well ECIS arrays consists of 40 active working electrodes in parallel (total area = 0.02 cm2), located at the bottom of the well, and a counter electrode. A non-invasive alternating current (<1 μA) is applied to the electrodes, which are electrically connected via the electrolytes of the cell culture medium above the cells. The ECIS device reads the associated voltage drop across the system (in-phase with the current) and determines the electrical resistance of the cell-covered electrodes. Preliminary experiments by our group have indicated that the resistance measured at an AC frequency of 4,000 Hz is most sensitive to monitor changes of transendothelial resistence in our experimental setup. Moreover, we carefully correlated changes in TER with changes in FITC-dextran flux and morphological changes in HDMEC cells under well characterized conditions such as activation and inhibition of Rho GTPases by bacterial toxins as described previously (Baumer et al. 2008). HDMEC were seeded in the wells of the electrode array and grown to confluence for 8–10 days. Before experiments, medium was exchanged (200 μl) and the arrays were plugged into the instrument. Then, a measurement of baseline transendothelial resistance was performed for 10 min to equilibrate monolayers. Afterwards, 200 μl of medium containing different mediators were applied to control wells.
Cytochemistry
HDMEC’s were grown to confluence on uncoated cover slips for 7–10 days. After incubation with mediators under various conditions as indicated above, culture medium was removed and monolayers were fixed for 10 min at room temperature (RT) with 2% formaldehyde (freshly prepared from paraformaldehyde) in PBS (consisting of 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, and 1.5 mM KH2PO4; pH 7.4). Afterwards, monolayers were treated with 0.1% Triton X-100 in PBS for 5 min. After rinsing with PBS at RT, HDMECs were preincubated for 30 min with 10% normal donkey serum (NDS) or normal goat serum (NGS) and 1% bovine serum albumin (BSA) in PBS at RT and incubated over night at 4°C with human VE-cadherin antibody (1:100 in PBS containing 0.05% NaN3; Santa Cruz, Heidelberg), a mouse monoclonal IgG directed against claudin 5 (1:100 in PBS; Zymed, CA, USA) or a mouse monoclonal IgG directed against Rac 1 (1:100 in PBS; Upstate, Schwalbach, Germany). After several rinses with PBS (3 × 5 min), monolayers were incubated for 60 min at RT with Cy3-labeled goat anti-rat IgG, Cy3-labeled goat anti-mouse IgG, Cy3-labeled goat anti-rabbit IgG or a donkey anti-goat IgG, respectively (all from Dianova, Hamburg, Germany, diluted 1:600 in PBS). For visualization of filamentous actin (F-actin), some monolayers were incubated with Alexa-phalloidin (Mobitec, Goettingen, Germany; diluted 1:60 in PBS, 1 h at RT). To visualize cortactin, an Alexa Fluor 555-conjugated cortactin mouse antibody was used (1:100, 1 h RT, Upstate). Cells incubated with antibodies or Alexa-phalloidin were rinsed with PBS (3 × 5 min). Cover slips were mounted on glass slides with 60% glycerol in PBS, containing 1.5% n-propyl gallate (Serva, Heidelberg,Germany) as anti-fading compound. Immunostained monolayers were photographed with a confocal microscope (LSM 510; Carl Zeiss Microimaging Inc, Göttingen, Germany) using identical exposure settings for all conditions (Pinhole: 112, detector gain: 684–710, amplifier offset: −0,021, amplifier gain: 1, scan speed 6). Fluorescence intensity and width (100 different reading points) of VE-cadherin and claudin 5 staining were evaluated by ImageJ.
Western blotting
HDMEC monolayers were dissolved in Laemmli sample buffer (Laemmli 1970) and subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE, 10 and 15% gels). Protein contents were quantified by protein estimation directly from SDS-PAGE loading buffer using 0.1% (w/v) Amidoschwarz (AppliChem, Darmstadt, Germany) in 25% (v/v) methanol/5% (v/v) acetic acid. For immunoblotting, proteins were transferred in Kyhse-Andersen transfer buffer (Kyhse-Andersen 1984) to Hybond nitrocellulose membranes (Amersham, Braunschweig, Germany) which were blocked with 5% (w/v) low fat milk in PBS (pH 7.4) and incubated overnight at 4°C with the respective primary antibodies (1:500 in PBS plus 5% low fat milk). The antibodies against VE-cadherin and claudin 5 as well as a polyclonal IgG raised against vasodilator-stimulated phosphoprotein (VASP) and a rabbit polyclonal IgG directed against phospho-S157-VASP (kind gift from U. Walter, University of Würzburg) were used at a dilution of 1:500. As secondary antibodies, the respective horseradish peroxidase-labelled goat anti-rabbit IgG, or a donkey anti-goat IgG (Santa Cruz, Heidelberg, Germany) were used (1:3,000 in PBS). Bound immunoglobulins were visualized by the enhanced chemiluminescence technique (ECL, Amersham). Densitometric analysis using Adobe Photoshop 7.0 was performed for quantification.
Rac 1 and Rho A activation assay
For measurement of Rac 1 or Rho A activity, the Rac G-Lisa Activation Assays Biochem Kit™ (Cytoskeleton Inc., Denver CO, USA) or the Rho G-Lisa Activation Assays Biochem Kit™ (Cytoskeleton Inc., Denver CO, USA), respectively was used according to manufacturers recommendations. HDMEC’s were grown to about 70% confluency in 35 mm dishes. Cells were incubated in presence or in absence of different mediators in endothelial growth medium. Afterwards, monolayers were washed with ice cold PBS to remove serum proteins. Ice-cold cell lysis buffer was added and cell lysates were harvested by centrifugation at 14,000 (∼15,000 g) rpm at 4°C for 2 min. An aliquot was kept on ice for protein concentration measurement and samples were snap frozen at −70°C. Following adjustment of protein concentration, cell lysates were thawed and 50 μl of lysate was added to the wells of the Rac 1 or Rho A binding plates, which are coated with Rac 1- or Rho A-binding domains. Additional wells were filled with lysis buffer or non-hydrolysable Rac 1 or Rho A as a negative or positive control, respectively. The plates were placed immediately on a cold orbital shaker at 400 rpm at 4°C for 30 min, washed twice with wash buffer at RT and 200 μl of antigen presenting buffer were added to each well for 2 min at RT. After washing three times, 50 μl of Rac 1 or Rho A primary antibody (diluted 1:200 in antibody dilution buffer) were added and incubated for 45 min on an orbital microplate shaker (400 rpm; RT). After washing 3 times, 50 μl of secondary anti HRP-labelled antibody (diluted 1:100) were added to each well on the orbital microplate shaker (RT; 45 min). The wells were then washed 3 times and 50 μl of horse radish peroxidase (HRP) detection reagent were added and incubated at 37°C for 15 min. Then, 50 μl of HRP stop buffer were added and the signal was measured immediately at 490 nm using a microplate spectrophotometer (Sunrise, Tecan GmbH, Crailsheim Germany). The results were analyzed using Microsoft Excel software.
