
Abstract
Purpose
Malignant optic glioma of adulthood is a rare, invasive neoplasm of the anterior visual pathway with 66 cases reported in the literature. It presents as anaplastic astrocytoma (WHO grade III) or glioblastoma (WHO grade IV). The present case series covers the spectrum of disease manifestations, discusses neuroradiological findings, and reviews the current literature.
Methods
Retrospective case series of five patients from three tertiary referral centers and literature review.
Results
Visual loss with or without pain was the presenting symptom in all patients (two women, three men). Two patients were initially misdiagnosed as optic neuritis, and one patient as atypical non-arteritic anterior ischemic optic neuropathy (NAION). A neoplastic disease was suspected in the two remaining patients. MRI features were iso- to hypointensity on T1-weighted native images, contrast enhancement, and hyperintensity on T2-weighted images. Biopsy was generally diagnostic; however, one patient required two biopsies for diagnosis. The series includes an exceptional case of intraocular tumor extension and vitreous spread. The disease was lethal within one to two years in all patients.
Conclusions
Malignant optic glioma is a diagnostic challenge and remains a devastating and lethal disease. Advances in the understanding of tumor biology have yet failed to translate into effective treatment regimens.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Malignant optic glioma of adulthood is a rare entity first defined and reviewed by Hoyt et al. in 1973 [1]. The tumor arises in the optic nerve, chiasm, or tract, and may present as a multifocal neoplasm. It is a high-grade astrocytoma, as such either an anaplastic astrocytoma (WHO grade III) or a glioblastoma (WHO grade IV) [2]. In addition to mitotic activity defining high-grade astrocytomas, the presence of necrosis or vascular proliferation is required for grade IV diagnosis. Table 1 gives an overview of the current WHO classification of CNS astrocytic tumors. Patients with malignant optic glioma usually suffer bilateral visual loss within a few weeks and die within one to two years. Upon initial presentation patients are frequently misdiagnosed with optic neuritis, anterior ischemic optic neuropathy, or primary retinal vascular occlusion. Intraocular signs range from a normal fundus to disc edema, disc pallor, and retinal vascular occlusions. Intraocular growth of the tumor is rare. Biopsies need to be obtained to confirm diagnosis. Lateral orbitotomy is performed for tumors affecting the orbital part of the optic nerve, whereas tumors with intracerebral extension are best accessed by pterional craniotomy [3].
The present case series covers the spectrum of disease manifestations including one exceptional case of intraocular tumor manifestation, and allows discussion of neuroradiologic findings.
Methods
Retrospective case series of five patients from three tertiary referral centers (1997 – 2011) and literature review. Three patients from the Zurich University Hospital, Switzerland, one patient from the Goldschleger Eye Institute Tel Aviv, Israel, and one patient from the Egas Moniz Hospital Lisbon, Portugal, are reported. All patients underwent biomicroscopy of the fundus and imaging of brain and orbits. In two patients, fluorescein angiography, and in one patient, more extensive work-up with an electroretinogram, genetic testing, and temporal artery biopsy was performed. Diagnosis was confirmed by biopsy in all patients. No ethical board approval was required from our institute when the data was collected (2012–2013).
Results
Three men and two women aged 54 to 76 years (mean 66.8 years) with malignant optic glioma were identified. Table 2 summarizes their disease manifestation and disease progression.
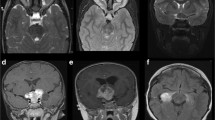
Patient 1 presented with left-sided visual loss to hand movement (HM) level for five weeks. On examination, the patient had a left relative afferent pupillary defect (RAPD), a blurred left disc margin, pain on retropulsion, and a junctional scotoma. Fluorescein angiography showed unspecific late leakage of the left disc. These findings suggested a retrobulbar and prechiasmatic lesion. MRI showed involvement of the left optic nerve, optic chiasm, thalamus, mesencephalon, and pons with enhancement on T1-weighted images, and hyperintensity on FLAIR and T2 images (Fig. 1). There was progression to disc edema with stasis retinopathy seven weeks later, and to optic atrophy another three months later. The right fundus was normal. Histopathology was obtained from a biopsy via left pterional craniotomy and revealed glioblastoma. The patient received combined temozolomide chemoradiotherapy with irradiation of the involved field (30 × 2 Gy planned, 24 × 2 Gy given); however, the patient died during therapy four and a half months after diagnosis, probably due to tumorous infiltration of the brainstem.

