
Abstract
Background
X-linked blue cone monochromatism (BCM) has to be differentiated from x-linked cone dystrophy and autosomal recessive rod monochromatism.
Methods
In nine male patients with congenital cone dysfunction (one family, six single cases; age range: 9–55 years), mutations in the red/green opsin gene cluster were confirmed. Clinical findings were analyzed retrospectively.
Results
In one family and three single cases, a single red-green hybrid gene was found carrying a Cys203Arg mutation. Two patients had multiple opsin genes, a red/green hybrid gene and at least one green pigment gene, all carrying the Cys203Arg mutation. In one patient, a large deletion of the locus control region and parts of the red pigment gene were detected. Two patients (ages: 45 and 55 years) complained about progression. Two patients presented with nystagmus. Refractive errors (+8.0 and −11.0 D) and visual acuity were variable (0.05–0.3). Only four patients had a visual acuity ≥0.1. In two patients, visual acuity could be improved using blue filter glasses. Four of five patients ≥25 years had dystrophic alterations in the macula. Severe color vision defects and relative central scotoma were present in all patients. In the electroretinogram, residual cone responses were detected in 2/8 patients.
Conclusions
Hybrid red/green opsin genes carrying the Cys203Arg mutation are a frequent cause of BCM in German patients. Molecular genetic evaluation is mandatory for adequate diagnosis of patients since from the clinical data only two patients were diagnosed as having BCM. In the other patients, either rod monochromatism or cone-rod dystrophy could not be excluded with certainty. The patients should be cautioned that macular dystrophy may develop in adults older than 30 years.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Blue cone monochromatism (BCM; MIM303700) is a rare disorder with x-linked inheritance; it was first identified by Blackwell and Blackwell [7]. Since then, the psychophysical characteristics have been described in several reports. The visual perception of affected males is limited to the input of short wavelength sensitive (blue) cones and rods. Due to the absence of blue cones in the foveola, the visual acuity is markedly reduced, and nystagmus is frequently observed [4, 26]. The course of BCM is regarded as stationary; however, macular dystrophy has been reported in some older patients [2, 3, 9, 17, 21]. Female carriers may show normal ocular findings, but in some carriers, macular pigment epithelial irregularities and specific ERG abnormalities have been reported [5]. BCM has been observed in Caucasian, Afro-American and Japanese patients [18, 22].
The clinical presentation of BCM is rather similar to autosomal recessive rod monochromatism (achromatopsia). If the family history is not informative, few features allow a clinical distinction between these two disorders. A specific color test has been designed to distinguish between rod monochromatism and BCM [4]. In addition, testing the visual acuity with the use of blue and yellow filtered glasses has been used to differentiate between the two disorders [26].
The molecular genetic basis of BCM has been identified by Nathans et al. [17]. BCM is associated with mutations that affect the genes encoding the apo-proteins of the middle wavelength-sensitive (green) and long wavelength-sensitive (red) cone photopigments. These two genes are located adjacently on the human x-chromosome, where they constitute a particular arrangement, including a locus control region, one red pigment gene and one or more green pigment genes [23]. Only a limited number of reports of BCM with identified genotype have been published [2, 3, 17, 18, 21].
Herein, we report the clinical and molecular genetic findings in a series of nine German patients with BCM to illustrate the variability of the disease and the value of molecular genetic testing.
Subjects and methods
The nine patients with congenital cone dysfunction or x-linked cone dystrophy were examined at the Department of Ophthalmology of the Benjamin Franklin University Clinic in Berlin by one author (UK). For all examinations, informed consent was obtained either by the patients or their parents. All examinations were done according to the tenets of the Declaration of Helsinki.
All patients underwent basic ophthalmologic evaluation. Visual field testing was done with Goldmann perimetry. The desaturated Panel D15 test was used for color vision testing using a standard daylight illumination lamp. If possible, tests with blue and yellow filtered glasses were performed as described by Zrenner et al. [26]. Berson’s specific color test for BCM was not available for examination [4]. Full-field ERGs were recorded according to the ISCEV standard [15]. The recording technique has been described in detail previously [12]. In short, recordings were done using a Nicolet Spirit and Ganzfeld (Nicolet, Madison, USA) with maximally dilated pupils (2.5% phenylephrine and 0.5% tropicamide). Stimulus duration was 0.1 ms. Following 30 min of dark adaptation, four stimuli with increasing intensity (maximum light intensity 10 cd s/m2) were used for recordings in the dark. Light-adapted recordings were performed after 10 min of light adaptation in the presence of white background light of 30 cd/m2 with white and chromatic stimuli of maximum light intensity. No averaging was done. Normal ranges for amplitudes and implicit times were determined by calculation of the median values and the 95% confidence intervals from single eyes of 50 age-similar probands.
