
Abstract
Aims
The hereditary spastic paraplegias (HSPs) are a heterogeneous group of inherited neurodegenerative disorders. Although, several genotype–phenotype studies have carried out on HSPs, the association between genotypes and clinical phenotypes remain incomplete since most studies are small in size or restricted to a few genes. Accordingly, this study provides the systematic meta-analysis of genotype–phenotype associations in HSP.
Methods and results
We retrieved literature on genotype–phenotype associations in patients with HSP and mutated SPAST, REEP1, ATL1, SPG11, SPG15, SPG7, SPG35, SPG54, SPG5. In total, 147 studies with 13,570 HSP patients were included in our meta-analysis. The frequency of mutations in SPAST (25%) was higher than REEP1 (3%), as well as ATL1 (5%) in AD-HSP patients. As for AR-HSP patients, the rates of mutations in SPG11 (18%), SPG15 (7%) and SPG7 (13%) were higher than SPG5 (5%), as well as SPG35 (8%) and SPG54 (7%). The mean age of AD-HSP onset for ATL1 mutation-positive patients was earlier than patients with SPAST, REEP1 mutations. Also, the tendency toward younger age at AR-HSP onset for SPG35 was higher than other mutated genes. It is noteworthy that the mean age at HSP onset ranged from infancy to adulthood. As for the gender distribution, the male proportion in SPG7-HSP (90%) and REEP1-HSP (78%) was markedly high. The frequency of symptoms was varied among patients with different mutated genes. The rates of LL weakness, superficial sensory abnormalities, neuropathy, and deep sensory impairment were noticeably high in REEP1 mutations carriers. Also, in AR-HSP patients with SPG11 mutations, the presentation of symptoms including pes cavus, Neuropathy, and UL spasticity was higher.
Conclusion
Our comprehensive genotype–phenotype assessment of available data displays that the mean age at disease onset and particular sub-phenotypes are associated with specific mutated genes which might be beneficial for a diagnostic procedure and differentiation of the specific mutated genes phenotype among diverse forms of HSP.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The hereditary spastic paraplegias (HSPs) include a diverse clinically and genetically heterogeneous group of inherited neurodegenerative disorders [1]. Affected individuals predominantly present bilateral progressive spasticity and weakness of the lower limbs. Mainly, HSP-related symptoms are associated with degeneration of the longest corticospinal nerves [2]. In the clinical perspective, HSPs have been classified into uncomplicated (or pure) and complicated (or complex) forms. The pure forms refer to a condition that most of the affected individuals display similar clinical characteristics including progressive lower extremity spastic weakness, urinary bladder symptoms and mild diminution of distal vibratory sensation. The complex form is characterized by additional features such as ataxia, seizures, intellectual disability, parkinsonism, cognitive impairment, extrapyramidal disturbance, and peripheral neuropathy, among others [1, 3].
In the field of neurogenetics, HSPs are conceivably the disorders with the most striking genetic heterogeneity, with all patterns of Mendelian inheritance reported. The autosomal dominant HSP (AD-HSP) is the predominant form of the disorder, responsible for up to 80% of all cases with pure HSP in developed societies. This is while autosomal recessive HSPs (AR-HSPs) are prevalent in populations with a high rate of inbreeding and frequently is associated with complex forms. However, there are reports of HSP cases with X-linked or mitochondrial transmission, but they are rare [1, 2, 4].
Thus far, more than 80 genes for HSPs have been identified and yet the genetic landscape of HSP is far from complete [5]. Mutations in SPAST (formerly known as SPG4) are the most common genetic findings in patients present with AD-HSP. In the autosomal recessive HSPs, thinning of the corpus callosum is the frequent abnormality of the disorder that is mostly due to pathogenic mutations in SPG11 [6, 7]. There is a relatively large list of other genes implicated in dominant and recessive HSP. Interestingly, genes like SPAST and SPG11 have seen with the transmission in both dominant and recessive models [8,9,10].
In general, HSP has a subtle onset and, therefore, the exact age of onset (AO) for symptoms is not always accurate. There is a significant difference between AOs among the distinct genetic subtypes of HSP. It is noteworthy that the disease can occur at any age, but it is more common in childhood or early adulthood [11].
There are different concomitant neurologic and non-neurologic symptoms such as intellectual disability, dysarthria, and ataxia in patients with AR-HSPs. A considerable number of reports support the existence of an association between the mutated genes and clinical features. It is most common in patients with mutated ATL1 to present distal atrophy or neuropathy. As for patients with mutated SPG7, the manifestation of ataxia is more prevalent. Despite the fact that several studies described the relationship between the phenotypes of HSP patients and their mutations by genotype–phenotype correlation analysis, the findings of a single study are mainly descriptive and also cannot be generalized due to the small cohort size [12,13,14,15]. Also, most records have used the sequences of a small number of genes to identify disease-causing mutations in index patients and their relatives, leading to a heterogeneous perspective of clinical genetic results in terms of the affected genes, the studied patient cohorts, and outcome variables.
Accordingly, in this study, we performed a more comprehensive systematic review on the mutation frequency, AO and the genotype–phenotype correlation in distinct HSP genetic subtypes via meta-analyzing the currently available literature on the genotype–phenotype association to sustain better-personalized treatment of HSP patients.
Methods
Search strategy and selection criteria
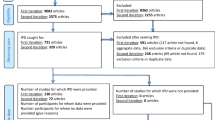
We performed a search on PubMed, Embase, Google Scholar, Scopus and Science Direct databases, for papers published in English up to March 2019, using a combination of key terms “Hereditary Spastic Paraplegia”, “HSP”, “AD-HSP”, “AR-HSP”,“SPAST”,“REEP1”, “ATL1”, “KIF5A”, “SPG4”, “SPG31”, “SPG3A”, “SPG10”, “SPG11”, “SPG15”, “ZFYVE26”, “SPG7”, “SPG35”, “FA2H”, “SPG54”, “DDHD2”, “SPG5A”, “CYP7B1”, “mutation”, “genetic”, “variant” and “genotype”. The lexicons of related risk factors were also searched for having a comprehensive study. Studies that evaluated genotype–phenotype correlation in HSP patients were selected. Furthermore, the abstract and title of the individual papers were perused thoroughly, and then studies of interest were selected for assessment of the full article. Case report studies, meta-analysis, reviews, abstract or conference papers, studies on cell line and animal models were excluded.