Coronal T2-weighted images of patient 1 with multifocal glioblastoma show hyperintensity of the left optic nerve (a), left chiasm (b) and infiltration of thalamus, mesencephalon, and pons (c). Four months later the tumor diffusely infiltrated the whole chiasm
Patient 2 presented with painful eye movements, scintillations, right-sided visual loss (20/30), right RAPD, and disc edema. The clinical picture and MRI scan with right optic nerve swelling, enhancement on T1-weighted images, and hyperintensity in T2-weighted images were interpreted as optic neuritis. The patient was placed on intravenous methylprednisolone and oral taper twice with resolution of pain, but had progressive visual loss to hand movements (HM) within two months due to a central scotoma in the right eye. The initial MRI scan had revealed a 2 cm lesion with abnormal signal intensity and a 2 mm enhancing spot in the right medial temporal gyrus of unknown etiology. Four months later, follow-up MRI showed progression of the latter. The subsequent cerebral biopsy allowed diagnosis of a multifocal glioblastoma. Despite combined temozolomide chemoradiotherapy with irradiation of the involved field (30 × 1.8 Gy), and bevacizumab salvage therapy, the patient experienced progressive tumor growth with bilateral involvement of the optic nerves, chiasm, tracts, as well as left thalamus und right temporal lobe. After seven months, vision in the left eye started to deteriorate, finally resulting in bilateral optic disc atrophy with no light perception (NLP) of the right eye and faint light perception (LP) of the left eye. The patient died within 18 months of disease onset.
Patient 3 reported painless right-sided visual loss overnight. Initial visual acuity of right eye HM and left eye 20/30 further deteriorated to right eye NLP and left eye 20/400 with temporal hemianopia within six weeks. Fundoscopy showed a membranous structure of the right optic disc (Fig. 2a) with progressive vitreous spread over a four months period (Fig. 2b-c). MRI demonstrated a homogeneously enhancing chiasmatic tumor (Fig. 2d) with extension to both optic tracts and optic nerves (right intraorbital and left intracranial portion). The tumor was hyperintense on T2-weighted images, and isointense on native T1 images. Biopsy via right pterional craniotomy allowed the diagnosis of anaplastic astrocytoma. The tumor progressed despite involved-field radiotherapy (34 × 1.8 Gy). The patient became comatose and died from pneumonia six months after diagnosis. Autopsy finally revealed pleomorphic, astrocytic tumor cells, pseudopalisading necrosis, and microvascular proliferation (Fig. 2e-f), consistent with the diagnosis of glioblastoma.

(patient 3): A membranous structure of the right optic disc (a) progressed in size (b) and resulted in vitreous spread (c) over a four months period. The left optic nerve was unremarkable. The axial (d) MRI scan shows impressive thickening of the chiasm with homogeneous contrast enhancement on T1-weighted images. Autopsy revealed diffuse infiltration of the optic nerve and chiasm with pleomorphic, astrocytic tumor cells, with pseudopalisading necrosis (e) and microvascular proliferation (f), the two features distinguishing glioblastoma from anaplastic astrocytoma
Patient 4 presented with painless unilateral visual blur (right 20/50, left 20/25), impaired color vision, and RAPD in the right eye. Both fundi were normal. Automated perimetry revealed superior constriction on the right and an inferior arcuate defect on the left. MRI was read as suspected vague enhancement of the right optic nerve. Lumbar puncture and laboratory work-up for inflammatory or hematological diseases were normal. Intravenous methylprednisolone for five days and oral taper was initiated without effect. Vision deteriorated to NLP in both eyes within six weeks. At that point an electroretinogram was obtained in order to rule out carcinoma-associated retinopathy. Genetic testing for Leber hereditary optic neuropathy was negative, temporal artery biopsy ruled out arteritis, and fluorescein angiography was normal. A follow-up MRI three months after onset of symptoms showed bilateral optic nerve enhancement on T1-weighted images, and hyperintensity in T2-weighted images. Optic nerve biopsy via lateral orbitotomy was unrevealing. MRI two months later showed progressive enlargement of the prechiasmatic optic nerves and chiasm suggestive for malignant optic glioma. Biopsy via pterional craniotomy confirmed glioblastoma. Combined temozolomide chemoradiotherapy was recommended. However, the patient decided against temozolomide. The tumor progressed despite involved-field irradiation (28 × 1.8 Gy), and the patient died one year after presentation.