cAMP activation assay
For quantification of intracellular cAMP levels, the commercial format A cyclic AMP “plus” enzyme immunoassay (Biomol, Germany) was used according to manufacturers recommendations. The assay is a competitive immunoassay that uses a polyclonal antibody to bind in competitive manner cAMP in samples and cAMP attached to alkaline phosphatase. HDMEC cells were grown to confluence in 35-mm dishes and incubated with medium or different mediators. After removing the media, cells were incubated for 10 min with 200 μl 0.1 M HCl to induce cell lysis and to stop endogenous phosphodiesterase activity. Cell lysates were centrifuged at 14,000 rpm for 5 min and 100 μl of the supernatant was added to the wells that are coated with a goat anti-rabbit IgG. Then, 50 μl of a solution of alkaline phosphatase conjugated with cAMP (cAMP conjugate) and 50 μl of the cAMP polyclonal rabbit antibody solution were added to the wells and incubated for 2 h on a plate shaker (500 rpm) at RT. After washing the wells (3 × 200 μl wash solution), 200 μl of the p-NPP substrate (solution of p-nitrophenyl phosphate in buffer) was added and incubated for 60 min. Thereafter, 50 μl stop solution was added and the plate was read immediately at 405 nm using a microplate spectrophotometer (Sunrise, Tecan GmbH, Crailsheim Germany). The measured optical density was used to calculate the concentration of cAMP using a standard curve. Concentrations of cAMP in pmol/ml of each control experiment were set to 100% and compared to cAMP levels of cell lysates treated with thrombin.
Transfection of HDMECs
HDMECs were transfected with a pEGFP-Rac 1V12 constitutive active (pEGFP-Rac 1 CA) mutant 1 day after plating using the amaxa nucleofector technology according to manufacturers’ recommendations. The plasmids were kindly supplied by Kozo Kaibuchi (University of Nagoya, Japan). Forty-eight hours after transfection, cells were fixed for 10 min at room temperature (RT) with 2% formaldehyde (freshly prepared from paraformaldehyde) in PBS and immunostaining for cortactin and staining with Alexa-phalloidin were carried out as described above.
Statistics
Values throughout are expressed as mean ± standard error. Possible differences between groups were assessed using unpaired Student’s t test and non-parametric Mann–Whitney statistic. Statistical significance is assumed for P < 0.05.
Results
Forskolin/rolipram and O-Me-cAMP increased TER and caused linearization of adherens and tight junctions in microvascular endothelium
For this study, we used forskolin/rolipram (F/R) to increase intracellular cAMP and O-Me-cAMP, which is known to primarily stimulate the Epac/Rap 1 pathway (Christensen et al. 2003; Holz et al. 2006). First, to investigate whether cAMP under these conditions stabilized endothelial barrier functions in microvascular endothelium, we measured transendothelial electrical resistance (TER) with ECIS 1600. In human dermal microvascular endothelial cells (HDMEC) the mean baseline TER was 27 ± 1 Ω* cm² (n = 30). Both F/R and O-Me-cAMP increased TER within 5 min and reached maximal effects after 60 min whereas activation of Rho GTPases by CNF-1 had no effect during the first 60 min (Fig. 1a). At this time-point, F/R and O-Me-cAMP increased TER to a mean of 170 ± 18 and 141 ± 5% of controls, respectively (Fig. 1b). After 120 min, a time point where Rac 1, Cdc 42 and Rho A are significantly activated by CNF-1 in HDMECs (Baumer et al. 2008), CNF-1 increased TER to 121 ± 4% of controls (n = 5). To characterize whether, under the conditions used in our study, increased cAMP in response to F/R was effective to activate PKA and to prove that O-Me-cAMP did not significantly activate PKA, Western blotting for vasodilator-stimulated phosphoprotein (VASP) was used (Fig. 1c) because VASP is a substrate for PKA-mediated phosphorylation at serine S157 (Blume et al. 2007). As shown in the upper panel, VASP was detected as a single band of 46 kDa under control conditions. F/R (60 min) caused a partial shift of the VASP-specific band to 50 kDa, which is due to a mobility change of phosphorylated VASP and is indicative for phosphorylation at S157 (Halbrugge et al. 1990). In contrast, neither O-Me-cAMP (60 min) nor CNF-1 (120 min) caused a comparable mobility shift indicating that VASP was not phosphorylated by PKA under these conditions.