Data gathering and statistical analysis
Two independent reviewers assessed and selected the studies according to the predetermined inclusion criteria, and the consensus was achieved by a third reviewer. The following data were extracted from the selected studies: author name, year, sample size, patients age, gender, mutated genes, and clinical findings like upper limb (UL) spasticity, lower limb (LL) spasticity, LL weakness, pes cavus, bladder abnormalities, UL hyperreflexia, LL hyperreflexia, extensor plantar, intellectual disability (ID), dysarthria or speech disorder), peripheral neuropathy, epilepsy, ataxia, thin corpus callosum (TCC), white matter abnormalities (WMA), cataracts or visual problems, and superficial sensory abnormalities.
The meta-analysis was conducted as stated by the Preferred Reporting Items for Systematic reviews and Meta-Analysis (PRISMA) [16]. The “meta” package (version 4.9-5) in R (version-3.5.3, The R Foundation for Statistics Computing, March 11, 2019) was employed to fit meta-analysis models for the proportions and means. We used Q test to test for subgroup differences. Heterogeneity between trials was quantified using the Cochrane’s Q statistic and the I2 test and considered significant at I2 > 50% or P value < 0.1. In the case of significant heterogeneity, the random-effects model was used for the meta-analysis. Otherwise, a fixed-effect model was fitted. Chi-squared test and Fisher’s exact test were used to evaluate the association of clinical features with genotypes. Moreover, the Benjamini–Hochberg procedure (BH) was run to control the false discovery rate. A P value < 0.05 was considered statistically significant.
Results
Characteristics of included studies
Studies that met our inclusion criteria are listed in Table 1. More details about the included studies for AD-HSP and AR-HSP are summarized in Supplementary Table 1 and Table 2, respectively.
Mutation frequency of HSPs
To estimate pooled frequency of mutations in HSP patients, we performed proportion meta-analysis (metaprop). We included SPAST, REEP1 and ATL1 gene for AD-HSP and SPG11, SPG15, SPG7, SPG35, SPG54, SPG5 for AR-HSP. Table 2 provides details on gene mutation frequencies in HSP patients.
Mutation frequencies in AD-HSP patients
We found a total of 57 studies screened 5084 HSP patients, for mutation frequency of SPAST. A pooled frequency of 25.00% (95% CI 21.00–30.00) for SPAST mutations in HSP cases was found (Fig. 1). However, we found a non-significant difference regarding SPAST mutation frequencies among different populations (Q = 3.47, P = 1.00); the frequency of mutations in the Asian population (32.62%) was higher than that of Caucasians (23.07%) and Americans (24.83%) (Table 3).

Forest plot of SPAST mutation frequencies in HSP patients. A total of 25% of AD-HSP patients carry SPAST mutations. CI confidence interval
For REEP1, a total of 12 studies that investigated 1972 HSP patients revealed a mutation frequency of 3.00% (95% CI 2.00–5.00) (Fig. 2). A frequency of 5.00% (95% CI 3.00–9.00) related to ATL1 mutations was identified considering 26 studies, including 3107 HSP patients (Fig. 3). Moreover, the mutation frequency of ATL1 in the Caucasian population (8%) was higher than that of Asians and Americans (4% each) (Table 3).

Forest plot of REEP1 mutation frequencies in HSP patients. A total of 3% of HSP patients carry REEP1 mutations. CI confidence interval

Forest plot of ATL1 mutation frequencies in HSP patients. Five percent of HSP patients carry ATL1 mutations. CI confidence interval
Mutation frequencies in AR-HSP patients
For SPG11, a total of 27 studies with 2126 HSP cases were assessed, resulting in a frequency of 18.00% (95% CI 9.00–33.00%) (Fig. 4). Population sub-group analysis revealed that the mutation frequency of SPG11 in the Asian population was higher (87.98%) than that of Caucasians (10.55%), and Americans (24.23%) (Table 3). As for SPG15, a frequency of 7.00% (95% CI 3.00–15.00%) in five studies including 296 HSP cases was reported (Fig. 5a). Our analysis revealed a frequency of 13.00% (95% CI 5.00–30.00%) related to SPG7 mutations, in seven studies including 303 HSP cases (Fig. 5b). In sub-group analysis, we found a higher frequency of mutated SPG7 in Americans in comparison to Caucasians (18.97% vs. 13.29%) (Table 3). A total of four studies investigated 198 HSP patients for mutations in SPG35 and a pooled frequency of 8.00% (95% CI 1.00–35.00%) was observed (Fig. 5c). There were five studies tested 289 HSP patients for mutations in SPG54 and we found a frequency of 7.00% (95% CI 0.00–50.00%) (Fig. 5d). A total of five studies screened 195 HSP patients for mutations in SPG5 with an overall frequency of 5.00% (95% CI 3.00–9.00%) (Fig. 5e).

Forest plot of SPG11 mutation frequencies in HSP patients. A total of 18% of HSP patients carry SPG11 mutations. CI confidence interval

a Forest plot of SPG15 mutation frequencies in HSP patients. The frequency of SPG15 mutations among HSP patients was 7%. b Forest plot of SPG7 mutation frequencies in HSP patients. The frequency of SPG7 mutations among HSP patients was 13%. c Forest plot of SPG35 mutation frequencies in HSP patients. The frequency of mutations among HSP patients was 8%. d Forest plot of SPG54 mutation frequency in HSP patients. The frequency was as 7%. e Forest plot of SPG5 mutation frequencies in HSP patients. A total of five percent of HSP patients found to carry SPG5 mutations. CI confidence interval
Mean AO in HSP patients
The meta-analysis result of mean AO of both AD-HSP and AR-HSP is presented in Table 4.
Our result suggested that patients with ATL1 mutations have the earliest mean AO as 4.53 (95% CI 3.09–5.98) in comparison to other AD-HSP genes. As for SPAST and REEP1, the mean of AO was 24.79 (95% CI 21.00–28.58) and 17.00 (95% CI 11.18–22.82), respectively (Fig. 6) (Table 4). The assessment of mean AO of AD-HSP genes in different ethnicity indicated a significantly earlier onset of disease for ATL1-HSP in the Asian population as well as Caucasians (P = 0.018). It is also a significant tendency toward younger age at HSP onset for SPAST with a mean age of 21.88 (95% CI 17.71–26.06) in the Caucasians (P = 0.023) (Table 5).

Summaries of forest plots of mean age at HSP onset in HSP patients with mutations in different genes
With regard to the AO of AR-HSP, SPG7-HSP tends to manifest in older years than HSP cases due to mutations in SPG11, SPG15, SPG35 and SPG5 group (Table 4) (Fig. 6). The mean age at the onset of SPG7-HSP patients was 37.17 (95% CI 33.31–41.02), for the SPG11-HSPs was 13.10 (95% CI 11.30–14.90), for the SPG15-HSP group was 14.67 (95% CI 11.44–17.91) and in the SPG35-HSP was 13.89 (95% CI 0.00–28.92) years, as well. It was 20.07 (95% CI 10.38–29.76) in SPG5 mutation-positive patients group (Fig. 6). The mean age at HSP onset in the SPG11 group is similar among patients with different ethnicities (Table 5).
