
Abstract
Background
NAP, an 8-amino acid peptide (NAPVSIPQ=Asn-Ala-Pro-Val-Ser-Ile-Pro-Gln) derived from activity-dependent neuroprotective protein (ADNP), plays an important role in neuronal differentiation and the survival of neurons in different pathological situations. We already discovered that NAP increases the survival of retinal ganglion cells (RGC) in vitro, and supports neurite outgrowth in retinal explants at femtomolar concentrations. The aim of this study was to investigate the effects of NAP on RGC survival after transient retinal ischemia and optic nerve crush.
Methods
RGC of male Wistar rats were labelled retrogradely with 6 l FluoroGold injected stereotactically into both superior colliculi. Seven days later, retinal ischemia was induced by elevating the intraocular pressure to 120 mm Hg for 60 minutes or by crushing one optic nerve for 10 s after a partial orbitotomy. NAP was either injected intraperitoneally in the concentration of 100 mg/kg 1 day before, directly after, and on the first and the second days after damage, or intravitreally (0.05 or 0.5 μg/eye) directly after the optic nerve crush. Controls received the same concentrations of a control peptide. Densities of surviving RGC and activated microglial cells (AMC) were quantified in a masked fashion 10 days after damage by counting FluoroGold-labelled cells.
Results
After retinal ischemia, intraperitoneal injections of NAP increased the number of surviving RGC by 40% (p < 0.005) compared to the control group. After optic nerve crush, NAP raised the number of surviving RGC by 31% (p = 0.07) when injected intraperitoneally and by 54% (p < 0.05) when administered intravitreally.
Conclusions
NAP acts neuroprotectively in vivo after retinal ischemia and optic nerve crush, and may have potential in treating optic nerve diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The apoptotic loss of retinal ganglion cells (RGC) is a key characteristic of various optic nerve diseases such as glaucoma, ischaemic or traumatic optic neuropathy or optic neuritis. Glaucoma is the second most common cause of blindness in the USA [48]. Apart from the reduction of intraocular pressure in glaucoma, there is still no adequate therapy for the other optic neuropathies. Because of this desperate therapeutic situation, numerous in vitro and in vivo in vitro- and in vivo investigations have been performed to prevent RGC apoptosis. The spectrum of tested substances comprises antagonists of excitatory amino acids [32, 53], openers of KATP-sensitive potassium channels [31], alpha-2-adrenoceptor agonists [29], caspase inhibitors [8], free radical scavengers [25], and neurotrophic factors such as brain-derived neurotrophic factor (BDNF) [54], to name a few. Although many of these pharmacological approaches have been successful in the laboratory, few have succeeded in clinical practice. Encouraging trial results have been published for chronic neurodegenerative diseases such as stroke or Alzheimer’s disease [15, 45]. Nevertheless, there is exciting research taking place to identify more specific and potent neuroprotective substances.
The differentiation and survival of neurons depend on neurotrophic polypeptides and proteins. These are endogenous substances, such as nerve growth factor [37], ciliary neurotrophic factor [39], BDNF [35], neurotrophin-3 [13], and neurotrophin-4/5 (NT-4/5) [22]. Another endogenous substance with neurotrophic properties is vasoactive intestinal peptide (VIP), widely distributed in the nervous system [19] and retina [12]. Its neuroprotective properties were first described in spinal cord cell cultures, with maximum effect at concentrations as low as 0.1 nM [9]. It has been shown that some of VIP’s neuroprotective action is mediated by two different glial-derived proteins: activity-dependent neurotrophic factor (ADNF) [11] and activity-dependent neuroprotective protein (ADNP) [4]. ADNP was also found essential for brain formation during embryogenesis when ADNP-knockout mice exhibited defective neuronal tube closure and a lack of PAX6-gene expression in the developing brain [44]. Neuroprotective ADNF- and ADNP-active sites have been identified and synthesised as the short peptides ADNF-9 (single letter code: SALLRISPA [10]), a 9-amino acid peptide, and NAP, an 8-amino acid peptide (single letter code: NAPVSIPQ). Remarkably, both peptides act at concentrations in the femtomolar range [4, 18]. In cultures of isolated RGC, NAP enhanced cell survival and stimulated dendritic outgrowth [33]. It increased the survival rate of isolated RGC to 167% compared to untreated controls, and extended neurite outgrowth to 126%.