Transendothelial electrical resistance was significantly increased by treatment with F/R and O-Me-cAMP. Measurements of transendothelial electrical resistance (TER) were used to assess endothelial barrier functions. a Representative experiments are shown for the different conditions. No changes in TER were observed in the control graph as well as after 60 min of treatment with CNF-1 (300 ng/ml). In contrast, treatment with both F/R (5/10 μM) and O-Me-cAMP (200 μM) increased TER over 60 min. b Mean TER values following 60 min of incubation are shown for F/R (n = 5) and O-Me-cAMP (n = 7) as well as following 120 min for CNF-1 (n = 5). c Western blotting was used to demonstrate that F/R but not O-Me-cAMP led to activation of PKA-dependent signalling. Upper panel: In controls as well as after treatment with O-Me-cAMP (60 min) and CNF-1 (120 min), the 46 kDa VASP band was detected. In contrast, treatment with F/R (60 min) caused a partial shift of the VASP band to 50 kDa, which is characteristic for PKA-dependent VASP phosphorylation at serine 157 (S157). Lower panel F/R, but not O-Me-cAMP and CNF-1 caused phosphorylation of VASP at S157. Experiments shown are representative (n = 3)
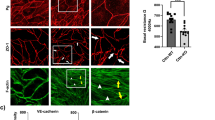
To determine whether the barrier-stabilizing effects of F/R and O-Me-cAMP would be paralleled by reorganization of endothelial adherens and tight junctions, immunostaining for VE-cadherin and claudin 5 was performed. In controls, immunostaining revealed continuous distribution of VE-cadherin and claudin 5 along cell borders with filopodia-like processes (asterisks) and partially wedge-shaped cellular overlap (arrows) (Fig. 2a, b). However, staining for claudin 5 was rather weak compared to the strong staining for VE-cadherin. Following treatment with F/R, VE-cadherin staining appeared to be linearized (Fig. 2c) and claudin 5 staining intensity was profoundly enhanced (Fig. 2d). Similar effects were observed following treatment with O-Me-cAMP (Fig. 2e, f). However, VE-cadherin staining appeared to be stronger compared to both controls and monolayers treated with F/R. We quantified the effects on width and intensity of VE-cadherin and claudin 5 immunostaining (Fig. 3a–c). Both, F/R and O-Me-cAMP significantly reduced the width of VE-cadherin staining to about 50% of control values (49 ± 2 and 47 ± 1%, respectively) indicating that adherens junctions were linearized whereas CNF-1 had no effect. In parallel, O-Me-cAMP increased VE-cadherin immunostaining intensity up to 176 ± 42% whereas F/R and CNF-1 did not affect VE-cadherin staining intensitiy.

Effects of F/R, O-Me-cAMP and CNF-1 on immunostaining of VE-cadherin and claudin 5. In HDMEC controls, immunostaining displayed continuous but uneven immunostaining of VE-cadherin and claudin 5 along cell borders with filopodia-like processes (asterisks) and apparently wedge-shaped cellular overlap (arrows) (a, b). Following treatment with F/R (5/10 μM, 60 min) or O-Me-cAMP (200 μM, 60 min), VE-cadherin and claudin 5 staining appeared to be more linearized (c–f). In addition, claudin 5 staining was enhanced under both conditions (d, f) whereas VE-cadherin staining appeared to be stronger only following treatment with O-Me-cAMP (e). Following treatment with CNF-1 (300 ng/ml, 120 min), staining of VE-cadherin was similar to controls (g) whereas claudin 5 staining appeared to be stronger and more linearized along some parts of the intercellular junctions (h). Note that under all conditions cell nuclei were also labelled by claudin 5 staining. Scale bar 20 μm for all panels

Effects of cAMP, O-Me-cAMP, and CNF-1 on width and intensity of immunostaining for VE-cadherin and claudin 5 at cell junctions. Width and intensity of VE-cadherin (a) and claudin 5 (b) staining were measured. The mean width and the mean peak fluorescence intensity are indicated in c. F/R (5 μM/10 μM, 60 min) and O-Me-cAMP (200 μM, 60 min) significantly reduced the width of VE-cadherin staining compared to controls. In addition, O-Me-cAMP also increased staining intensity for VE-cadherin. In contrast, CNF-1 (300 ng/ml, 120 min) had no effect on VE-cadherin staining. All three mediators reduced the width of claudin 5 distribution and enhanced staining intensity of claudin 5. Significant changes compared to controls are indicated by asterisks. d Western blot analyses revealed no changes of the band specific for VE-cadherin (115 kDa) or claudin 5 (24 kDa) under the different conditions indicating that total protein levels were unaffected by increased cAMP or activation of Epac/Rap 1. Experiment shown is a representative of three
In addition to the effects on adherens junctions, F/R, O-Me-cAMP and CNF-1 caused linearization of tight junctions which was detectable as reduced width of claudin 5 immunstaining compared to controls (29 ± 1, 32 ± 1 and 38 ± 1%, respectively) and also enhanced intensity of claudin 5 immunostaining to 219 ± 11, 216 ± 9 and 144 ± 9% of controls, respectively. It has to be emphasized that the changes in the width of immunostaining reveales junctional reorganization whereas the exact width of intercellular junctions under the different conditions cannot be estimated from these measurements. Western blotting demonstrated that augmented immunostaining intensities for VE-cadherin (O-Me-cAMP) and claudin 5 (F/R, O-Me-cAMP and CNF-1) were not caused by increased protein levels (Fig. 3d). Taken together, these data demonstrate that the barrier stabilizing effects of cAMP were paralleled by reorganization of endothelial adherens and tight junctions.
F/R and O-Me-cAMP significantly activated Rac 1 but had no effect on Rho A activity
Next, we analyzed the effects of F/R and O-Me-cAMP on activity of Rho family GTPases. We performed G-LISA assays specific for Rho A and Rac 1 (Fig. 4). F/R activated Rac 1 to 153 ± 16% of control levels but had no significant effect on Rho A activity (88 ± 6%). Similarly, O-Me-cAMP activated Rac 1 to 281 ± 65% of control levels, again without affecting Rho A activity (102 ± 6%). As a positive control, we used CNF-1, which we recently found to activate Rac 1, Cdc42 and Rho A in HDMEC under conditions where it significantly stabilized barrier functions (Baumer et al. 2008). Here, CNF-1 increased Rac 1 activity up to 231 ± 59% and Rho A activity up to 144 ± 14% of controls. These data indicate that cAMP activates Rac 1 and that this effect possibly contributes to the barrier-stabilizing effects of cAMP. In contrast, because Rho A activity was not affected, it is not likely that cAMP regulates barrier functions via Rho A, at least under resting conditions.