Patient 5 noticed painless visual loss in the right eye for five weeks. Visual acuity was 20/100 with impaired color vision and RAPD in the right eye, and 20/30 with normal color vision in the left eye. Fundoscopy revealed optic disc swelling with hemorrhages and exudates in the right eye. An arteritic cause could be ruled out, and a diagnosis of atypical NAION was made. Four months later, the patient was referred with progressive deterioration to LP in the right eye and visual loss in the left eye to finger counting level. Fundoscopy revealed shunt vessels and a combined retinal artery and venous occlusion in the right eye, whereas the left fundus was normal. Visual fields were not tested because vision was too low. The MRI was suggestive for malignant optic glioma with bilateral thickening and enhancement of the prechiasmatic optic nerves, chiasm, and tract on T1-weighted images, T1 iso- to hypointensity on native images, and hyperintensity on T2-weighted images (Fig. 3a-c). The diagnosis of glioblastoma was confirmed by right optic nerve biopsy via pterional craniotomy. The patient underwent involved-field radiotherapy. However, the patient died within seven months of symptom onset. Precise information about radiotherapy dose could not be retrieved from the records.

(patient 5): The coronal MRI scans show bilateral thickening of the optic nerve with hyperintensity in T2-weighted images (a), iso- to hypointensity in native T1-weighted images (b) and bilateral enhancement (c) five months after onset of symptoms
Discussion
Malignant optic glioma of adulthood is a rare, invasive neoplasm of the anterior visual pathway with 66 cases reported. By 2004, Wabbels et al. [4] reviewed 45 cases. Including our five patients, 21 additional cases have been published since (Tables 2 and 3) [5–18]. Mean age of onset of all 66 cases is 57 years (standard deviation ±15; range 22 – 83), with women and men almost equally affected (30 females and 36 males). It rarely occurs in paediatric populations, either as a primary high-grade glioma [19–22] or as malignant transformation of low-grade gliomas [20, 23–25].
Patients suffer from rapidly progressive visual acuity and visual field loss, usually leading to blindness. Depending on tumor localization and extension, visual field defects might be unspecific or show localizing patterns.
With onset of symptoms, all our patients experienced visual loss within one to two months in at least one eye. Patient 2 had a delayed involvement of the fellow eye after seven months. Patient 1 initially seemed to have a strictly unilateral infiltration of the chiasm (Fig. 1) with preserved visual function of the fellow eye. However, there was neuroradiologic evidence of diffuse chiasmatic infiltration and extension to both optic tracts within four months of disease onset, two weeks before the patient passed away. Interestingly, purely unilateral involvement of the anterior visual pathway has been reported in one patient described by Wabbels et al. [4] with a follow-up period of 12 months until death.
At an early stage, clinical findings might suggest anterior ischemic optic neuropathy (patient 5) or inflammatory neuropathy (patient 2 and 4) with minimal neuroradiologic findings and transient responsiveness to steroids. However, progressive visual acuity and visual field deterioration, progressive dyschromatopsia, subsequent retinal vascular occlusions (patient 1), ocular ischemia as well as ocular pain, headaches, ophthalmoplegia, proptosis, and other neurological deficits depending on tumor localization and extension [4] point towards a possible malignant infiltrative disease, and a follow-up MRI should be obtained.
Neuroradiologic findings are unspecific, usually described as contrast enhancement and eventual thickening of the optic nerve, chiasm or tract in T1-weighted images [4, 26], with iso- to hypointensity on native T1 images [27, 28]. T2 hyperintensity of the affected anterior visual pathway is a matter of debate [27]. It was first described by Albers et al. [29] in one patient. Friedman et al. [28] presented a second case, and suggested T2 hyperintensity as a possible distinguishing feature of malignant optic glioma versus sarcoidosis with its tendency to hypointensity on T2-weighted images. However, T2 signal intensity is variable in sarcoidosis [30]. Most authors did not comment on neuroradiologic findings of T2-weighted images in malignant optic glioma. In two more recent cases [5, 15] T2 hyperintensity was described, whereas Wabbels et al. [4] explicitly did not find T2 hyperintensity of the optic nerve and chiasm. Our case series supports T2 or FLAIR hyperintensity as a characteristic, albeit unspecific finding of malignant optic glioma, since it was evident in all five patients. This is consistent with neuroradiologic findings in the better-characterized cerebral high-grade gliomas [31]. With regard to imaging features, differential diagnosis of a suspected malignant optic glioma still includes demyelinating, infectious, granulomatous, vasculitic, and infiltrative optic neuropathies [30]. CT imaging is not helpful in diagnosing malignant optic glioma.