Two formulations of NAP are currently being tested in Phase II trials, one in Alzheimer’s disease-mild cognitive impairment (AL-108; intranasal), and a second one in mild cognitive impairment associated with coronary artery bypass graft surgery (AL-208, intravenous) and cognitive impairment associated with schizophrenia (AL-108). These studies are being conducted by Allon Therapeutics Inc. Since optic nerve diseases pose a major clinical problem and cannot be treated yet, we believe there is an obvious rationale for testing NAP with respect to ophthalmic indications. Due to the proven neuroprotective effects on RGC in vitro [33], and neuroprotective properties in other animal models of acute or chronic neurodegeneration [6, 36, 57], we chose to evaluate the properties of NAP on the survival of RGC in vivo after retinal ischemia and optic nerve crush in rats.
Materials and methods
Animals
Adult male Wistar rats (200–250 g) were obtained from Charles River, Sulzfeld, Germany. Rats had free access to food and water and were held in cages in temperature-controlled rooms with a controlled 12-hour light–dark cycle. All animal studies were conducted in accordance with the ARVO Statement for Use of Animals in Ophthalmic and Vision Research, and procedures were approved by the Committee on Animal Care of the University of Freiburg. Efforts were made to minimise the suffering and number of animals used. Operations and manipulations were performed under general anaesthesia with Isoflurane/O2. During recovery from anaesthesia, animals were placed in separate cages, and antibiotic ointment (Refobacin®, Merck, Darmstadt, Germany) was applied on the ocular surface and skin wounds. Body temperature was maintained at 37 ± 0.5° C via a heating pad and monitored with a rectal thermometer probe.
Drug treatment
When administered intraperitoneally, NAP (Bachem custom-synthesised for Allon Therapeutics Inc.) was given 1 day before, immediately after and the first and second days after surgery in the dosage 100 μg/kg per injection. Control animals received the same concentration of a control peptide VLGGGSALL [14]. When adminstererd intravitreally, 5 μl of either 10 μg/ml or 100 μg/ml NAP soluted in BSS were injected directly after the optic nerve crush. Control animals received 100 μg/ml of the control peptide.
Retinal Ischemia
Rats were anaesthetised as described above, and the anterior chamber was cannulated with a 30-gauge needle connected to a reservoir containing 0.9% NaCl. Intraocular pressure in the left eyes was increased to 120 mm Hg for 60 minutes. Retinal ischemia was confirmed by retinal oedema and stases in retinal arteries. The needle was removed at the end of the period of elevated IOP. Rats that were not reperfused within 5 min, or those with incomplete ischemia, were excluded from the experiments. To prevent potential infection, Refobacin® was applied topically after the procedure.
Optic nerve crush
The optic nerve of the left eye was approached by orbitotomy, with partial removal of the lacrimal gland and transsection of superior rectus and obliquus muscle. The retractor bulbi muscle was separated, and the optic nerve exposed by blunt dissection. The nerve with its sheath was compressed at a distance of approximately 2 mm from the eye for 10 s, using a specifically built forceps with a residual aperture of 0.1 mm. An intact retinal perfusion was confirmed within 3 min after the crush.
Morphological quantification of RGC loss and glial response
RGC were retrogradely labelled by stereotactic injection of the fluorescent tracer FluoroGold® (Fluorochrome, Denver, CO, USA) dissolved in dimethylformamide into the superior colliculi of anaesthetised rats using a stereotactic device (Stoelting, Germany), 7 days before retinal ischemia or optic nerve crush. Rats were sacrificed 17 days after labelling by an overdose of chloralhydrate, and then perfused with 4% paraformaldehyde in PBS for 10 minutes. The eyes were removed and fixed again for 30 minutes. The retinae were then dissected, flatmounted on gelatin-coated glass slides, and embedded. After ischemia and optic nerve crush, cells stained due to phagocytosis of fluorescent RGC particles appeared. We named these cells “activated microglial cells” (AMC); they were easily distinguishable from RGC due to characteristic differences in their size, morphology and intracellular distribution in the fluorescent dye [21]. The number of FluoroGold-positive RGC and AMC were quantified under a fluorescence microscope (AxioImager, Carl Zeiss, Jena, Germany) counting them in 12 distinct areas of 40.000 μm2 each in blinded fashion.
Statistical analysis
All values are expressed as means and their 95%-confidence intervals (CI95) for direct indication of significance. Statistical significance was analysed by paired t-test or one-way ANOVA. Values of p < 0.05 were considered significant. RGC and AMC were correlated by regression analysis with Prism 4.0 (GraphPad Software Inc, San Diego, CA, USA).