All molecular genetic analyses were performed in the Molecular Genetics Laboratory at the University Eye Hospital of Tübingen. In some patients (nos. 1,251 and 1,812), mutations in the CNGA3, CNGB3 and GNAT2 genes, all known to be associated with rod monochromatism, had been excluded [13, 14, 15, 25].
Molecular genetic analysis of the red/green opsin gene cluster was done as follows. In a first series of experiments, the presence or absence of specific parts of the gene cluster was analyzed by PCR amplification (primers and their sequences in brackets): (1) the locus control region (FEO 207:5’- TTA GGA GTA GTC GCA TTA GAG A; FEO 208:5’- CAG AGG AGG AGT GGG GTG TC), (2) the red opsin gene promoter (FEO 204:5’- GCC TCT TCA CCT TAA AAG CT; FEO 206:5’- GGC GGA CGC AGG ACA GTA GAA), (3) the green opsin gene promoter (FEO 204: see above; FEO 205:5’- ACC TCC GCC TCC CAG ATT CAA G), exon 4 of the red and green opsin gene (FEO 26:5’- AGG AGT CTC AGT GGA CTC AT; FEO 27:5’- ACA AAC CCC ACC CGA GTT AG) and exon 5 of the red and green opsin gene (FEO 201:5’- TCC AAC CCC CGA CTC ACT ATC; FEO 202:5’- ACG GTA TTT TGA TGT GGA TCT GCT). Standard 50-µl PCR reactions included 50 ng genomic DNA, 10 pmol each primer, 200 µM each dNTP and 1 U Taq polymerase in 10 mM Tris-HCl pH 8.9, 50 mM KCl and 1.5–3 mM MgCl2. Typical thermal cycling protocols included an initial denaturation for 4 min at 94°C, 30 cycles of 20 s at 94°C, 30 s at 58°C and 30 s at 72°C, and a final extension for 7 min at 72°C.
For the differentiation of red and green opsin gene sequences, exon 5 PCR amplification products (see above) were digested with RsaI [18] and separated on agarose gels. Similarly, a BstUI RFLP assay on exon 4 PCR products (see above) was used for the detection of the Cys203Arg mutation [18]. The presence of the mutation was additionally confirmed by DNA sequencing (Big Dye Terminator Chemistry; Applera, Darmstadt) of exon 4 PCR products using FEO 26 as sequencing primer.
Results
Mutations in the red/green opsin gene cluster were identified in three patients from a family with x-linked recessive inheritance (family no. 124, pedigree displayed in Fig. 1) and six unrelated patients with non-informative family history. In family no. 124 and three other unrelated patients, a single red-green hybrid opsin gene was found that harbored a Cys203Arg mutation (Fig. 2). Two patients had a red/green hybrid opsin gene and one or more green opsin gene(s), all carrying the Cys203Arg mutation. In one patient, a large deletion including the locus control region and at least parts of the red pigment gene were detected.

Pedigree family no. 124. Numbers indicate examined family members

Scheme on the BCM genotypes found in this study. Grey, white and half grey/white arrows represent the red, green and red/green hybrid opsin genes, respectively. The block box upstream of the opsin genes represents the locus control region (LCR), which controls the expression of the opsin genes
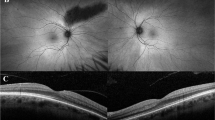
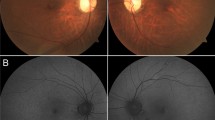
The age at examination ranged between 9 and 55 years. All patients reported low visual acuity from childhood on. Two patients (age: 45 and 55 years) complained about progression after 20 years of age; one patient (age 25 years) reported increased problems with photophobia. Ophthalmoscopy revealed normal findings in three patients (Table 1). In family no. 124, both older brothers displayed macular dystrophy, and even the 9-year-old cousin had fine pigment irregularities in the foveal area, possibly a precursor to macular dystrophy. One 13-year-old patient with normal refraction had mid-peripheral areas of choriocapillaris atrophy. In most patients (4/5) older than 25 years, dystrophic alterations in the macula were present (Fig. 3).