So far, obtaining a biopsy is mandatory for diagnosis, and in case of an unspecific inflammatory histopathologic result, the biopsy might have to be repeated in a patient with a progressive, presumably neoplastic disease as in our patient 4 [13]. Interestingly, patient 3 had documented progression of a grade III glioma (biopsy) to a grade IV glioma (autopsy), which is unique in the current literature on adult malignant optic glioma. However, it might also simply reflect the tumor heterogeneity with sampling bias.
Considering the aggressively invasive behavior of malignant astrocytomas, intraocular tumor extension is surprisingly rare. While a mechanical barrier at the level of the lamina cribrosa seems plausible, a biological barrier influencing local tumor growth might be suspected as well. Intravitreal seeding of malignant optic glioma of adulthood has not been reported before and makes our case 3 unique. Dumas-Stoeckel et al. [12] presented one patient in the French literature with subretinal tumor extension and combined central retinal vein and artery occlusion. However, neither in this latter nor in our patient was intraocular growth confirmed histologically.
The standard of care for newly diagnosed high-grade astrocytomas consists of surgery or biopsy as feasible followed by radiotherapy alone (WHO grade III) or temozolomide chemoradiotherapy (WHO grade IV). The introduction of temozolomide increased the median survival of glioblastoma patients by 2–3 months and the likelihood of 2-year survival from 10 % to 26 % [32]. In contrast, the standard radiotherapy regimen of 54–60 Gy administered in 1.8-2 Gy fractions has remained essentially unaltered over the last decades. Risk structures such as optic nerves, chiasm, or brain stem commonly receive no more than 54 Gy. Recurrence or progression may be treated with re-resection, a second course of radiotherapy, or most commonly, using systemic alkylating agent chemotherapy or the VEGF antibody, bevacizumab [33]. The course of disease has not considerably improved over the last century [1], and malignant optic glioma remains lethal within one to two years [4]. The latter also holds true for our patients.
Advances in the understanding of tumor biology have yet failed to translate into effective treatment regimens [34]. However as research evolves, it is our hope that patients affected from a disease as devastating as malignant optic glioma will benefit from early diagnosis and treatment in the future.
All authors certify that they have NO affiliations with or involvement in any organization or entity with any financial interest (such as honoraria, educational grants, participation in speakers’ bureaus, membership, employment, consultancies, stock ownership, or other equity interest, and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
References
Hoyt WF, Meshel LG, Lessell S, Schatz NJ, Suckling RD (1973) Malignant optic glioma of adulthood. Brain 96:121–132
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO Classification of tumours of the central nervous system. IARC, Lyon
Winn HR (2011) Youmans Neurological Surgery, 6th edn. Elsevier Saunders, Philadelphia
Wabbels B, Demmler A, Seitz J, Woenckhaus M, Bloss HG, Lorenz B (2004) Unilateral adult malignant optic nerve glioma. Graefes Arch Clin Exp Ophthalmol 242:741–748
Hahn U, Ritz R, Ernemann U (2004) Glioblastoma multiforme des Nervus opticus. Röfo 176:1700–1701
Danesh-Meyer HV, Savino PJ, Bilyk JR, Sergott RC (2005) Aggressive glioma of adulthood simulating ischemic optic neuropathy. Arch Ophthalmol 123:694–700
Hartel PH, Rosen C, Larzo C, Nestor S (2006) Malignant optic nerve glioma (glioblastoma multiforme): A case report and literature review. W V Med J 102:29–31
Dinh TT, Wang YY, Rosenfeld JV, Cherny M (2007) Glioblastoma of the optic chiasm. J Clin Neurosci 14:502–505
Romano LM, Gaspari M, Guagnini M (2007) Astrocitoma óptico maligno bilateral del adulto. Neurologia 22:389–390