Results
Quantification of RGC and AMC with and without RGC damage
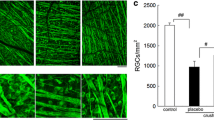
RGC were labelled retrogradely by injection of FluoroGold into the superior colliculus. After 17 days, the mean density of labelled RGC in control flat mounted retinae was 2456 cells/mm2, CI95[2313, 2596], n = 45. As shown in Fig. 1a, no AMC were observed in untreated retinae. Ten days after retinal ischemia, there was an obvious drop in the number of RGC. Furthermore, AMC appeared stained with FluoroGold by phagocytosis of RGC, and could be differentiated morphologically from RGC (Figs. 1b). Ten days after optic nerve crush, the number of RGC was diminished and the number of AMC was increased compared to retinal ischemia (Fig. 1c).

Typical fluorescence images of RGC and AMC in whole mounted retinae. RGC were retrogradelly labelled with FluoroGold applied to the superior colliculi 1 week prior to surgery. Animals were processed 17 days after labelling. a Control eye with only RGC of different sizes (asterisks; also in b, c). b,c After 60 minutes of retinal ischemia and after 10 sec of optic nerve crush, microglial phagocytes containing fluorescent RGC fragments become visible (arrows). Due to their bright, thin and often ramified appearance, AMC can easily be distinguished frorm the RGC. It is obvious from the figure that ischemia and even more pronounced optic nerve crush resulted in an increased density of AMC, together with a reduction in RGC density. Bar a-c: 100 μm
Correlation of RGC and AMC
The coefficient of determination r 2 was determined to evaluate the relationship between RGC and AMC. As shown in Fig. 2, RGC and AMC correlated significantly, when all groups were compared, with r 2 = 0.731 (p < 0.0001).

Correlation between RGC and AMC. Scatter plot shows RGC density as a function of AMC density in 115 animals in eight different treatment groups. The regression line demonstrates good correlation of r 2 = 0.731 (p < 0.001) between both cell populations
The neuroprotective effects of NAP
As shown in Fig. 3, after 60 min of retinal ischemia the intraperitoneal injection of NAP (n = 16) increased the number of surviving RGC by 40.2% (p < 0.005) from 1271 cells/mm2, CI95[967, 1572] in the control group (n = 11) to 1781 cells/mm2, CI95[1548, 2015]. Additionally, NAP reduced the AMC density by 45.5% (p < 0.0005) from 374 cells/mm2, CI95[309, 440] to 204 cells/mm2, CI95[144, 264] After optic nerve crush, the intraperitoneal administration of NAP (Fig. 4) increased the number of surviving RGC from the control group (n = 10) with 344 cells/mm2, CI95[267, 421], to the NAP group (n = 9) with 449 cells/mm2, CI95[350, 548]. This difference was not significant (p = 0.07). Furthermore, the values for the density of AMC fell significantly (p < 0.0001) from the 591.06 cells/mm2, CI95[529, 653], by 29.1% to 420 cells/mm2, CI95[371, 467] due to NAP. The intravitreal administration of NAP (Fig. 5) increased the number of surviving RGC from 551 cells/mm2, CI95[433, 671], in the control group (n = 9) to 757 cells/mm2, CI95[591, 923] with NAP 0.05 μg/eye (n = 8) and to 848 cells/mm2, CI95[640, 1056] with NAP 0.5 μg/eye (n = 7).