Gender distribution in HSP patients
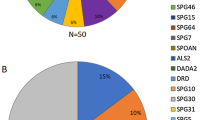
Frequently, males were more reported to have HSP due to mutations in SPG5, SPG11, SPG7, ATL1, REEP1, and SPAST, while there was an equal gender distribution among patients with mutated SPG35. We found that male to female ratio for SPAST–HSP patients was 1.27 (171 male out of 305 patients). This ratio for SPG7-HSP patients was 9.00 (18 male out of 20 patients). Also, 67% of SPG5-HSP, 54% of SPG11-HSP, 65% of ATL1-HSP, and 78% of REEP1-HSP patients were male (Fig. 7).

Gender proportion in HSP patients with mutations in different genes. Numerically males are significantly higher in AR-HSP patients with SPG7 mutations (90%) and in AD-HSP patients with REEP1 mutations (78%) than females. The gender distribution among patients with mutated SPG35 was even
The male/female ratio was significantly higher in SPG7-HSP patients compared to those with mutation in SPG5 (P = 0.0001), SPG11 (P < 0.0001), and SPG35 (P < 0.0001). As for AD-HSP patients, numerically the males were significantly higher in REEP1 mutation carriers in comparison with SPAST-HSP patients (P = 0.0015).
Genotype–phenotype correlation in HSP patients
However, the lower limbs weakness and spasticity are considered to be the predominant clinical characteristics; there are significant differences showing special characteristics with regard to the underlying gene. Figure 8a, b represents the relative frequency of finding the clinical features in AR-HSP and AD-HSP, respectively.

a Relative frequency of findings of the clinical features in AR-HSP patients. b Relative frequency of findings of the clinical features in AD-HSP patients. The color gray indicates not available data
Findings in neurological examination
We were able to only include SPG11 and SPG15 to investigate the association of AR-HSP-related genes with LL weakness. The results evidenced that all the patients (n = 8) with SPG15 mutations presented LL weakness which was significantly higher than those with SPG11-HSP (P < 0.0001) (Fig. 8a). In the case of AD-HSP, we found 25 out of 29 patients (86%) with REEP1-related HSP were discerned with LL weakness. This frequency is significantly higher than that observed in HSP cases due to ATL1 mutations (50%, 8 out of 16 patients) and SPAST mutations (47%, 113 out of 241 patients) (Fig. 8b).
The frequency of UL spasticity was 69% (11 out of 16 patients) in SPG11-HSP patients, while 3/5 of patients with mutations in SPG54 and 3/7 of SPG15-HSP cases were reported to have UL spasticity. In the case of AD-HSP patients, UL spasticity was reported in 13% of SPAST-HSP (23 out of 178 patients) and 11% of REEP1-HSP patients (2 out of 19 patients). The UL spasticity was not observed in patients with ATL1 mutations.
In general, UL hyperreflexia is a more common finding in AR-HSP than AD-HSP. All included HSP patients with mutated SPG15, SPG7, and SPG54 to manifest UL hyperreflexia; however, 90% of SPG11-HSP showed such lesions (Fig. 8a) (Supplementary Table 3). In AD-HSP, almost 37% of SPAST-HSP (102 out of 274 patients) and 33% of REEP1-HSP (6 out of 18 patients) also showed UL hyperreflexia, which was higher than in patients with ATL1 mutations (1 out of 15 patients).
For hyperreflexia in the lower limbs, we found data for AD-HSP-related genes. Data evidenced that all the ATL1-HSP patients have diagnosed with LL hyperreflexia (14 out of 14 patients). Moreover, LL hyperreflexia is a frequent finding in SPAST-HSP patients, with a rate of 96% (220 out of 228 patients) and also in REEP1-HSP patients was 86% (24 out of 28 mutation-positive patients (Fig. 8b).
For a number of neurological findings like superficial sensory abnormalities, deep sensory impairments and extensor plantar we found eligible data only for AD-HSPs. A greater percentage of patients with REEP1 mutations showed disorders such as superficial sensory abnormalities (50%), deep sensory impairments (41%) in comparison to SPAST- and ALT1-related cases. All the AD-HSP patients with mutated ATL1 were identified with extensor plantar. Moreover, the diagnosis of extensor plantar was significantly higher in REEP1-HSP patients than those with mutated SPAST (P < 0.0001) (Fig. 8b) (Supplementary Table 4).
Intellectual disability
As can be seen from Fig. 8a, intellectual disability was more often observed in SPG54-HSP than in the other AR-HSPs. The relative frequency of intellectual disability frequency in HSP patients in order of highest to lowest was as follows: SPG54 (89%), SPG11 (86%), SPG15 (78%), SPG35 (71%) and SPG7 (8%) (Fig. 8a).
As for AD-HSP, SPAST-HSP patients were most often diagnosed with intellectual disability. However, there was no report of intellectual disability in patients with ATL1 mutations (Fig. 8b).
Ataxia
Data evidenced that 39% of SPG11-HSP patients manifested with ataxia (37 out of 96 patients). Also, ataxia was reported in 71% of SPG35-HSP (5 out of 7 patients), in 62% of SPG7-HSP patients (5 out of 8 patients), and 25% of SPG15-HSP (2 out of 8 patients) (Fig. 8a).
Cataract
In the case of cataract, data showed that ~ 67% (4/6) with SPG54 mutations and 39% (19/60) of patients with SPG11 mutations were diagnosed with cataract. Also, cataract was reported in two out of nine (~ 22%) SPG11-HSP patients. There was an evident association between SPG7 mutations and cataract, as five out of six SPG7-HSP patients were identified with cataract (Fig. 8a).
White matter abnormalities (WMA)
We found all the included cases for HSP-SPG54 (7/7), 78% of SPG15-HSP cases (7/9), 73% of SPG11-HSP patients (85/116) and 20% of SPG7-HSP patients (3/15) manifested WMA (Fig. 8a).
Thin corpus callosum (TCC)
In the case of SPG11-HSP, 101 out of 114 patients (89%) reported having TCC. All HSP cases due to mutations in SPG15 (8 out of 8) and SPG54 (8 out of 8) reported showing TCC. None of the SPG7-HSP patients identified with TTC (Fig. 8a).
Pes cavus
Pes cavus was observed in 33% (15/46) of HSP cases due to mutations in SPG11, in 20% (1/5) of patients with mutations in SPG54, and 12% (1/8) of cases with mutations in SPG15. It seems that patients with mutated SPG11 were at significantly higher risk of developing pes cavus (Fig. 8a).