F/R and O-Me-cAMP activated Rac 1 but had no effect on Rho A activity. Rac 1 G-LISA showed that F/R (5/10 μM, 60 min) and O-Me-cAMP (200 μM, 60 min) increased Rac 1 activity to 153 ± 16 or 281 ± 65% of control levels, respectively. Rho A activity measured by Rho G-LISA was unaffected. Under similar conditions, treatment with CNF-1 (300 ng/ml, 120 min) increased Rac 1 activity to 231 ± 59% and slightly augmented Rho A activity to 144 ± 14% (n = 5)
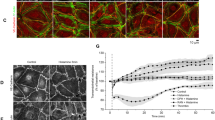
Therefore, to investigate whether cAMP-mediated Rac 1 activation is sufficient to induce Rac 1-dependent cytoskeletal reorganization, we characterized the effects of F/R and O-Me-cAMP on the localization of Rac 1 and cortactin, a downstream effector of Rac 1, as well as on the actin cytoskeleton (Fig. 5). In controls, few stress fibers were seen, Rac 1 was primarily located in the perinuclear region and cortactin was detected both in the cytoplasm as well as along some parts of the cell membrane (Fig. 5A a, B a, b). F/R, O-Me-cAMP and CNF-1 caused partial Rac 1 translocation to the cell membrane (Fig. 5A b–d). Under same conditions, F/R (Fig. 5B c, d) and O-Me-cAMP (Fig. 5B e, f) reduced the number of stress fibers and led to recruitment of cortactin to cell junctions. When CNF-1 was used to directly activate Rac 1, cortactin recruitment was even more pronounced whereas strengthening of the cortical actin cytoskeleton was similar (Fig. 5B g, h). Increased stress fiber formation following treatment with CNF-1 (Fig. 5 B g) was presumably due to simultaneous activation of Rho A (Fig. 4). To specifically test the effects of Rac 1 activation, HDMECs were transfected with constitutive active Rac 1 (pEGFP-Rac 1 CA, asterisks in Fig. 5B i,j). Transfected cells displayed a more prominent peripheral actin band (arrows in Fig. 5B i), less stress fibers and stronger junction-associated cortactin staining (arrow-heads in Fig. 5B j) compared to untransfected cells. Taken together, these results indicate that the cAMP-induced cytoskeletal rearrangement may be mediated by Rac 1-dependent mechanisms.

F/R, O-Me-cAMP and CNF-1 strengthened peripheral actin and recruited Rac 1 as well as cortactin to cell junctions. A In controls, Rac 1 was diffusely distributed in the cytoplasm (a). F/R (5 μM/10 μM, 60 min) (b), O-Me-cAMP (200 μM, 60 min) (c) and CNF-1 (300 ng/ml, 120 min) (d) induced translocation of Rac 1 to cell junctions. Scale bar 20 μm for all panels. B In controls, few stress fibres were observed in the cytoplasm (a) and cortactin was primarily located in the cytoplasm (b). F/R (c) and O-Me-cAMP (e) led to a loss of stress fibers and strengthened the peripheral actin band. In addition, F/R (d), O-Me-cAMP (f) and CNF-1 (h) caused a change in cortactin localization to a continuous linear staining pattern indicating recruitment of cortactin to cell junctions. Parallel to its effects on the peripheral actin band, CNF-1 induced stress fiber formation indicating simultaneous activation of Rho A (g). HDMECs transfected with constitutively active Rac 1 (pEGFP-Rac 1 CA, marked by asterisks) displayed are more prominent peripheral actin band (arrows) and less stress fibers (i) as well as stronger junction-associated cortactin staining compared to controls (j). Scale bar 20 μm for all panels
Increased cAMP blocked thrombin-induced Rac 1 inactivation and Rho A activation
To investigate whether cAMP-mediated Rac 1 activation would also be relevant under conditions when barrier functions are challenged by a physiologic mediator, we investigated the effects of F/R and O-Me-cAMP on thrombin-induced barrier break-down. In HDMEC monolayers, mean baseline TER in monolayers for all experiments using thrombin (n = 15) was 35 ± 2 Ω* cm². Thrombin caused a transient drop of TER (Fig. 6). The figure shows the graphs obtained from five individual experiments. TER was significantly reduced after 5 min of thrombin and reached a minimum of 58 ± 3% of controls following 9.4 min after addition of thrombin (n = 5). After 60 min, baseline TER levels were re-established. We investigated the effects of thrombin on activity of Rac 1 and Rho A as well as on intracellular cAMP levels (Fig. 7). Control lysates contained 8 ± 1.2 pmol cAMP (per mg/ml of total cellular protein). We found that after 5 min, when TER was reduced, Rac 1 activity was strongly decreased to 47 ± 8% of baseline levels whereas Rho A was activated to 181 ± 15% of controls. However, at that time, total cellular cAMP was not significantly different from controls. After 15 min of thrombin addition, Rac 1 was still significantly inactivated (57 ± 7%) and Rho A was activated (169 ± 15%). In addition, total cAMP levels were significantly reduced to 59 ± 13% of controls. After 60 min, activity of Rho GTPases and cAMP levels returned to baseline levels. Thrombin-induced Rac 1 inactivation (91 ± 6% of controls) and Rho A activation (75 ± 6% of controls) was blocked by F/R. Under these conditions, total cellular cAMP was increased to 139 ± 12% of controls. O-Me-cAMP also completely prevented Rho A activation (108 ± 15% of controls) but only partially inhibited Rac 1 inactivation (74 ± 3% of controls). Taken together, reduction of cAMP as well as inactivation of Rac 1 and activation of Rho A closely paralleled thrombin-mediated barrier breakdown and therefore both events likely are involved in the mechanisms underlying the thrombin response.