Effect of the intraperitoneal administration of NAP on RGC and AMC density after retinal ischemia. RGC were retrogradely labelled with FluoroGold 1 week prior to surgery; retinas were processed 10 days later. Retinal section of the non-treated right eye (a). Rats received 100 μg/kg of a control peptide as a vehicle (b) or 100 μg/kg of NAP (c) 1 day before, right after and the first and the second days after damage. d Bar histograms represent mean densities with corresponding CI95 of FluoroGold-labelled RGC and AMC of the experimental and control retinae. In comparison to control, the density of RGC after retinal ischemia increased significantly due to NAP. This was accompanied by the decrease in the density of AMC. Bar a+b: 100 μm

Effect of the intraperitoneal administration of NAP on RGC survival after optic nerve crush. RGC were retrogradely labelled with FluoroGold 1 week prior to surgery, retinae were processed 10 days later. Retinal section of the non-treated right eye (a). Rats received 100 μg/kg of a control peptide as a vehicle (b) or 100 μg/kg of NAP (c) 1 day before, right after and the first and the second days after damage. d Bar histogram represents mean densities with corresponding CI95 of FluoroGold-labelled RGC and AMC for the experimental compared to control retinae. The treatment with NAP increases the number of surviving RGC (p = 0.069) and decreases the number of AMC (p < 0.001). Bar a+b: 100 μm

Effect of intravitreal administration of NAP on RGC survival after optic nerve crush. RGC were retrogradely labelled with FluoroGold 1 week prior to surgery, retinae were processed 10 days later. Rats received 0.5 μg/eye of a control peptide as a vehicle, and 0.05 μg/eye or 0.5 μg/eye of NAP direct after the optic nerve crush. Bar histogram represents mean densities with corresponding CI95 of FluoroGold-labelled RGC and AMC. The intravitreal treatment with NAP increases significantly the number of surviving RGC by 37% and 54%, depending on concentration. The decrease of the number of AMC due to NAP was not significant
The neuroprotective effect of NAP given intravitreally was significant for the major concentration (p < 0.05). Again, the density of AMC was decreased in both NAP-treated groups. This effect did not reach significance. Three rats developed cataract due to the injection, and were excluded. NAP application was very well tolerated in all experiments.
Discussion
The aim of this study was to translate the known neuroprotective properties of the neuropeptide NAP into in vivo models of RGC loss, optic nerve crush and transient retinal ischemia. These two distinct models were chosen to address the fact that the cause of RGC loss lies in either the axon (i.e., traumatic or ischemic optic neuropathy, glaucoma, optic nerve tumors) or the ganglion cell itself (i.e., hereditary optic neuropathy, arterial retinal infarction) [42, 43]. Both models correspond to pathological events—transient ischemia to the reduced perfusion of RGC during angle-block glaucoma, and optic nerve crush to traumatic optic neuropathy.
Neuroprotection is typically determined by quantifying the degree of cell loss. In the retina, the most common method used today is retrograde labelling of RGC with a tracer molecule such as FluoroGold prior to insult with subsequent counting of surviving RGC. This procedure has been applied in several models such as ischemia reperfusion [29], partial optic nerve crush [55], optic nerve axotomy [21], excitotoxin injection [53], and chronic elevation of intraocular pressure [41]. In our hands, the number of RGC of untreated eyes was 2456 cells/mm2 17 days after labelling the optic nerve axons by stereotactical injection of FluoroGold in the superior colliculus. These results concur well with the data published by other authors showing numbers between 1800 and 2664 cells/mm2, depending on the rat strain, manner of applying FluoroGold, and evaluation period [3, 29, 38, 41]. The percentage of dying cells after retinal ischemia in different studies is scarcely comparable. Variables include methods of induction of retinal ischemia (e.g., vessel ligature [30], increasing intraocular pressure [24]), methods of detecting RGC (i.e. staining with cresyl violet, Brn-3b, FluoroGold or diAsp [24, 34]), and the time of observation after ischemia [46]. In this study, 60 min of retinal ischemia reduced the number of RGC in control animals to 1271 cells/mm2, which was 48% of the right non-treated eyes, corroborating the results by Selles-Navarro et al. [46].
Optic nerve crush mimics traumatic optic neuropathy, and is a commonly used model for studying RGC death. We found that it led in control animals to a distinct reduction in RGC to 344 cells/mm2, or 14% of the RGC in the non-treated eyes. The survival rate of RGC after optic nerve crush in rats depends on conditions such as severity and time of injury [17]. Yoles and Schwartz [55] showed that even moderate injury leads to more than 90% loss of surviving RGC within 5 days. We recently demonstrated that hardly any functional activity is detected after optic nerve crush [23]. Furthermore, active retrograde transport is not detectable (own unpublished data) after optic nerve crush. We conclude that compared to retinal ischemia, optic nerve crush is such a destructive model that effects from a neuroprotective substance might be harder to discern.
After damage, another cell type appeared to be stained in addition to the RGC. These cells had multiple cell processes, were intensively labelled by FluoroGold, and were similar in structure to microglial or macrophage cells [26, 50]. We classified these cells as AMC since they were microglia cells stained by phagocytosis of injured RGC, as shown by Bodeutsch and Thanos [7]. The mean cell density of AMC in control animals was 374 cells/mm2 after retinal ischemia and 591 cells/mm2 after optic nerve crush. Microglia activation is described as a sensor for pathological events in the central nervous system [28]. In the retina, it has been demonstrated in various pathologic conditions such as diabetes [59], retinal dystrophy [49], axotomy of the optic nerve [47, 51] and glaucoma [56]. As far as we are aware, the response of the retinal microglial cells to ischemia and optic nerve crush has not yet been quantified. AMC and RGC showed a highly significant negative correlation of r 2 = 0.731. However, the relation between RGC and AMC is not strictly proportional, since microglial cells migrate through retinal tissue, and may phagocytose RGC to a different extent at different points in time. Furthermore, certain drugs may influence the presence and motility of the microglial cells. The quantification of AMC may therefore provide additional information about substance effects in neuroprotective studies.