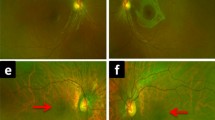
Patient no. 587: a macular pigment epithelial irregularities. No. 1,812: macular dystrophy (b) and retinal autofluorescence (c)
At the time of the examination, two patients presented with nystagmus (Table 2). Refractive errors varied between +8.0 and −11.0 D; however, even within family no. 124, refractive errors varied between +8.0 and −4.0 D. Visual acuity was between 0.05 and 0.3, but only four patients had a visual acuity better than 0.1. In four patients, blue filtered glasses were tested, but only in two patients could visual acuity be improved slightly. One patient preferred a light blue (Schott BG23), the other patient a dark blue filter (Schott BG28). Visual field testing revealed relative central scotomata in all seven patients tested. In addition, a mild concentric narrowing was detected in all three members of family no. 124. Severe color vision defects were present in all eight patients tested.
Full-field ERG recordings could be obtained in 8/9 patients (Table 3). Due to lack of cooperation, only one eye could be recorded in patient no. 1,485. In most patients, cone function could be detected neither by single flash white, chromatic nor 30-Hz flicker stimulation. In two patients, residual cone responses could be recorded when stimulating with single white flashes. However, no responses could be elicited to 30-Hz flicker stimulation. Interestingly, within family no. 124, the 45-year-old patient had residual cone function, whereas the older and younger patients did not show measurable cone function (Fig. 4). The b-wave amplitude for the maximum mixed rod-cone response was reduced in all patients, but the implicit time was slightly delayed only in one eye. The amplitudes of rod responses to dim white or blue flashes were reduced in about half of the eyes tested. The implicit times were normal in the majority of patients.

Family no. 124: ERG DA dark-adapted responses with rod blue stimulus (upper row), rod white stimulus (second row), maximum rod cone combined response (third row), single-flash cone response (fourth row), 30-Hz flicker response (last row). a Normal ERG, b patient no. 600 (9 years), c patient no. 582 (45 years), d patient no. 587 (55 years)
Only in the two patients with increased visual acuity using blue filter glasses was BCM diagnosed clinically. In patient no. 1,525, the visual acuity of 0.3 was suggestive of BCM, but blue filtered glasses did not improve the visual acuity. In family no. 124, due to the reported subjective progression, the macular dystrophy, the constricted fields and the residual cone function in one patient, x-linked cone dystrophy was suspected. In the remaining three patients (nos. 880, 1,251 and 1,485), rod monochromatism was diagnosed. There was no obvious correlation between the mutations in the red/green opsin gene cluster and the phenotype in the patients of this study.
Discussion
Blue cone monochromatism results from the simultaneous loss or defect of the red and the green opsin gene. Basically, two basic molecular mechanisms can be distinguished: (1) deletions that include both genes and/or the locus control region that is necessary for the expression of the downstream opsin genes and (2) point mutations that result in functionally defective pigments. The latter can occur either on a single leftover opsin gene or be simultaneously present on all opsin genes in the array [2, 3, 8, 17, 18]. Among the few BCM-associated point mutations reported so far, the Cys203Arg mutation is by far the most frequent [17, 18, 21, 23]. Recent in vitro experiments have shown that the Cys203Arg mutation disrupts the folding, half life of the mutant protein and its light absorption and G protein activation capabilities [11]. In our sample of German BCM patients, all but one were associated with the Cys203Arg mutation. This is in line with previous findings in other German and Northern European patients included in the study by Nathans and colleagues [18], but contrasts with a recent report in American BCM patients, in which deletions were observed in most families [3]. Presently, it appears that the Cys203Arg mutation is the most prevalent cause of BCM in German patients.
The clinical and functional findings in patients with BCM are variable. Visual acuity can be as good as 0.3. However, it was about 0.1 in our series and in the majority of reported cases. In young patients, the disorder appears to be stationary. Two of our older patients as well as patients in other studies reported progression, and progressive macular dystrophy with severe atrophy of macular pigment epithelium and choriocapillaris atrophy could be documented during long-term observation in two males [2, 17]. The majority of affected males with macular dystrophy were older than 30 years of age. In our series, four of five patients older than 25 years of age presented with macular pigment epithelium alterations. The measurement of retinal pigment epithelium autofluorescence in patient no. 1,812 revealed an increased autofluorescence at the upper and lower borders of the atrophic lesion. The increased autofluorescence is in accordance with an accumulation of lipofuscin, which is indicative of a progressive dystrophic process [6]. Macular alterations have been more frequently observed in patients with mutations of the locus control region [2, 3, 9, 17]. However, as reported here (Table 1) and in a previous study [21], macular changes may also occur in patients with the Cys203Arg mutation.