Thrombin significantly reduced TER. Thrombin treatment resulted in a rapid and reversible decrease of TER in all experiments. Thrombin (10 U/ml) significantly reduced TER during the first 5 min. Minimal TER values of 58 ± 3% of baseline values were measured after 9.4 min of thrombin addition. After 60 min, baseline values were re-established in all experiments (103 ± 2% of controls) (n = 5)

F/R and O-Me-cAMP significantly reduced the effects of thrombin on Rac 1 and Rho A activity. Activity of Rac 1 was reduced whereas Rho A activity was augmented after 5 min and 15 min of thrombin treatment, as revealed by G-LISA. The concentration of global cAMP was significantly reduced after 15 min of thrombin (10 U/ml) treatment. After 60 min, activities of Rac 1 and Rho A as well as cAMP concentration returned to baseline levels (n ≥ 5). F/R (5 μM/10 μM) completely abolished inactivation of Rac 1, activation of Rho A and decrease of cAMP in response to thrombin. In contrast, O-Me-cAMP (200 μM) abrogated the effect of thrombin on Rho A activity but not completely on Rac 1 activity (n ≥ 6). Significant changes compared to controls are indicated by asterisks. Significant effects compared to experiments using thrombin alone are indicated by hash key
Increased cAMP blocked thrombin-induced endothelial barrier breakdown
Immunostaining of F-actin and VE-cadherin demonstrated that the effects of thrombin on endothelial barrier functions were closely accompanied by formation of intercellular gaps (Fig. 8). Compared to controls (Fig. 8a, b), after 5 and 15 min of thrombin treatment, large intercellular gaps were observed at sites where VE-cadherin staining was fragmented (arrows in Fig. 8c–f) and stress fibers were increased. After 60 min, no gaps were observed but stress fibers were still more numerous than in controls (Fig. 8g, h). Both F/R and O-Me-cAMP completely blocked thrombin-induced gap formation and largely reduced stress fiber formation (Fig. 8i–l). Finally, thrombin-induced drop of TER was also completely abolished by F/R or O-Me-cAMP and TER (Fig. 9).

F/R and O-Me-cAMP completely blocked thrombin-induced intercellular gap formation. Compared to HDMEC controls (a, b), stress fiber formation was observed after 5 and 15 min of thrombin (10 U/ml) (c, e) and VE-cadherin distribution was fragmented at cell junctions (d, f). Large intercellular gaps were visible under these conditions (arrows in c–f). 60 min after thrombin addition, stress fibers were only slightly increased compared to controls (g) and VE-cadherin distribution closely resembled controls (h). Pretreatment of HDMEC with F/R (5/10 μM) or O-Me-cAMP (200 μM) for 60 min completely abolished the effects of thrombin which was added for additional 5 min (i–l). Scale bar 20 μm for all panels

F/R and O-Me-cAMP completely blocked thrombin-induced drop of TER. The effect of F/R and O-Me-cAMP on thrombin-induced drop of TER was investigated. Thrombin (10 U/ml) resulted in a transient reduction of TER. TER was significantly reduced after 5 and 15 min after thrombin was added but was not different from controls after 60 min. Following pretreatment for 60 min with F/R (5 μM/10 μM) or O-Me-cAMP (200 μM), thrombin did not significantly reduce TER (n = 5 for each condition). Significant changes compared to controls are indicated by asterisks. Significant effects compared to experiments using thrombin alone are indicated by hash key
Discussion
The present study extends our investigations on the role of cAMP and Rac 1 in the regulation of endothelial barrier functions. We provide evidence that cAMP, under conditions where it stabilized endothelial barrier functions and induced reorganization of endothelial adherens and tight junctions, is a potent activator of Rac 1 in microvascular endothelium. Moreover, similar to direct pharmacologic activation of Rac 1 or expression of constitutively active Rac 1, increased cAMP strengthened the peripheral actin cytoskeleton and lead to cortactin recruitment to cell junctions.
cAMP-mediated barrier stabilization was paralleled by reorganization of adherens and tight junctions
It is well established that cAMP reduces both increased permeability in response to inflammatory mediators as well as baseline permeability under resting conditions in vivo and in vitro (Adamson et al. 2002, 1998, 2008, 2003; Schlegel et al. 2008). Accordingly, we found that increased cAMP in microvascular endothelium stabilized endothelial barrier functions as revealed by a significant increase in TER. Enhanced barrier function was accompanied by strengthening of the peripheral, junction-associated actin band, linearization of adherens junctions and increased immunostaining intensity of the tight junction marker claudin 5. These results are supported by previous in vitro studies in the literature (Cullere et al. 2005; Fukuhara et al. 2005; Kooistra et al. 2005) and also by a recent in vivo investigation, which demonstrated that O-Me-cAMP blocked the PAF-mediated redistribution of VE-cadherin (Adamson et al. 2008). However, it is not entirely clear at present how these effects of increased cAMP and O-Me-cAMP affect the ultrastructure of adherens and tight junctions. Linearization of adherens and tight junctions was detected by a reduced width of immunostaining for VE-cadherin and claudin 5, respectively. This could be due to a reduction of the degree of cellular interdigitations with filopodial processes and changes of wedge-shaped intercellular overlap toward more perpendicular orientation of intercellular clefts. On the other hand, increased staining intensity of claudin 5 in response to F/R, O-Me-cAMP and CNF-1 as well as of VE-cadherin, which was only observed following treatment with O-Me-cAMP, could be caused by higher amounts of adherens and tight junction components within the intercellular cleft. Because Western blotting revealed that the total cellular amounts of VE-cadherin and claudin 5 were not increased, it can be speculated that cAMP rather may regulate the incorporation of these proteins into cell junctions. More refined studies using electron microscopy are required to address these important issues of junctional reorganization in response to increased cAMP.
cAMP enhanced endothelial barrier functions via regulation of Rho GTPases
It is possible that cAMP via Epac/Rap 1 regulates VE-cadherin-mediated adhesion by direct effects on adherens junctions because this effect was reported to be paralleled by recruitment of afadin 6 (Cullere et al. 2005; Fukuhara et al. 2005; Kooistra et al. 2005). Afadin 6 and Rap 1 have been shown to regulate E-cadherin-based adherens junctions by inhibition of E-cadherin endocytosis (Hoshino et al. 2005). However, because Rho GTPases are well known regulators of the cytoskeleton and of cell adhesion (Bustelo et al. 2007; Jaffe and Hall 2005) and Rac 1 is required for maintenance of VE-cadherin binding whereas Rho A via Rho kinase negatively regulates VE-cadherin-mediated adhesion under resting conditions (Baumer et al. 2008; Waschke et al. 2004a), it is conceivable that some of the protective effects of cAMP on endothelial barrier functions could be mediated via Rho GTPases.