The rationale for choosing NAP as a potential protective agent in this study was its neuroprotective properties on RGC in vitro [33] and in models of cerebral ischemia [36] or severe head injury [6] in vivo. The intraperitoneal injection of NAP leads in the ischemia model to a significant increase in surviving RGC by 40%, reducing the number of AMC by 46%. Leker et al. [36] demonstrated that a single post-injury intravenous injection of NAP (3 μg/kg) had durable protective effects in a model of focal irreversible cerebral ischemia. Like other neuroprotective substances, NAP cannot save all neurons. However, it was at least as efficient after retinal ischemia as the NMDA-receptor-antagonist memantine in a similar experimental setup [32]. NAP therefore provides a promising therapeutic opportunity in treating different ischemia-related RGC diseases such as anterior ischemic optic neuropathy and central artery occlusion.
When given intraperitoneally, NAP increased the number of surviving RGC by 31% after optic nerve crush; however, this increase did not reach significance. Since we observed a significant drop in AMC density by 29% compared to control, we assume that NAP given intraperitoneally does exert neuroprotective effects. We further assume that the effect on RGC is less pronounced compared to the retinal ischemia, because of the severity of the insult. When given intravitreally after optic nerve crush, NAP increased the number of surviving RGC by 37% and 54% depending on dosage. As expected, NAP was more effective after intravitreal compared to systemical administration. Since the risk of hitting the lens with the complications of cataract formation and/or undesirable neuroprotective effects by lens crystallines [16] is elevated due to the low intraocular pressure after anterior chamber cannulation, we did not apply NAP intravitreally following ischemia. Recently, neuroprotective effects by NAP after intravitreal injection were also shown after laser photocoagulation induced retinal damage [5]. For acute traumatic or ischemic insults like central artery occlusion or anterior optic neuropathy in patients, the intravitreal administration of NAP appears to be most sensible. For more chronic diseases like glaucoma topical application (eye drops) or a retrobulbar depot could be developed and tested in further animal experiments.
The number of AMC as a fraction of dying RGC was significantly lower in the NAP-treated rats than in the control, in both experimental models. These results indicate that less phagocytosis took place, which underlines the improved survival of RGC with NAP.
How does NAP protect optic nerve axons after ischemia or optic nerve compression? NAP structure allows membrane penetration, followed by tubulin binding and facilitation of microtubule assembly towards cellular protection in astrocytes and neurons [14]. This primary active mechanism, providing protection through interaction with microtubule,s may lead to protection against apoptosis in vivo (e.g., in a rat model of stroke as measured by numbers of caspase-3-positive cells and fragmented DNA staining [36]). Furthermore, NAP acts as a peptide chaperone, protecting against toxic protein aggregation [2] and reducing the level of p53, a key regulator of cellular apoptosis [20]. Other potential signal transduction pathways include cGMP production [1] and interference with inflammatory mechanisms, tumour necrosis factor-alpha, and MAC1-related changes [57]. In addition, NAP, like the classic growth factors, enhances poly-ADP ribosylation [52], which may in turn be associated with microtubule assembly through the kinesin superfamily protein 4 [27, 40]. Further data showed that while NAP may provide direct neuronal protection in the absence of glial cells, the presence of glial cells increases NAP potency [58]. Thus, the NAP protective effect on glial cells that is related to microtubule function may contribute to the neuroprotection that is afforded by this compound. As the tubulin cytoskeleton is a key component of the maintenance and stability of the nervous tissue, the results presented above explains in part the breadth and potency of the protective effect of NAP.
In this study we demonstrated that the neuropeptide NAP results in significant neuronal protection against retinal ischemia and traumatic optic neuropathy in vivo. We conclude that NAP deserves further investigation in the treatment of ocular diseases, in particular when they are associated with RGC loss due to ischaemic conditions such as optic nerve infarction, central artery occlusion, or acute glaucoma. Since optic nerve diseases pose a major clinical problem and are not yet successfully treatable, these findings may help in the quest for new therapeutic strategies.