Dysarthria
Dysarthria was reported in both AD and AR-HSPs. For AD-HSP patients, only a number of those with mutated SPAST were diagnosed with dysarthria. Data evidenced that 4% of SPAST-HSP (11 out of 270 patients) has dysarthria (Fig. 8b).
In AR-HSPs, 62% (61/99) of SPG11-HSP patients, 83% (5/6) of SPG54-HSPs and 56% of SPG7-HSP cases (9/16) were found to have dysarthria (Fig. 8a).
Neuropathy
From 90, 46 patients with SPG11-HSP showed neuropathy. This is less frequent in SPG15-HSP (38%, 3 out of 8 patients), and in patients with mutations in SPG7 (15%, 2 out of 13 patients) (Fig. 8a). Neuropathy was more often observed in the SPG11 group than in two other groups.
In addition, in AD-HSP groups neuropathy was more often observed in REEP1-HSP patients (17%, 4 out of 24 patients) than those with mutated ATL1 (11%, 2 out of 18) and SPAST (2%, 6 out of 260 patients) (Fig. 8b).
The total number of mutation-positive patients with HSP who were diagnosed with other reported abnormalities was also derived from the studies; however, there was no significant association between mutated genes and the sub-phenotypes. A summary of the results of the association of sub-phenotypes with mutated genes is presented in Supplementary Tables 3–6.
Discussion
HSP denotes a degenerative, genetically heterogeneous group of neurological disorders that primarily affect the upper motor neurons [1]. Thus far, multiple investigations have demonstrated the frequency of distinct mutations in individual cases, families, and cohorts of various sizes, and analyzed phenotype–genotype associations [108,109,110]. The growing application of next-generation sequencing is not only leading to the improvement of diagnosis processes but also leading to the unremitting identification of new causal genes for HSP [2]. To establish robust associations between genotypes and clinically relevant phenotypes and also increase the statistical power, having conclusive data on larger cohorts is indispensable. Accordingly, to strengthen diagnosis, we pooled altogether 147 studies encompassing as many as 13,570 HSP patients according to the stringent quality criteria. The potentiality of evidence for specific genotype–phenotype species and the existence of a predictable relevance between the essential clinical outcomes with distinct genotypes were ascertained by the current meta-analysis. Hitherto, a total of 12 genes for AD-HSP and 41 genes for AR-HSP have been discerned [6]. We encompassed the following genes (SPAST, REEP1, ATL1, SPG11, SPG15, SPG7, SPG35, SPG54, and SPG5) in the study for which an adequate number of individuals for evaluation was feasible.
We are aware that there are some limitations regarding the current study. In some cases, we could not evaluate the relationship of the reported gene mutations and specific risk factors because of the unavailability of relatable data. Therefore, further studies are necessary to evaluate the predictive power of genotyping. Moreover, differences in genotyping methods, ethnical background of patients and study design could potentially impose bias into the obtained results. Nevertheless, the advantage of this study is its large sample size which enables statistical power to determine genotype–phenotype associations.
The weight of mutations in AD-HSP patients was on SPAST with a frequency of 25%, which is noticeably higher than REEP1 (3%) and ATL1 (5%). These findings are compatible with previous reports using a narrative methodology to review evidence of mutation frequency in HSPs [2].
However, it is not statically significant; the Asians showed a higher frequency of mutations in the SPAST gene (32.62%) in comparison to the Caucasians and Americans. Also, we found a higher frequency for mutations in the ATL1 gene in the Caucasian HSP patients (8%) in comparison to the Asian and American populations (4%). In descriptive reviews, it has been reported that mutations in SPAST account for nearly 40% of AD-HSP cases [6].
As for AR-HSP patients, the contribution of SPG11 mutations (18%) is higher than that of SPG15 (11%), SPG7 (13%), SPG35 (8%), SPG54 (7%), and SPG5 (5%). Almost the obtained results are in line with previous findings that showed SPG11 as the most common mutated gene in AR-HSP [99]. Interestingly, we found a high rate of SPG11 mutation in Asian HSP patients (87.98%). This further emphasizes differences in the genetic architecture of different ethnic groups. There are reports tried to provide explanations for the high rate of SPG11 mutations in populations. It has been determined that repeated Alu elements are one of the factors related to the SPG11 locus instability which might lead to substantial gene rearrangements of the genomic region [99].
Our result also suggested that the age of onsets varied among patients regardless of the mutated genes. It has been determined that the age of onset can be ranged from 0 to 73 [111]. The results of this meta-analysis suggested that HSP-AD patients with ATL1 mutations were younger than 10 years at the onset of the disease which are similar to the findings of the study carried by Namekawa et al. [81]. On the other hand, AD-HSP patients with mutated SPAST were older than 20 years old when they presented the disease. However, it has been reported that the average onset of AD-HSP in SPAST patients is mostly when they are in their 30s [6]. Among AR-HSP patients, SPG7 mutated ones presented the disease in the fourth decade of their life, which are older at diagnosis than AR-HSP patients with SPG11, SPG15, SPG35, and SPG5 mutations. Also, our results displayed a significant tendency toward younger age at HSP onset for Caucasian patients with mutated SPAST and Asian patients with mutated ATL1.
Since HSPs are diseases with diverse range for age at symptom onset, symptoms progression rate and level of disability even in patients from families with the same mutations [6], genotype–phenotype relationships are not straightforward. Several factors contribute to a high level of phenotype variability in HSPs.
In contrast to the general conception that HSP affects males and females to the same degree, our findings represented the predominance of male patients in some specific mutations, notably SPG7 in AR-HSP group and REEP1 in AD-HSP group, suggesting the possibility of sex-dependent penetrance or intensity of the disease [112]. This could be attributed to unknown modifying factors. In some cases (SPG4-related HSP), age for symptom onset and penetrance is dependent on the patient gender [6].
Regarding the outcome and disease course, there is considerable variability between different subtypes of HSPs. AD-HSP patients with REEP1 mutations more often present LL weakness, superficial sensory abnormalities, neuropathy, and deep sensory impairment. It has been reported that the manifestation of rare complicating features in REEP1-HSP patients is principally related peripheral nerve involvement [13]. Hence, as it has been determined, most REEP1 patients represent pure spastic paraplegia [113]. Additionally, AD-HSP patients with SPAST mutations presented more often with related symptoms including UL spasticity, UL hyperreflexia, ID, and dysarthria than patients with ATL1 mutations.
HSP-SPAST patients experience progressive degeneration of axons which lead to the weakness of lower limbs [114].