Abou-Zeid A, Duplessis D, Gnanalingham KK (2008) Blindness from multiple cerebral gliomas mimicking metastatic brain disease. Br J Neurosurg 22:772–773
Chacko JG, Lam BL, Adusumilli J, Dubovy SR (2010) Multicentric malignant glioma of adulthood masquerading as optic neuritis. Br J Ophthalmol 94(782–3):812
Dumas-Stoeckel S, Gambrelle J, Cornut PL, El Chehab H, Vighetto A, Denis P (2010) Central retinal vein and artery occlusions related to intraocular involvement of an anaplastic optochiasmatic glioma. J Fr Ophtalmol 33:564–567
Matloob S, Fan JC, Danesh-Meyer HV (2011) Multifocal malignant optic glioma of adulthood presenting as acute anterior optic neuropathy. J Clin Neurosci 18:974–977
Simao LM, Dine Sultan EN, Hall JK, Reardon DA, Bhatti MT (2011) Knee deep in the nerve. Surv Ophthalmol 56:362–370
Lincoff NS, Chung C, Balos L, Corbo JC, Sharma A (2012) Combing the globe for terrorism. J Neuroophthalmol 32:82–85
Kang JJ, Hou JH, Bui KM, Michals E, Valyi-Nagy T, Koshy M, Munson T, Charbel FT, Villano JL, Moss HE (2012) malignant optic chiasm glioma with initial clinical response to steroids. Neuroophthalmology 36:59–63
Ashur-Fabian O, Blumenthal DT, Bakon M, Nass D, Davis PJ, Hercbergs A (2013) Long-term response in high-grade optic glioma treated with medically induced hypothyroidism and carboplatin: a case report and review of the literature. Anticancer Drugs 24:315–323
Colpak AI, Isikay I, Mut M, Soylemezoglu F, Kansu T, Foroozan R (2014) Acute visual loss: Just the beginning? Surv Ophthalmol 59:548–552
Cirak B, Unal O, Arslan H, Cinal A (2000) Chiasmatic glioblastoma of childhood. A case report. Acta Radiol 41:375–376
Wong JY, Uhl V, Wara WM, Sheline GE (1987) Optic gliomas. A reanalysis of the University of California, San Francisco experience. Cancer 60:1847–1855
Safneck JR, Napier LB, Halliday WC (1992) Malignant astrocytoma of the optic nerve in a child. Can J Neurol Sci 19:498–503
Brooks WH, Parker JCJ, Young AB, Mortara RH (1976) Malignant gliomas of the optic chiasm in adolescents. Clin Pediatr (Phila) 15:557–561
Wilson WB, Feinsod M, Hoyt WF, Nielsen SL (1976) Malignant evolution of childhood chiasmal pilocytic astrocytoma. Neurology 26:322–325
de Keizer RJ, de Wolff-Rouendaal D, Bots GT, Thomeer RT, Brouwer OF, Vielvoye GJ (1989) Optic glioma with intraocular tumor and seeding in a child with neurofibromatosis. Am J Ophthalmol 108:717–725
Zoeller GK, Brathwaite CD, Sandberg DI (2010) Malignant transformation of an optic pathway glioma without prior radiation therapy. J Neurosurg Pediatr 5:507–510
Miller NR (2004) Primary tumours of the optic nerve and its sheath. Eye (Lond) 18:1026–1037
Chong VF (2006) The orbits in cancer imaging. Cancer Imaging 6:S27–S31
Friedman DP, Hollander MD (1998) Neuroradiology case of the day. Malignant optic glioma of adulthood. Radiographics 18:1046–1048
Albers GW, Hoyt WF, Forno LS, Shratter LA (1988) Treatment response in malignant optic glioma of adulthood. Neurology 38:1071–1074
Becker M, Masterson K, Delavelle J, Viallon M, Vargas MI, Becker CD (2010) Imaging of the optic nerve. Eur J Radiol 74:299–313
Clarke JL, Chang SM (2012) Neuroimaging: diagnosis and response assessment in glioblastoma. Cancer J 18:26–31
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Weller M, van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, Cohen-Jonathan-Moyal E, Frappaz D, Henriksson R, Balana C, Chinot O, Ram Z, Reifenberger G, Soffietti R, Wick W (2014) EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol 15:e395–e403
Weller M, Stupp R, Hegi M, Wick W (2012) Individualized targeted therapy for glioblastoma: fact or fiction? Cancer J 18:40–44
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Traber, G.L., Pangalu, A., Neumann, M. et al. Malignant optic glioma – the spectrum of disease in a case series. Graefes Arch Clin Exp Ophthalmol 253, 1187–1194 (2015). https://doi.org/10.1007/s00417-015-3045-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-015-3045-8