For Rho A, we found that both F/R and O-Me-cAMP did not modulate Rho A activity in the resting state. In contrast, F/R and O-Me-cAMP completely blocked thrombin-induced Rho A activation. These data support previous studies, which demonstrated that increased cAMP via PKA partially inhibited thrombin-induced Rho A activation in microvascular endothelial cells (Qiao et al. 2003). However, it was not investigated in this study whether cAMP also influences baseline Rho A activity. In this study, a mechanism has been proposed by which cAMP could interfere with thrombin-induced Rho A activation. PKA was shown to phosphorylate the guanine nucleotide dissociation inhibitor (GDI) for Rho A which resulted in reduced dissociation of Rho A from GDI which is normally triggered by thrombin via PKC-mediated GDI phosphorylation and thereby cAMP interfered with Rho A activation (Mehta et al. 2001; Qiao et al. 2003). Alternatively, because thrombin was shown to also cause Rho A activation via p115 Rho GEF (Majumdar et al. 1999), it is possible that cAMP could interfere with this mechanism. Both mechanisms would also explain why cAMP does not affect Rho A activity in the resting state. More recently, it was reported that cAMP via Epac/Rap 1 also interfered with thrombin-induced Rho A activation and that basal Rho A activity was not changed (Cullere et al. 2005). Taken together, these data indicate that cAMP might inhibit the barrier-compromising effects of thrombin in part via blocking thrombin-induced Rho A activation. However, the reduction of permeability under resting conditions cannot be simply ascribed to a mechanism involving a predominant role of Rho A.
Therefore, we also characterized whether cAMP regulates the activity of Rac 1. We found that F/R as well as O-Me-cAMP activated Rac 1 similar to direct activation of Rac 1 by CNF-1. Moreover, cAMP-mediated Rac 1 activation was sufficient to strengthen the cortical actin cytoskeleton and to recruit the Rac 1 downstream effector cortactin to cell junctions, similar to experiments where cells were transfected with constitutively active Rac 1. It is likely that recruitment of cortactin contributed to cAMP-mediated barrier stabilization under resting conditions, because it has been shown that Rac 1 via cortactin stabilized barrier functions as revealed by increased TER (Jacobson et al. 2006). This has been suggested by Fukuhara and colleagues who also observed reorganization of the cortical actin cytoskeleton and recruitment of cortactin to cell junctions (Fukuhara et al. 2005).
Because in a previous study we found that increased cAMP blocked inactivation of Rac 1 caused by the bacterial toxin LT, which directly inhibits Rac 1 by glucosylation (Waschke et al. 2004b), we studied whether increased cAMP would be effective to modulate Rac 1 inactivation in response to a physiologic mediator such as thrombin. In our study, increased cAMP in response to F/R completely blocked thrombin-induced Rac 1 inactivation whereas O-Me-cAMP only partially (about 50%) inhibited this effect. Nevertheless, both F/R and O-Me-cAMP were equally effective to prevent thrombin-induced barrier breakdown as well as thrombin-mediated Rho A activation. Thus, these data indicate that the effect of cAMP on Rac 1 activity may be less important compared to the effect on Rho A to stabilize barrier properties under conditions of mediator-induced barrier destabilization, at least when the mediator, like thrombin, is a strong activator of Rho A.
The effects of cAMP on Rac 1 and Rho A activity seem to be mediated by both PKA-dependent and Epac/Rap 1-dependent mechanisms
Since it has been discovered that O-Me-cAMP primarily stimulates the Epac/Rap 1 pathway but not PKA (Christensen et al. 2003), it has been revealed that some effects that were previously thought to be mediated via PKA were in fact dependent on Epac/Rap 1. In order to study whether PKA is strictly required for Rac 1 activation, we increased cAMP by F/R to activate both PKA-dependent and -independent pathways and applied O-Me-cAMP to primarily stimulate the Epac/Rap 1 pathway. Both approaches caused significant activation of Rac 1. We studied phosphorylation of VASP at S157, which is typically mediated by PKA (Halbrugge et al. 1990), under these conditions to prove that F/R led to significant activation of PKA whereas O-Me-cAMP did not. Therefore it is safe to conclude that PKA is not required for activation of Rac 1 and thus, Rac 1 activation by increased cAMP could be solely induced by the Epac/Rap 1 pathway. However, it has to be emphasized that it is unclear at present whether F/R under the conditions used in our study, was potent to stimulate the Epac/Rap 1 pathway and therefore the role of PKA-independent pathways is not entirely clear at present.
F/R, O-Me-cAMP and CNF-1 similarly augmented the staining intensity of claudin 5 and caused linearization of claudin 5 staining indicating that these effects could be mediated by the Epac/Rap 1/Rac 1 pathway, possibly by Rap 1-mediated activation of the Rac GEFs Tiam and Vav2 (Kooistra et al. 2007). On the other hand, F/R but not O-Me-cAMP completely blocked thrombin-induced Rac 1 inactivation suggesting that PKA-dependent mechanisms were also involved. This is in line with our previous study, which showed that F/R-mediated inhibition of Rac 1 inactivation by LT was prevented by the PKA-inhibitor peptide PKI (Waschke et al. 2004b). Since Rac 1, in contrast to Rho A, lacks a PKA phosphorylation site, the effect of PKA is more likely to be indirect by activation of the Rac GEFs Tiam-1 or Trio (Dormond and Ruegg 2003; O’Connor and Mercurio 2001). Interestingly, F/R and O-Me-cAMP but not CNF-1 in addition caused linearization of adherens junctions. Therefore it is unclear whether this effect was also due to Rac 1 activation or rather was caused by other Rap 1-dependent mechanisms. Moreover, O-Me-cAMP but not F/R also increased immunostaining intensity for VE-cadherin. This could be due to stronger activation of Epac/Rap 1 by O-Me-cAMP compared to treatment with F/R. Taken together, all these data indicate that activation of Rac 1 could at least in part be mediated by Epac/Rap 1.