Due to the limited number of patients, the risk for development of macular dystrophy cannot be calculated. Not all older BCM patients develop visible macular alterations. On the other hand, many BCM patients do not undergo long-term follow-up when the condition is considered stationary. Some authors consider BCM a slowly progressive disorder [9]. In this respect, two findings are of interest. Pinckers [19] obtained similar results for testing with the Berson test for blue cone monochromatism when he compared BCM patients and four patients with x-linked inherited cone dystrophy. It could be possible that some families diagnosed as having x-linked cone dystrophy (similar to our family no. 124) may have mutations in the red/green opsin gene cluster.
Reichel and co-workers reported macular dystrophy to occur in older patients in one family with x-linked cone dystrophy distinct from BCM. Although this family segregates a deletion in the red opsin gene, a causal relationship still remains to be established [20]. In addition, macular alterations can be found in some obligate female carriers of BCM [5].
Color vision was severely abnormal in our series and in the majority of reported patients. Occasionally, BCM patients that performed exceptionally well in color tests like the Panel D15 test have been reported [3]. Similar variability has been reported for the full-field ERG. As in our patients, cone responses were absent in the majority of patients, but in some patients the single flash cone response was clearly present [3]. The origin of these residual cone responses remains unclear [3]. The longer cone b-wave implicit time in our patients compared to healthy subjects indicates blue cones as a likely origin for these responses [10]. Although the cone b-wave implicit times appear to be increased judging from figures in previous reports, unfortunately exact data for these implicit times have not been given. It could be speculated that the number of blue cones varies between individuals, and therefore measurable cone responses can be obtained only in some patients. Responses at dark adaptation and maximum light intensity were reduced due to the (largely) missing cone contribution. Rod response amplitudes to dim stimuli were subnormal in half of the recordings, but the implicit times were normal. Previously, rod responses were reported to be in the lower normal range [3], mildly reduced [9] or normal [1]. The variability of ERG findings is apparent in family no. 124, with residual cone responses in one patient and variably reduced rod responses (Fig. 3). Similar ERG variability within families has been observed by Ayyagri et al. [3]
Based on our clinical data, only two patients were correctly diagnosed as having BCM. In the other patients, either rod monochromatism or x-linked cone dystrophy were suspected and could not be excluded with certainty. In our experience, clinical findings in BCM patients overlap with findings in patients with rod monochromatism [13]. In the majority of our patients, molecular genetic evaluation was necessary for the correct diagnosis and adequate counseling of patients and families. It has to be noted that in addition to the BCM patients reported here and patients with rod monochromatism in a previous study [13], we have observed seven patients with clinical and functional features in accordance with either blue-cone monochromatism or rod monochromatism without detecting mutations in the red green pigment gene cluster, the CNGA3, CNGB3 or GNAT2 genes. This indicates, that additional genes may be associated with the clinical characteristics of monochromatism.
References
Andreasson S, Tornquist K (1991) Electroretinograms in patients with achromatopsia. Acta Ophthalmol (Copenh) 69:711–716
Ayyagari R, Kakuk LE, Coats CL, Bingham EL, Toda Y, Felius J, Sieving PA (1999) Bilateral macular atrophy in blue cone monochromacy (BCM) with loss of the locus control region (LCR) and part of the red pigment gene. Molecular Vision 5:13
Ayyagari R, Kakuk LE, Bingham EL, Szczesny JJ, Kemp J, Toda Y, Felius J, Sieving PA (2000) Spectrum of color gene deletions and phenotype in patients with blue cone monochromacy. Hum Genet 107:75-82
Berson EL, Sandberg MA, Rosner B, Sullivan PL (1983) Color plates to help identify patients with blue cone monochromatism. Am J Ophthalmol 95:741–747
Berson EL, Sandberg MA, Maguire A, Bromley WC, Roderick TH (1986) Electroretinograms in carriers of blue cone monochromatism. Am J Ophthalmol 102:254–261