Abbreviations
- RGC:
-
retinal ganglion cell
- AMC:
-
activated microglial cell
- NAP:
-
NAPVSIPQ
- CI95 :
-
95% confidence interval
References
Ashur-Fabian O, Giladi E, Furman S, Steingart RA, Wollman Y, Fridkin M, Brenneman DE, Gozes I (2001) Vasoactive intestinal peptide and related molecules induce nitrite accumulation in the extracellular milieu of rat cerebral cortical cultures. Neurosci Lett 307:167–170
Ashur-Fabian O, Segal-Ruder Y, Skutelsky E, Brenneman DE, Steingart RA, Giladi E, Gozes I (2003) The neuroprotective peptide NAP inhibits the aggregation of the beta-amyloid peptide. Peptides 24:1413–1423
Bakalash S, Kipnis J, Yoles E, Schwartz M (2002) Resistance of retinal ganglion cells to an increase in intraocular pressure is immune-dependent. Invest Ophthalmol Vis Sci 43:2648–2653
Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, Bassan H, Blat C, Gibney G, Glazner G, Brenneman DE, Gozes I (1999) Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem 72:1283–1293
Belokopytov M, Shulman S, Dubinsky G, Gozes I, Belkin M, Rozner M (2006) Neuroprotective treatment with NAP reduces laser-induced retinal damage in rats. Invest Ophthalmol Vis Sci 47:E-Abstract 4820
Beni-Adani L, Gozes I, Cohen Y, Assaf Y, Steingart RA, Brenneman DE, Eizenberg O, Trembolver V, Shohami E (2001) A peptide derived from activity-dependent neuroprotective protein (ADNP) ameliorates injury response in closed head injury in mice. J Pharmacol Exp Ther 296:57–63
Bodeutsch N, Thanos S (2000) Migration of phagocytotic cells and development of the murine intraretinal microglial network: an in vivo study using fluorescent dyes. Glia 32:91–101
Braun JS, Tuomanen EI, Cleveland JL (1999) Neuroprotection by caspase inhibitors. Expert Opin Investig Drugs 8:1599–1610
Brenneman DE, Eiden LE (1986) Vasoactive intestinal peptide and electrical activity influence neuronal survival. Proc Natl Acad Sci USA 83:1159–1162
Brenneman DE, Glazner G, Hill JM, Hauser J, Davidson A, Gozes I (1998) VIP neurotrophism in the central nervous system: multiple effectors and identification of a femtomolar-acting neuroprotective peptide. Ann N Y Acad Sci 865:207–212
Brenneman DE, Gozes I (1996) A femtomolar-acting neuroprotective peptide. J Clin Invest 97:2299–2307
Cellerino A, Arango-Gonzalez B, Pinzon-Duarte G, Kohler K (2003) Brain-derived neurotrophic factor regulates expression of vasoactive intestinal polypeptide in retinal amacrine cells. J Comp Neurol 467:97–104
Cheng B, Mattson MP (1994) NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res 640:56–67
Divinski I, Mittelman L, Gozes I (2004) A femtomolar acting octapeptide interacts with tubulin and protects astrocytes against zinc intoxication. J Biol Chem 279:28531–28538
Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Ruther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Siren AL (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8:495–505
Fischer D, Pavlidis M, Thanos S (2000) Cataractogenic lens injury prevents traumatic ganglion cell death and promotes axonal regeneration both in vivo and in culture. Invest Ophthalmol Vis Sci 41:3943–3954
Gellrich NC, Schramm A, Rustemeyer J, Schon R, Theodor Eysel U (2002) Quantification of the neurodegenerative impact on the visual system following sudden retrobulbar expanding lesions—an experimental model. J Craniomaxillofac Surg 30:230–236
Glazner GW, Boland A, Dresse AE, Brenneman DE, Gozes I, Mattson MP (1999) Activity- dependent neurotrophic factor peptide (ADNF9) protects neurons against oxidative stress- induced death. J Neurochem 73:2341–2347
Gozes I, Avidor R, Yahav Y, Katznelson D, Croce CM, Huebner K (1987) The gene encoding vasoactive intestinal peptide is located on human chromosome 6p21—-6qter. Hum Genet 75:41–44
Gozes I, Steingart RA, Spier AD (2004) NAP mechanisms of neuroprotection. J Mol Neurosci 24:67–72
Heiduschka P, Thanos S (2000) Aurintricarboxylic acid promotes survival and regeneration of axotomised retinal ganglion cells in vivo. Neuropharmacology 39:889–902
Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, McMahon SB, Armanini MP et al (1993) Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature 363:266–270
Jehle T, Kunze D, Wingert K, Bach M, Largreze WA (2007) Modulation of contrast depth and flash frequencies: Effects on visual evoked potentials recorded in awake, freely moving brown Norway rats. Invest Ophthalmol Vis Sci 48:E-Abstract 3756
Jehle T, Lagreze WA, Blauth E, Knorle R, Schnierle P, Lucking CH, Feuerstein TJ (2000) Gabapentin-lactam (8-aza-spiro[5,4]decan-9-on; GBP-L) inhibits oxygen glucose deprivation- induced [3H]glutmate release and is a neuroprotective agent in amodel of acute retinal ischemia. Naunyn Schmiedebergs Arch Pharmacol 362:74–81
Kacza J, Mohr C, Pannicke T, Kuhrt H, Dietzel J, Fluss M, Richt JA, Vahlenkamp TW, Stahl T, Reichenbach A, Seeger J (2001) Changes of the organotypic retinal organization in Borna virus-infected Lewis rats. J Neurocytol 30:801–820
Kacza J, Seeger J (1997) Transcellular labelling of activated retinal microglia following transection of the optic nerve. Inflamm Res 46:430–433
Kaplan DR, Miller FD (2006) When a motor goes bad: a kinesin regulates neuronal survival. Cell 125:224–226
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318
Lafuente MP, Villegas-Perez MP, Mayor S, Aguilera ME, Miralles de Imperial J, Vidal-Sanz M (2002) Neuroprotective effects of brimonidine against transient ischemia-induced retinal ganglion cell death: a dose response in vivo study. Exp Eye Res 74:181–189
Lafuente MP, Villegas-Perez MP, Sobrado-Calvo P, Garcia-Aviles A, Miralles de Imperial J, Vidal-Sanz M (2001) Neuroprotective effects of alpha(2)-selective adrenergic agonists against ischemia-induced retinal ganglion cell death. Invest Ophthalmol Vis Sci 42:2074–2084
Lagreze WA (2001) [Neuroprotection. Bases and possibilities of a future clinical use]. Ophthalmologe 98:235–236
Lagreze WA, Knorle R, Bach M, Feuerstein TJ (1998) Memantine is neuroprotective in a rat model of pressure-induced retinal ischemia. Invest Ophthalmol Vis Sci 39:1063–1066
Lagreze WA, Pielen A, Steingart R, Schlunck G, Hofmann HD, Gozes I, Kirsch M (2005) The peptides ADNF-9 and NAP increase survival and neurite outgrowth of rat retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci 46:933–938
Leahy KM, Ornberg RL, Wang Y, Zhu Y, Gidday JM, Connor JR, Wax MB (2004) Quantitative ex vivo detection of rodent retinal ganglion cells by immunolabeling Brn-3b. Exp Eye Res 79:131–140
Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA (1989) Molecular cloning and expression of brain-derived neurotrophic factor. Nature 341:149–152
Leker RR, Teichner A, Grigoriadis N, Ovadia H, Brenneman DE, Fridkin M, Giladi E, Romano J, Gozes I (2002) NAP, a femtomolar-acting peptide, protects the brain against ischemic injury by reducing apoptotic death. Stroke 33:1085–1092
Levi-Montalcini R, Angeletti PU (1968) Nerve growth factor. Physiol Rev 48:534–569
Levkovitch-Verbin H, Quigley HA, Martin KR, Zack DJ, Pease ME, Valenta DF (2003) A model to study differences between primary and secondary degeneration of retinal ganglion cells in rats by partial optic nerve transection. Invest Ophthalmol Vis Sci 44:3388–3393
Lin LF, Mismer D, Lile JD, Armes LG, Butler ET 3rd, Vannice JL, Collins F (1989) Purification, cloning, and expression of ciliary neurotrophic factor (CNTF). Science 246:1023–1025
Midorikawa R, Takei Y, Hirokawa N (2006) KIF4 motor regulates activity-dependent neuronal survival by suppressing PARP-1 enzymatic activity. Cell 125:371–383
Naskar R, Wissing M, Thanos S (2002) Detection of early neuron degeneration and accompanying microglial responses in the retina of a rat model of glaucoma. Invest Ophthalmol Vis Sci 43:2962–2968
Nickells RW (2004) The molecular biology of retinal ganglion cell death: caveats and controversies. Brain Res Bull 62:439–446
Osborne NN, Chidlow G, Layton CJ, Wood JP, Casson RJ, Melena J (2004) Optic nerve and neuroprotection strategies. Eye 18:1075–1084
Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, Servoss SJ, Brenneman DE, Gozes I (2003) Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res 144:83–90
Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ (2003) Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348:1333–1341
Selles-Navarro I, Villegas-Perez MP, Salvador-Silva M, Ruiz-Gomez JM, Vidal-Sanz M (1996) Retinal ganglion cell death after different transient periods of pressure-induced ischemia and survival intervals. A quantitative in vivo study. Invest Ophthalmol Vis Sci 37:2002–2014
Sobrado-Calvo P, Vidal-Sanz M, Villegas-Perez MP (2007) Rat retinal microglial cells under normal conditions, after optic nerve section, and after optic nerve section and intravitreal injection of trophic factors or macrophage inhibitory factor. J Comp Neurol 501:866–878
Sommer A, Tielsch JM, Katz J, Quigley HA, Gottsch JD, Javitt JC, Martone JF, Royall RM, Witt KA, Ezrine S (1991) Racial differences in the cause-specific prevalence of blindness in east Baltimore. N Engl J Med 325:1412–1417
Thanos S (1992) Sick photoreceptors attract activated microglia from the ganglion cell layer: a model to study the inflammatory cascades in rats with inherited retinal dystrophy. Brain Res 588:21–28
Thanos S, Kacza J, Seeger J, Mey J (1994) Old dyes for new scopes: the phagocytosis- dependent long-term fluorescence labelling of microglial cells in vivo. Trends Neurosci 17:177–182
Thanos S, Mey J, Wild M (1993) Treatment of the adult retina with microglia-suppressing factors retards axotomy-induced neuronal degradation and enhances axonal regeneration in vivo and in vitro. J Neurosci 13:455–466
Visochek L, Steingart RA, Vulih-Shultzman I, Klein R, Priel E, Gozes I, Cohen-Armon M (2005) PolyADP-ribosylation is involved in neurotrophic activity. J Neurosci 25:7420–7428
Vorwerk CK, Lipton SA, Zurakowski D, Hyman BT, Sabel BA, Dreyer EB (1996) Chronic low- dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Invest Ophthalmol Vis Sci 37:1618–1624
Watanabe M, Fukuda Y (2002) Survival and axonal regeneration of retinal ganglion cells in adult cats. Prog Retin Eye Res 21:529–553
Yoles E, Wheeler LA, Schwartz M (1999) Alpha2-adrenoreceptor agonists are neuroprotective in a rat model of optic nerve degeneration. Invest Ophthalmol Vis Sci 40:65–73
Yuan L, Neufeld AH (2001) Activated microglia in the human glaucomatous optic nerve head. J Neurosci Res 64:523–532
Zaltzman R, Alexandrovich A, Trembovler V, Shohami E, Gozes I (2005) The influence of the peptide NAP on Mac-1-deficient mice following closed head injury. Peptides 26:1520–1527
Zemlyak I, Furman S, Brenneman DE, Gozes I (2000) A novel peptide prevents death in enriched neuronal cultures. Regul Pept 96:39–43
Zeng XX, Ng YK, Ling EA (2000) Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci 17:463–471
Acknowledgment
This work was supported by a grant from the Berta and Ernst Grimmke Stiftung and the Forschungskommission of the University of Freiburg. We thank Allon Therapeutics Inc, Canada, for supplying NAP. We also thank Mr. H. Graner for excellent technical support and Dr. Alistair Stewart for his critical review of the article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the Ernst und Berta Grimmke Stiftung, Germany. IG is the incumbent of the Lily and Avraham Gildor Chair for the Investigation of Growth Factors and the Director of the Adams Super Center for Brain Research at Tel Aviv University and is the Chief Scientific Officer of Allon Therapeutics Inc., Vancouver, Canada.
An erratum to this article can be found at http://dx.doi.org/10.1007/s00417-008-0867-7
Rights and permissions
About this article
Cite this article
Jehle, T., Dimitriu, C., Auer, S. et al. The neuropeptide NAP provides neuroprotection against retinal ganglion cell damage after retinal ischemia and optic nerve crush. Graefes Arch Clin Exp Ophthalmol 246, 1255–1263 (2008). https://doi.org/10.1007/s00417-007-0746-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-007-0746-7