AR-HSP patients with SPG11 mutation had symptoms including pes cavus, neuropathy, and UL spasticity more often than other patients. Furthermore, the rate of SPG7 patients with cataract is higher than others. Klebe et al. demonstrated the optic abnormalities observed in all SPG7-positive patients [115]. Moreover, patients with mutated SPG54 present WMA more often, which could be due to the fact that SPG54 mutations were accompanied by accumulation of lipids [107]. Cognitive abnormalities, dysarthria, and TCC are most often manifested in SPG54 patients. All in all, it can be inferred from these findings that each specific mutations may have different phenotypes among HSP patients.
Here, we generated the extensive dataset available on genotype–phenotype associations in HSP. Our data exhibit the frequency of each symptom associated with the specific gene mutations which might have prognostic and therapeutic values. In the case of REEP1 mutations, a higher likelihood for unfavorable outcome including LL weakness, superficial sensory abnormalities, deep sensory impairment, and neuropathy was observed. In addition, AR-HSP patients with SPG11 mutations would expect symptoms such as pes cavus, neuropathy, and UL spasticity more often. However, we could not evaluate the relationship of some of the reported gene mutations and specific risk factors because of the unavailability of relatable data. Therefore, further studies are necessary to evaluate the predictive power of genotyping. Moreover, various genotyping methods or ethnical background of patients and also the evaluation of studies with different study design are limitations of this study. Also, the unavailability of clinical information of some patients, the genetic and clinical heterogeneity of the disease, and the limited number of studies regarding some countries impede the demonstration of more explicit genotype–phenotype relationships.
References
Noreau A, Dion PA, Rouleau GA (2014) Molecular aspects of hereditary spastic paraplegia. Exp Cell Res 325(1):18–26
Boutry M, Morais S, Stevanin G (2019) Update on the genetics of spastic paraplegias. Curr Neurol Neurosci Rep 19(4):18
Tesson C, Koht J, Stevanin G (2015) Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum Genet 134(6):511–538
Schlang KJ, Arning L, Epplen JT, Stemmler S (2008) Autosomal dominant hereditary spastic paraplegia: novel mutations in the REEP1 gene (SPG31). BMC Med Genet 9(1):71
D’Amore A, Tessa A, Casali C, Dotti MT, Filla A, Silvestri G et al (2018) Next generation molecular diagnosis of Hereditary Spastic Paraplegias: an Italian cross-sectional study. Front Neurol. 9:981
Giudice TL, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A (2014) Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 261:518–539
Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70(16):1384–1389
Chrestian N, Dupre N, Gan-Or Z, Szuto A, Chen S, Venkitachalam A et al (2017) Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol Genet 3(1):e122
Balicza P, Grosz Z, Gonzalez MA, Bencsik R, Pentelenyi K, Gal A et al (2016) Genetic background of the hereditary spastic paraplegia phenotypes in Hungary—an analysis of 58 probands. J Neurol Sci 364:116–121
Morais S, Raymond L, Mairey M, Coutinho P, Brandao E, Ribeiro P et al (2017) Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. EJHG 25(11):1217–1228
Adam M, Ardinger H, Pagon R, Wallace S, Bean L, Stephens K, et al. Hereditary spastic paraplegia overview—GeneReviews®
Faber J, Fonseca LM (2014) How sample size influences research outcomes. Dent Press J Orthodont 19(4):27–29
Beetz C, Schüle R, Deconinck T, Tran-Viet K-N, Zhu H, Kremer BP et al (2008) REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 131(4):1078–1086
Zhao G-h, Liu X-m (2017) Clinical features and genotype-phenotype correlation analysis in patients with ATL1 mutations: a literature reanalysis. Transl Neurodegen 6(1):9
Fink JK (2013) Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 126(3):307–328
Moher D, Liberati A, Tetzlaff J, Altman DG (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med 151(4):264–269
Orlacchio A, Patrono C, Borreca A, Babalini C, Bernardi G, Kawarai T (2008) Spastic paraplegia in Romania: high prevalence of SPG4 mutations. J Neurol Neurosurg Psychiatry 79(5):606–607
Erichsen A, Inderhaug E, Mattingsdal M, Eiklid K, Tallaksen C (2007) Seven novel mutations and four exon deletions in a collection of Norwegian patients with SPG4 hereditary spastic paraplegia. Eur J Neurol 14(7):809–814
Polymeris AA, Tessa A, Anagnostopoulou K, Rubegni A, Galatolo D, Dinopoulos A et al (2016) A series of Greek children with pure hereditary spastic paraplegia: clinical features and genetic findings. J Neurol 263(8):1604–1611
Martinuzzi A, Montanaro D, Vavla M, Paparella G, Bonanni P, Musumeci O et al (2016) Clinical and paraclinical indicators of motor system impairment in hereditary spastic paraplegia: a pilot study. PLoS One 11(4):e0153283
Magariello A, Muglia M, Patitucci A, Ungaro C, Mazzei R, Gabriele AL et al (2010) Mutation analysis of the SPG4 gene in Italian patients with pure and complicated forms of spastic paraplegia. J Neurol Sci 288(1–2):96–100
Sulek A, Elert E, Rajkiewicz M, Zdzienicka E, Stepniak I, Krysa W et al (2013) Screening for the hereditary spastic paraplaegias SPG4 and SPG3A with the multiplex ligation-dependent probe amplification technique in a large population of affected individuals. Neurol Sci 34(2):239–242
Mészárosová AU, Putzová M, Čermáková M, Vávrová D, Doležalová K, Smetanová I et al (2016) SPAST mutation spectrum and familial occurrence among Czech patients with pure hereditary spastic paraplegia. J Hum Genet 61(10):845
Tang B, Zhao G, Xia K, Pan Q, Luo W, Shen L et al (2004) Three novel mutations of the spastin gene in Chinese patients with hereditary spastic paraplegia. Arch Neurol 61(1):49–55
Boukhris A, Stevanin G, Feki I, Denora P, Elleuch N, Miladi M et al (2009) Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 75(6):527–536