Birnbach CD, Jarvelainen M, Possin DE, Milam AH (1994) Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 101:1211–1219
Blackwell HR, Blackwell OM (1957) Blue mono-cone monochromacy: a new color vision defect. J Optical Soc America 47:338–341
Deeb SS, Kohl S (2003) Genetics of color vision deficiencies. Dev Ophthalmol 37:170–187
Fleischman JA, O’Donnell FE (1981) Congenital x-linked incomplete achromatopsia. Evidence for slow progression, carrier fundus findings, and possible genetic linkage with glucose-6-phosphate dehydrogenase locus. Arch Ophthalmol 99:468–472
Gouras P, MacKay CJ (1990) Electroretinographic responses of the short-wavelength-sensitive cones. Invest Ophthalmol Vis Sci 31:1203–1209
Kazmi MA, Sakmar TP, Ostrer H (1997) Mutation of a conserved cysteine in the X-linked cone opsins causes color vison deficiencies by disrupting protein folding and stability. Invest Ophthalmol Vis Sci 38:1074–1081
Kellner U, Kraus H, Heimann H, Helbig H, Bornfeld N, Foerster MH (1998) Electrophysiologic evaluation of visual loss in Müller cell sheen dystrophy. Br J Ophthalmol 82:650–654
Kellner U, Wissinger B, Kohl S, Kraus H, Foerster MH (2004) Molekulargenetische Ergebnisse bei Patienten mit kongenitalen Zapfenfunktionsstörungen: Mutationen in den Genen CNGA3, CNGB3 oder GNAT2. Ophthalmologe, in press
Kohl S, Baumann B, Broghammer M et al (2000) Mutations in the CNGB3 gene encoding the β-subunit of the cone photoreceptor cGMP gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 9:2107–2116
Kohl S, Baumann B, Rosenberg T, Kellner U, Lorenz B, Vadala M, Anastasia M, Jacobson SG, Wissinger B (2002) Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet 71:422–425
Marmor MF, Zrenner E (1999) Standard for clinical electroretinography (1999 update) Doc Ophthalmol 97:143–156
Nathans J, Davenport CM, Maumenee IH, Lewis RA, Hejtmancik JF, Litt M, Lovrien E, Weleber R, Bachynski B, Zwas F, Klingaman R, Fishman G (1989) Molecular genetics of human blue cone monochromacy. Science 245:831–838
Nathans J, Maumenee IH, Zrenner E, Sadowski B, Sharpe LT, Lewis RA, Hansen E, Rosenberg T, Schwartz M, Heckenlively JR, Traboulsi E, Klingaman R, Bech-Hansen NT, LaRoche GR, Pagon RA, Murphey WH, Weleber RG (1993) Genetic heterogeneity among blue-cone monochromats. Am J Hum Genet 53:987–1000
Pinckers A (1992) Berson test for blue monochromatism. Int Ophthalmol 16:185–186
Reichel E, Bruce AM, Sandberg MA, Berson EL (1989) An electroretinographic and molecular genetic study of x-linked cone degeneration. Am J Ophthalmol 108:540–547
Reyniers E, Van Thienen MN, Meire F, De Boulle K, Devries K, Kestelijn P, Willems PJ (1995) Gene conversion between red and defective green opsin gene in blue cone monochromacy. Genomics 29:323–328
Terasaki H, Miyake Y (1992) Japanese family with blue cone monochromacy. Jpn J Ophthalmol 36:132–141
Vollrath D, Nathans J, Davis RW (1988) Tandem array of human visual pigment genes at Xq28. Science 240:1669–1672
Winderickx J, Sanocki E, Lindsey DT, Teller DY, Motulsky AG, Deeb SS (1992) Defective color vision associated with a missense (cys-203-arg) mutation in the human green visual pigment gene. Nat Genet 1:251–256
Wissinger B, Gamer D, Jägle H, Giorda R, Marx T, Mayer S, Tippmann S, M. Broghammer M, Jurklies B, Rosenberg T, Jacobson SG, Cumhur Sener E, Tatlipinar S, Hoyng C, Castellan C, Bitoun P, Andreasson S, Rudolph G, Kellner U, Lorenz B, Wolff G, Verellen-Dumoulin C, Krastel H, Akarsu N, Schwartz M, Cremers F, Apfelstedt-Sylla E, E. Zrenner E, Salati R, Sharpe LT, Kohl S (2001) CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet 69:722–737
Zrenner E, Magnussen S, Lorenz B (1988) Blauzapfenmonochromasie: Diagnose, genetische Beratung und optische Hilfsmittel. Klin Monatsbl Augenheilkd 193:510–517
Acknowledgements
The molecular genetic analyses were supported in part by a grant from the Deutsche Forschungsgemeinschaft (SFB 430/A5).
Author information
Authors and Affiliations
Corresponding author
Additional information
Ulrich Kellner and Bernd Wissinger contributed equally to this work and should be considered joint first authors. Parts of the results were presented at the meeting of the Deutsche Ophthalmologische Gesellschaft 2003.
Rights and permissions
About this article
Cite this article
Kellner, U., Wissinger, B., Tippmann, S. et al. Blue cone monochromatism: clinical findings in patients with mutations in the red/green opsin gene cluster. Graefe's Arch Clin Exp Ophthalmol 242, 729–735 (2004). https://doi.org/10.1007/s00417-004-0921-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-004-0921-z