We found that F/R and O-Me-cAMP did not modulate Rho A activity in the resting state but both blocked thrombin-mediated Rho A activation similar to what has been shown previously (Cullere et al. 2005; Fukuhara et al. 2005; Kooistra et al. 2005; Qiao et al. 2003). It is likely that cAMP blocks Rho A activation both via PKA- and via Epac/Rap 1-dependent mechanisms. PKA can inhibit Rho A activation in endothelial cells by blocking the thrombin-induced dissociation of Rho-GDI from Rho A, a step required for proper activation of the GTPase (Qiao et al. 2003). This effect is possibly mediated by direct phosphorylation of Rho A at serine 188 (Ellerbroek et al. 2003). Rap 1 on the other hand has been shown to inactivate Rho A by stimulation of the Rho GTPase-activating protein (Rho GAP) Arap 3 (Kooistra et al. 2007). Taken together, cAMP likely regulates endothelial barrier properties via regulation of Rac 1 and of Rho A. The underlying mechanisms possibly involve both PKA-dependent mechanisms as well as the Epac/Rap 1 pathway, although the relative contribution of the two is yet to be determined.
Thrombin likely induces endothelial barrier breakdown via cAMP, Rho A and Rac 1 signalling
Finally, our study provides some new insight in the mechanisms involved in thrombin-mediated endothelial barrier breakdown. In our study, the barrier compromising effects of thrombin closely correlated with inactivation of Rac 1 as well as with activation of Rho A. The activity of Rac 1 was reduced whereas Rho A was activated 5 and 15 min after thrombin addition, i.e., at a time-point were large gaps were visible in the monolayers and TER was significantly reduced. However, after 60 min of thrombin action, when barrier functions were restored, activities of Rac 1 and Rho A also had returned to baseline levels. The effect of thrombin on Rho A is well established (Essler et al. 1998; Wojciak-Stothard and Ridley 2002). However, Rac 1 inactivation in response to thrombin was found in some studies (Vouret-Craviari et al. 2002) but not in others (Qiao et al. 2003).
Because we found that increased cAMP not only abolished barrier-destabilizing effects of thrombin but also completely inhibited the effects of thrombin on Rac 1 and Rho A activity, we measured the effect of thrombin on intracellular cAMP levels. Intracellular cAMP was significantly reduced after 15 min of thrombin treatment and returned to baseline levels after 60 min, i.e., when barrier functions were restored. These results suggest that thrombin primarily decreased intracellular cAMP which in turn was responsible for inactivation of Rac 1 and activation of Rho A. Based on the above discussed important role of cAMP in the regulation of Rho A and Rac 1, this scenario is possible because it has been shown that thrombin via Ca2+-induced inhibition of adenylyl cyclase 6 can reduce cAMP levels (Cioffi et al. 2002). These results are supported by very recent data which showed that atrial natriuretíc peptide (ANP) blocked thrombin-mediated endothelial barrier dysfunction via PKA and Epac/Rap-1-dependent mechanisms (Birukova et al. 2007). Moreover, because most inflammatory mediators cause an increase of intracellular Ca2+ and this is thought to reduce cAMP by various mechanisms (Curry 2002), the relevance of this pathway may not be confined to thrombin, which is not a typical inflammatory mediator.
Therefore, more refined studies are required to more closely resolve the temporo-spatial association of the effects of inflammatory mediators on intracellular Ca2+, cAMP levels and Rho GTPase activities. This is especially important because it is known that inflammatory mediators only modestly change levels of total cellular cAMP levels (Cioffi et al. 2002) and therefore our result that levels of total cAMP were significantly reduced after 15 min of thrombin but not after 5 min does not rule out that the primary mechanism underlying thrombin-induced barrier breakdown is the reduction of cAMP.
References
Adamson RH, Liu B, Fry GN, Rubin LL, Curry FE (1998) Microvascular permeability and number of tight junctions are modulated by cAMP. Am J Physiol 274:H1885–H1894
Adamson RH, Curry FE, Adamson G, Liu B, Jiang Y, Aktories K, Barth H, Daigeler A, Golenhofen N, Ness W, Drenckhahn D (2002) Rho and rho kinase modulation of barrier properties: cultured endothelial cells and intact microvessels of rats and mice. J Physiol 539:295–308
Adamson RH, Zeng M, Adamson GN, Lenz JF, Curry FE (2003) PAF- and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. Am J Physiol Heart Circ Physiol 285:H406–H417
Adamson RH, Ly JC, Sarai RK, Lenz JF, Altangerel A, Drenckhahn D, Curry FR (2008) Epac/Rap1 pathway regulates microvascular hyperpermeability induced by PAF in rat mesentery. Am J Physiol Heart Circ Physiol (in press)
Baumer Y, Burger S, Curry FE, Golenhofen N, Drenckhahn D, Waschke J (2008) Differential role of Rho GTPases in endothelial barrier regulation dependent on endothelial cell origin. Histochem Cell Biol 129:179–191
Bazzoni G, Dejana E (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84:869–901
Birukova AA, Zagranichnaya T, Alekseeva E, Bokoch GM, Birukov KG (2007) Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J Cell Physiol 215(3):715–724
Blume C, Benz PM, Walter U, Ha J, Kemp BE, Renne T (2007) AMP-activated protein kinase impairs endothelial actin cytoskeleton assembly by phosphorylating vasodilator-stimulated phosphoprotein. J Biol Chem 282:4601–4612
Bustelo XR, Sauzeau V, Berenjeno IM (2007) GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29:356–370
Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Doskeland SO (2003) cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem 278:35394–35402
Cioffi DL, Moore TM, Schaack J, Creighton JR, Cooper DM, Stevens T (2002) Dominant regulation of interendothelial cell gap formation by calcium-inhibited type 6 adenylyl cyclase. J Cell Biol 157:1267–1278
Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN (2005) Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105:1950–1955
Curry FE (2002) Microvascular injury: mechanisms and modulation. Int J Angiol 11:1–6