Depienne C, Tallaksen C, Lephay J-Y, Bricka B, Poea-Guyon S, Fontaine B et al (2006) Spastin mutations are frequent in sporadic spastic paraparesis and their spectrum is different from that observed in familial cases. J Med Genet 43(3):259–265
Proukakis C, Auer-Grumbach M, Wagner K, Wilkinson P, Reid E, Patton M et al (2003) Screening of patients with hereditary spastic paraplegia reveals seven novel mutations in the SPG4 (Spastin) gene. Hum Mutat 21(2):170
McDermott C, Burness C, Kirby J, Cox L, Rao D, Hewamadduma C et al (2006) Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology. 67(1):45–51
Proukakis C, Hart PE, Cornish A, Warner TT, Crosby AH (2002) Three novel spastin (SPG4) mutations in families with autosomal dominant hereditary spastic paraplegia. J Neurol Sci 201(1–2):65–69
Patrono C, Scarano V, Cricchi F, Melone MA, Chiriaco M, Napolitano A et al (2005) Autosomal dominant hereditary spastic paraplegia: DHPLC-based mutation analysis of SPG4 reveals eleven novel mutations. Hum Mutat 25(5):506
Burguez D, Polese-Bonatto M, Scudeiro LAJ, Björkhem I, Schöls L, Jardim LB et al (2017) Clinical and molecular characterization of hereditary spastic paraplegias: a next-generation sequencing panel approach. J Neurol Sci 383:18–25
Orsucci D, Petrucci L, Ienco EC, Chico L, Simi P, Fogli A et al (2014) Hereditary spastic paraparesis in adults. A clinical and genetic perspective from Tuscany. Clin Neurol Neurosurg 120:14–19
Lynch DS, Koutsis G, Tucci A, Panas M, Baklou M, Breza M et al (2016) Hereditary spastic paraplegia in Greece: characterisation of a previously unexplored population using next-generation sequencing. Eur J Hum Genet 24(6):857
Elert-Dobkowska E, Stepniak I, Krysa W, Rajkiewicz M, Rakowicz M, Sobanska A et al (2015) Molecular spectrum of the SPAST, ATL1 and REEP1 gene mutations associated with the most common hereditary spastic paraplegias in a group of Polish patients. J Neurol Sci 359(1–2):35–39
Crippa F, Panzeri C, Martinuzzi A, Arnoldi A, Redaelli F, Tonelli A et al (2006) Eight novel mutations in SPG4 in a large sample of patients with hereditary spastic paraplegia. Arch Neurol 63(5):750–755
Vandebona H, Kerr N, Liang C, Sue C (2012) SPAST mutations in A ustralian patients with hereditary spastic paraplegia. Internal Med J 42(12):1342–1347
Ishiura H, Takahashi Y, Hayashi T, Saito K, Furuya H, Watanabe M et al (2014) Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses. J Hum Genet 59(3):163
Park H, Kang S-H, Park S, Kim SY, Seo SH, Lee SJ et al (2015) Mutational spectrum of the SPAST and ATL1 genes in Korean patients with hereditary spastic paraplegia. J Neurol Sci 357(1–2):167–172
Yabe I, Sasaki H, Tashiro K, Matsuura T, Takegami T, Satoh T (2002) Spastin gene mutation in Japanese with hereditary spastic paraplegia. J Med Genet 39(8):e46-e
Meijer IA, Hand CK, Cossette P, Figlewicz DA, Rouleau GA (2002) Spectrum of SPG4 mutations in a large collection of North American families with hereditary spastic paraplegia. Arch Neurol 59(2):281–286
Ivanova N, Löfgren A, Tournev I, Rousev R, Andreeva A, Jordanova A et al (2006) Spastin gene mutations in Bulgarian patients with hereditary spastic paraplegia. Clin Genet 70(6):490–495
Lindsey J, Lusher M, McDermott C, White K, Reid E, Rubinsztein D et al (2000) Mutation analysis of the spastin gene (SPG4) in patients with hereditary spastic paraparesis. J Med Genet 37(10):759–765
Loureiro JL, Brandão E, Ruano L, Brandão AF, Lopes AM, Thieleke-Matos C et al (2013) Autosomal dominant spastic paraplegias: a review of 89 families resulting from a portuguese survey. JAMA Neurol 70(4):481–487
Svenstrup K, Bross P, Koefoed P, Hjermind LE, Eiberg H, Born AP et al (2009) Sequence variants in SPAST, SPG3A and HSPD1 in hereditary spastic paraplegia. J Neurol Sci 284(1–2):90–95
Nanetti L, Baratta S, Panzeri M, Tomasello C, Lovati C, Azzollini J et al (2012) Novel and recurrent spastin mutations in a large series of SPG4 Italian families. Neurosci Lett 528(1):42–45
Travaglini L, Aiello C, Stregapede F, D’Amico A, Alesi V, Ciolfi A et al (2018) The impact of next-generation sequencing on the diagnosis of pediatric-onset hereditary spastic paraplegias: new genotype-phenotype correlations for rare HSP-related genes. Neurogenetics 19:111–121
Loureiro J, Miller-Fleming L, Thieleke-Matos C, Magalhães P, Cruz V, Coutinho P et al (2009) Novel SPG3A and SPG4 mutations in dominant spastic paraplegia families. Acta Neurol Scand 119(2):113–118
Braschinsky M, Tamm R, Beetz C, Sachez-Ferrero E, Raukas E, Lüüs S-M et al (2010) Unique spectrum of SPAST variants in Estonian HSP patients: presence of benign missense changes but lack of exonic rearrangements. BMC Neurol 10(1):17
Franca MC Jr, Dogini DB, D’Abreu A, Teive HA, Munhoz RP, Raskin S et al (2014) SPG4-related hereditary spastic paraplegia: frequency and mutation spectrum in Brazil. Clin Genet 86(2):194–196
McCorquodale DS 3rd, Ozomaro U, Huang J, Montenegro G, Kushman A, Citrigno L et al (2011) Mutation screening of spastin, atlastin, and REEP1 in hereditary spastic paraplegia. Clin Genet 79(6):523–530
Falco M, Scuderi C, Musumeci S, Sturnio M, Neri M, Bigoni S et al (2004) Two novel mutations in the spastin gene (SPG4) found by DHPLC mutation analysis. NMD 14(11):750–753
Lan MY, Fu SC, Chang YY, Wu-Chou YH, Lai SC, Chen RS et al (2012) Clinical and genetic analysis of four Taiwanese families with autosomal dominant hereditary spastic paraplegia. J Formosan Med Assoc Taiwan yi zhi. 111(7):380–385
Lan MY, Chang YY, Yeh TH, Lai SC, Liou CW, Kuo HC et al (2014) High frequency of SPG4 in Taiwanese families with autosomal dominant hereditary spastic paraplegia. BMC Neurol 14:216
Shoukier M, Neesen J, Sauter SM, Argyriou L, Doerwald N, Pantakani DV et al (2009) Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. EJHG 17(2):187–194
Zhao N, Sui Y, Li XF, Liu W, Lu YP, Feng WH et al (2015) Mutation analysis of four Chinese families with pure hereditary spastic paraplegia: pseudo- X-linked dominant inheritance and male lethality due to a novel ATL1 mutation. GMR 14(4):14690–14697
Ribai P, Depienne C, Fedirko E, Jothy AC, Viveweger C, Hahn-Barma V et al (2008) Mental deficiency in three families with SPG4 spastic paraplegia. EJHG 16(1):97–104
Fei QZ, Tang WG, Rong TY, Tang HD, Liu JR, Guo ZL et al (2011) Two novel mutations in the Spastin gene of Chinese patients with hereditary spastic paraplegia. Eur J Neurol 18(9):1194–1196
Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, et al. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long non-coding RNA. Diabetes. 2014:DB_140298
Battini R, Fogli A, Borghetti D, Michelucci A, Perazza S, Baldinotti F et al (2011) Clinical and genetic findings in a series of Italian children with pure hereditary spastic paraplegia. Eur J Neurol 18(1):150–157
Basri R, Yabe I, Soma H, Takei A, Nishimura H, Machino Y et al (2006) Four mutations of the spastin gene in Japanese families with spastic paraplegia. J Hum Genet 51(8):711–715
de Bot ST, van den Elzen RT, Mensenkamp AR, Schelhaas HJ, Willemsen MA, Knoers NV et al (2010) Hereditary spastic paraplegia due to SPAST mutations in 151 Dutch patients: new clinical aspects and 27 novel mutations. J Neurol Neurosurg Psychiatry 81(10):1073–1078
Sauter S, Miterski B, Klimpe S, Bonsch D, Schols L, Visbeck A et al (2002) Mutation analysis of the spastin gene (SPG4) in patients in Germany with autosomal dominant hereditary spastic paraplegia. Hum Mutat 20(2):127–132
Park SY, Ki CS, Kim HJ, Kim JW, Sung DH, Kim BJ et al (2005) Mutation analysis of SPG4 and SPG3A genes and its implication in molecular diagnosis of Korean patients with hereditary spastic paraplegia. Arch Neurol 62(7):1118–1121
Kim TH, Lee JH, Park YE, Shin JH, Nam TS, Kim HS et al (2014) Mutation analysis of SPAST, ATL1, and REEP1 in Korean patients with hereditary spastic paraplegia. J Clin Neurol (Seoul, Korea) 10(3):257–261
Kawarai T, Montecchiani C, Miyamoto R, Gaudiello F, Caltagirone C, Izumi Y et al (2017) Spastic paraplegia type 4: a novel SPAST splice site donor mutation and expansion of the phenotype variability. J Neurol Sci 380:92–97
Alvarez V, Sanchez-Ferrero E, Beetz C, Diaz M, Alonso B, Corao AI et al (2010) Mutational spectrum of the SPG4 (SPAST) and SPG3A (ATL1) genes in Spanish patients with hereditary spastic paraplegia. BMC Neurol 10:89
Chelban V, Tucci A, Lynch DS, Polke JM, Santos L, Jonvik H et al (2017) Truncating mutations in SPAST patients are associated with a high rate of psychiatric comorbidities in hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry 88(8):681–687
Lu X, Cen Z, Xie F, Ouyang Z, Zhang B, Zhao G et al (2014) Genetic analysis of SPG4 and SPG3A genes in a cohort of Chinese patients with hereditary spastic paraplegia. J Neurol Sci 347(1–2):368–371
Luo Y, Chen C, Zhan Z, Wang Y, Du J, Hu Z et al (2014) Mutation and clinical characteristics of autosomal-dominant hereditary spastic paraplegias in China. Neuro-degen Dis 14(4):176–183
Depienne C, Fedirko E, Forlani S, Cazeneuve C, Ribai P, Feki I et al (2007) Exon deletions of SPG4 are a frequent cause of hereditary spastic paraplegia. J Med Genet 44(4):281–284
Lynch DS, Koutsis G, Tucci A, Panas M, Baklou M, Breza M et al (2016) Hereditary spastic paraplegia in Greece: characterisation of a previously unexplored population using next-generation sequencing. EJHG 24(6):857–863
de Bot ST, Veldink JH, Vermeer S, Mensenkamp AR, Brugman F, Scheffer H et al (2013) ATL1 and REEP1 mutations in hereditary and sporadic upper motor neuron syndromes. J Neurol 260(3):869–875
Beetz C, Schule R, Deconinck T, Tran-Viet KN, Zhu H, Kremer BP et al (2008) REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 131(Pt 4):1078–1086
Goizet C, Depienne C, Benard G, Boukhris A, Mundwiller E, Sole G et al (2011) REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum Mutat 32(10):1118–1127
Elsayed LE, Mohammed IN, Hamed AA, Elseed MA, Johnson A, Mairey M et al (2016) Hereditary spastic paraplegias: identification of a novel SPG57 variant affecting TFG oligomerization and description of HSP subtypes in Sudan. EJHG 25(1):100–110
Travaglini L, Aiello C, Stregapede F, D’Amico A, Alesi V, Ciolfi A et al (2018) The impact of next-generation sequencing on the diagnosis of pediatric-onset hereditary spastic paraplegias: new genotype-phenotype correlations for rare HSP-related genes. Neurogenetics. 19(2):111–121
Smith BN, Bevan S, Vance C, Renwick P, Wilkinson P, Proukakis C et al (2009) Four novel SPG3A/atlastin mutations identified in autosomal dominant hereditary spastic paraplegia kindreds with intra-familial variability in age of onset and complex phenotype. Clin Genet 75(5):485–489
Durr A, Camuzat A, Colin E, Tallaksen C, Hannequin D, Coutinho P et al (2004) Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia. Arch Neurol 61(12):1867–1872
Meszarosova AU, Grecmalova D, Brazdilova M, Dvorackova N, Kalina Z, Cermakova M et al (2017) Disease-causing variants in the ATL1 gene are a rare cause of hereditary spastic paraplegia among Czech patients. Ann Hum Genet 81(6):249–257
Abel A, Fonknechten N, Hofer A, Durr A, Cruaud C, Voit T et al (2004) Early onset autosomal dominant spastic paraplegia caused by novel mutations in SPG3A. Neurogenetics 5(4):239–243
Namekawa M, Ribai P, Nelson I, Forlani S, Fellmann F, Goizet C et al (2006) SPG3A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology 66(1):112–114
Sauter SM, Engel W, Neumann LM, Kunze J, Neesen J (2004) Novel mutations in the Atlastin gene (SPG3A) in families with autosomal dominant hereditary spastic paraplegia and evidence for late onset forms of HSP linked to the SPG3A locus. Hum Mutat 23(1):98
Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L et al (2014) Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 137(Pt 7):1907–1920
Franca MC Jr, Yasuda CL, Pereira FR, D’Abreu A, Lopes-Ramos CM, Rosa MV et al (2012) White and grey matter abnormalities in patients with SPG11 mutations. J Neurol Neurosurg Psychiatry 83(8):828–833
Kim SM, Lee JS, Kim S, Kim HJ, Kim MH, Lee KM et al (2009) Novel compound heterozygous mutations of the SPG11 gene in Korean families with hereditary spastic paraplegia with thin corpus callosum. J Neurol 256(10):1714–1718