Dormond O, Ruegg C (2003) Regulation of endothelial cell integrin function, angiogenesis by COX-2, cAMP and Protein Kinase A. Thromb Haemost 90:577–585
Ellerbroek SM, Wennerberg K, Burridge K (2003) Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 278:19023–19031
Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M (1998) Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem 273:21867–21874
Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N (2005) Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Molec Cell Biol 25:136–146
Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D (2001) Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108:689–701
Halbrugge M, Friedrich C, Eigenthaler M, Schanzenbacher P, Walter U (1990) Stoichiometric and reversible phosphorylation of a 46-kDa protein in human platelets in response to cGMP- and cAMP-elevating vasodilators. J Biol Chem 265:3088–3093
Hartmann C, Zozulya A, Wegener J, Galla HJ (2007) The impact of glia-derived extracellular matrices on the barrier function of cerebral endothelial cells: an in vitro study. Exp Cell Res 313:1318–1325
Hastie LE, Patton WF, Hechtman HB, Shepro D (1997) H2O2-induced filamin redistribution in endothelial cells is modulated by the cyclic AMP-dependent protein kinase pathway. J Cell Physiol 172:373–381
Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG (2006) Cell physiology of cAMP sensor Epac. J Physiol 577:5–15
Hoshino T, Sakisaka T, Baba T, Yamada T, Kimura T, Takai Y (2005) Regulation of E-cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. J Biol Chem 280:24095–24103
Jacobson JR, Dudek SM, Singleton PA, Kolosova IA, Verin AD, Garcia JG (2006) Endothelial cell barrier enhancement by ATP is mediated by the small GTPase Rac and cortactin. Am J Physiol 291:L289–L295
Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Ann Rev of Cell Dev Biol 21:247–269
Kooistra MR, Corada M, Dejana E, Bos JL (2005) Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett 579:4966–4972
Kooistra MR, Dube N, Bos JL (2007) Rap1: a key regulator in cell–cell junction formation. J Cell Sci 120:17–22
Kyhse-Andersen J (1984) Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J Biochem Biophys Methods 10:203–209
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lorenowicz MJ, Fernandez-Borja M, Hordijk PL (2007) cAMP signaling in leukocyte transendothelial migration. Arteriosclerosis Thrombosis Vasc Biol 27:1014–1022
Majumdar M, Seasholtz TM, Buckmaster C, Toksoz D, Brown JH (1999) A rho exchange factor mediates thrombin and Galpha(12)-induced cytoskeletal responses. J Biol Chem 274:26815–26821
Mehta D, Malik AB (2006) Signaling mechanisms regulating endothelial permeability. Physiol Rev 86:279–367
Mehta D, Rahman A, Malik AB (2001) Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem 276:22614–22620
Michel CC, Curry FE (1999) Microvascular permeability. Physiol Rev 79:703–761
Nishikawa M, de Lanerolle P, Lincoln TM, Adelstein RS (1984) Phosphorylation of mammalian myosin light chain kinases by the catalytic subunit of cyclic AMP-dependent protein kinase and by cyclic GMP-dependent protein kinase. J Biol Chem 259:8429–8436
O’Connor KL, Mercurio AM (2001) Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem 276:47895–47900
Qiao J, Huang F, Lum H (2003) PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. Am J Physiol 284:L972–L980
Rosenberger P, Khoury J, Kong T, Weissmuller T, Robinson AM, Colgan SP (2007) Identification of vasodilator-stimulated phosphoprotein (VASP) as an HIF-regulated tissue permeability factor during hypoxia. Faseb J 21:2613–2621
Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J (2008) The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. Am J Physiol Cell Physiol 294:C178–C188
Stelzner TJ, Weil JV, O’Brien RF (1989) Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol 139:157–166
Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I (1992) Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci USA 89:7919–7923
van Nieuw Amerongen GP, van Hinsbergh VW (2002) Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul Pharmacol 39:257–272
Vouret-Craviari V, Bourcier C, Boulter E, van Obberghen-Schilling E (2002) Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. J Cell Sci 115:2475–2484
Waschke J, Baumgartner W, Adamson RH, Zeng M, Aktories K, Barth H, Wilde C, Curry FE, Drenckhahn D (2004a) Requirement of Rac activity for maintenance of capillary endothelial barrier properties. Am J Physiol Heart Circ Physiol 286:H394–H401
Waschke J, Curry FE, Adamson RH, Drenckhahn D (2005) Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol 288:H1296–H1305
Waschke J, Drenckhahn D, Adamson RH, Barth H, Curry FE (2004b) cAMP protects endothelial barrier functions by preventing Rac-1 inhibition. Am J Physiol Heart Circ Physiol 287:H2427–H2433
Wojciak-Stothard B, Ridley AJ (2002) Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol 39:187–199
Acknowledgments
We are grateful to Nadja Niedermeier and Lisa Bergauer for skilful technical assistance and to Wolfgang-Moritz Heupel and Nicolas Schlegel for helpful discussion and technical advice. We thank Moritz Bünemann and Ruth Werthmann (both Institute for Pharmacology and Toxicology, University of Würzburg) for using amaxa nucleofector technology. Cytotoxic necrotizing factor (CNF-1) was supplied by Gundula Schmidt (Department of Pharmacology and Toxicology, University of Freiburg). The phosho-S157-VASP antibody was a kind gift from Ulrich Walter (University of Wuerzburg; Institute for Clinical biochemistry and Pathobiochemistry). The EFGP-Rac 1 plasmids were a kind gift of Kozo Kaibuchi (University of Nagoya, Japan). These studies were supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 688, TP A4).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baumer, Y., Drenckhahn, D. & Waschke, J. cAMP induced Rac 1-mediated cytoskeletal reorganization in microvascular endothelium. Histochem Cell Biol 129, 765–778 (2008). https://doi.org/10.1007/s00418-008-0422-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-008-0422-y