Lee MJ, Cheng TW, Hua MS, Pan MK, Wang J, Stephenson DA et al (2008) Mutations of the SPG11 gene in patients with autosomal recessive spastic paraparesis and thin corpus callosum. J Neurol Neurosurg Psychiatry 79(5):607–609
Liao SS, Shen L, Du J, Zhao GH, Wang XY, Yang Y et al (2008) Novel mutations of the SPG11 gene in hereditary spastic paraplegia with thin corpus callosum. J Neurol Sci 275(1–2):92–99
Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70(16 Pt 2):1384–1389
Paisan-Ruiz C, Nath P, Wood NW, Singleton A, Houlden H (2008) Clinical heterogeneity and genotype-phenotype correlations in hereditary spastic paraplegia because of Spatacsin mutations (SPG11). Eur J Neurol 15(10):1065–1070
Samaranch L, Riverol M, Masdeu JC, Lorenzo E, Vidal-Taboada JM, Irigoyen J et al (2008) SPG11 compound mutations in spastic paraparesis with thin corpus callosum. Neurology 71(5):332–336
Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A et al (2008) Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131(Pt 3):772–784
Yoon G, Baskin B, Tarnopolsky M, Boycott KM, Geraghty MT, Sell E et al (2013) Autosomal recessive hereditary spastic paraplegia-clinical and genetic characteristics of a well-defined cohort. Neurogenetics 14(3–4):181–188
Boukhris A, Stevanin G, Feki I, Denis E, Elleuch N, Miladi MI et al (2008) Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia: SPG11, SPG15, and further genetic heterogeneity. Arch Neurol 65(3):393–402
Crimella C, Arnoldi A, Crippa F, Mostacciuolo ML, Boaretto F, Sironi M et al (2009) Point mutations and a large intragenic deletion in SPG11 in complicated spastic paraplegia without thin corpus callosum. J Med Genet 46(5):345–351
Denora PS, Smets K, Zolfanelli F, Ceuterick-de Groote C, Casali C, Deconinck T et al (2016) Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions. Brain 139(Pt 6):1723–1734
de Bot ST, Burggraaff RC, Herkert JC, Schelhaas HJ, Post B, Diekstra A et al (2013) Rapidly deteriorating course in Dutch hereditary spastic paraplegia type 11 patients. EJHG 21(11):1312–1315
Du J, Hu YC, Tang BS, Jiang H, Shen L (2018) Identification of novel SPG11 mutations in a cohort of Chinese families with hereditary spastic paraplegia. Int J Neurosci 128(2):146–150
Erichsen AK, Stevanin G, Denora P, Brice A, Tallaksen CM (2008) SPG11–the most common type of recessive spastic paraplegia in Norway? Acta Neurol Scand Suppl 188:46–50
Conceicao Pereira M, Loureiro JL, Pinto-Basto J, Brandao E, Margarida Lopes A, Neves G et al (2012) Alu elements mediate large SPG11 gene rearrangements: further spatacsin mutations. Genet Med 14(1):143–151
Goizet C, Boukhris A, Maltete D, Guyant-Marechal L, Truchetto J, Mundwiller E et al (2009) SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology 73(14):1111–1119
Bhattacharjee S, Beauchamp N, Murray BE, Lynch T (2017) Case series of autosomal recessive hereditary spastic paraparesis with novel mutation in SPG 7 gene. Neurosciences (Riyadh, Saudi Arabia) 22(4):303–307
Soehn AS, Rattay TW, Beck-Wodl S, Schaferhoff K, Monk D, Dobler-Neumann M et al (2016) Uniparental disomy of chromosome 16 unmasks recessive mutations of FA2H/SPG35 in 4 families. Neurology 87(2):186–191
Liao X, Luo Y, Zhan Z, Du J, Hu Z, Wang J et al (2015) SPG35 contributes to the second common subtype of AR-HSP in China: frequency analysis and functional characterization of FA2H gene mutations. Clin Genet 87(1):85–89
Citterio A, Arnoldi A, Panzeri E, D’Angelo MG, Filosto M, Dilena R et al (2014) Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J Neurol 261(2):373–381
Kumar KR, Wali GM, Kamate M, Wali G, Minoche AE, Puttick C et al (2016) Defining the genetic basis of early onset hereditary spastic paraplegia using whole genome sequencing. Neurogenetics 17(4):265–270
Gonzalez M, Nampoothiri S, Kornblum C, Oteyza AC, Walter J, Konidari I et al (2013) Mutations in phospholipase DDHD2 cause autosomal recessive hereditary spastic paraplegia (SPG54). EJHG 21(11):1214–1218
Schuurs-Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, Ben-Salem S, de Bot ST, Nijhof B et al (2012) Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet 91(6):1073–1081
Helbig KL, Hedrich UB, Shinde DN, Krey I, Teichmann AC, Hentschel J, et al. A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann Neurol. 2016;80(4):638–642
Kumar KR, Blair NF, Sue CM (2015) An update on the hereditary spastic paraplegias: new genes and new disease models. Mov Disord Clin Pract 2(3):213–223
Faber I, Branco LMT, Júnior F, Cavalvante M (2016) Cognitive dysfunction in hereditary spastic paraplegias and other motor neuron disorders. Dement Neuropsychol 10(4):276–279
Schüle R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S et al (2016) Hereditary spastic paraplegia: clinicogenetic lessons from 608 patients. Ann Neurol 79(4):646–658
Fjermestad KW, Kanavin ØJ, Næss EE, Hoxmark LB, Hummelvoll G (2016) Health survey of adults with hereditary spastic paraparesis compared to population study controls. Orphanet J Rare Dis 11(1):98
Züchner S, Wang G, Tran-Viet K-N, Nance MA, Gaskell PC, Vance JM et al (2006) Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet 79(2):365–369
Solowska JM, Rao AN, Baas PW (2017) Truncating mutations of SPAST associated with hereditary spastic paraplegia indicate greater accumulation and toxicity of the M1 isoform of spastin. Mol Biol Cell 28(13):1728–1737
Klebe S, Depienne C, Gerber S, Challe G, Anheim M, Charles P et al (2012) Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 135(10):2980–2993
Acknowledgements
The authors thank the Shahid Beheshti University of Medical Sciences for continuous support.
Funding
The presented article is financially supported by “Research Department of the School of Medicine Shahid Beheshti University of Medical Sciences”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Erfanian Omidvar, M., Torkamandi, S., Rezaei, S. et al. Genotype–phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J Neurol 268, 2065–2082 (2021). https://doi.org/10.1007/s00415-019-09633-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09633-1