
Abstract
Impulse control disorders (ICDs) occur in a subset of patients with Parkinson’s disease (PD) who are receiving dopamine replacement therapy. In this study, we aimed to investigate structural abnormalities within the mesocortical and limbic cortices and subcortical structures in PD patients with ICDs. We studied 18 PD patients with ICDs, 18 PD patients without ICDs and a group of 24 age and sex-matched healthy controls. Cortical thickness (CTh) and subcortical nuclei volume analyses were carried out using the automated surface-based analysis package FreeSurfer (version 5.3.0). We found significant differences in MRI measures between the three groups. There was volume loss in the nucleus accumbens of both PD patients with ICDs and without ICDs compared to the control group. In addition, PD patients with ICDs showed significant atrophy in caudate, hippocampus and amygdala compared to the group of healthy controls. PD patients with ICDs had significant increased cortical thickness in rostral anterior cingulate cortex and frontal pole compared to PD patients without ICDs. Cortical thickness in rostral anterior cingulate and frontal pole was increased in PD patients with ICDs compared to the control group, but the differences failed to reach corrected levels of statistical significance. PD patients with ICDs showed increased cortical thickness in medial prefrontal regions. We speculate that these findings reflect either a pre-existing neural trait vulnerability to impulsivity or the expression of a maladaptive synaptic plasticity under non-physiological dopaminergic stimulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Impulse control disorders (ICDs) are characterized by a loss of voluntary control over impulses or drives that lead to repetitive, excessive and self-destructive behaviors of sufficient magnitude to interfere adversely with daily functioning [1]. ICDs have been estimate to occur in up to 14 % of Parkinson’s disease (PD) patients and are associated with dopamine replacement therapy particularly oral dopamine agonist use as well as high levels of trait impulsivity [2]. The mechanisms underlying the development of ICDs in PD are still poorly understood and their management can be challenging [3].
Positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) studies have demonstrated mesocortical and limbic network dysfunction during valuation (risk–reward processing), motivation, decision making and inhibitory control in PD patients with ICDs [4–9]. Using serial [11C]raclopride PET scanning to acquire indirect measures of levodopa-induced striatal dopamine release, it was shown that medicated PD patients with ICDs had higher ventral striatal dopamine release during a reward-related cue reactivity paradigm compared to PD patients without ICDs [4, 5]. A single photon emission computed tomography (SPECT) connectivity analysis found specific functional disconnection between anterior cingulate cortex (ACC) and striatum in PD patients with pathological gambling [6]. Administration of dopamine agonists reduced regional cerebral blood flow as measured by H2O15 PET in inhibitory frontolimbic network in PD patients with pathological gambling [7]. In PD patients with pathological gambling or compulsive shopping, it was shown that dopamine agonist exposure determines faster gain (reward) learning and greater choice bias for gain cues and is accompanied by increased ventral striatal-related blood-oxygen level-dependent activity on fMRI [8]. In PD patients with hyperlibidinous behavior, exposure to erotic images resulted in enhanced activation of ventral striatum and the cingulate and orbitofrontal cortices compared to PD controls [9]. These findings suggest that there is dysregulated reward processing in PD patients with ICDs including enhanced ventral striatum activity and poor cortical inhibitory control.
Antecedent anatomical differences might underlie an increased vulnerability to ICDs [10]. If this is the case, structural MRI studies may help in clarifying why some patients are at greater risk for impulsivity. Alternatively, they may reflect the impact of non-physiological dopaminergic stimulation on the brain in sensitized individuals [11]. Recent data demonstrated reduced local gyrification index and gray matter atrophy in orbitofrontal cortex of PD patients with pathological gambling [12] and significant cortical thinning in fronto-striatal circuitry of PD patients with ICDs [13].
In this study, we have used high-resolution structural 3-T MRI and FreeSurfer automated reconstruction of cortical thickness (CTh) and subcortical nuclei to investigate macrostructural alterations in mesocorticolimbic regions in medicated PD patients with ICDs relative to PD patients without ICDs and healthy control participants. We hypothesized that the presence of ICDs would be associated with structural alterations in brain reward circuitry.
Methods
Participants and clinical characteristics
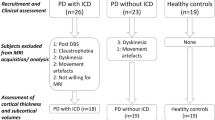
Thirty-six non-demented patients with Parkinson’s disease (PD) who fulfilled the Queen Square Brain Bank criteria [14] were recruited from specialist movement disorders clinics at Imperial College Healthcare NHS Trust and King’s Health Partners (Table 1). Eighteen of these satisfied the diagnostic criteria for one or more current ICDs according the Questionnaire for Impulsive–Compulsive Disorders in Parkinson’s Disease (QUIP) [15] and a formal diagnosis of ICD was made on the basis of the Diagnostic and Statistical Manual of Mental Disorders IV Text Revision (DSM-IV-TR) [16] following a semi-structured interview. Different types of ICDs including: pathological gambling, compulsive shopping, hypersexuality, binge eating, dopamine dysregulation syndrome, and problematic internet use were described by the patients and half had more than one ICD (Table 2). The other 18 PD patients had no history of ICDs. Twenty-four healthy controls (HC) matched for age and sex with no history of neurological or psychiatric disorders served as the control group.
Motor symptom severity was assessed with the Unified Parkinson’s Disease Rating Scale (UPDRS) part-III [17] (UPDRS-III) and staged with Hoehn and Yahr scale [18] (H&Y). Global cognitive status was assessed by Mini-Mental State Examination [19] (MMSE) in all participants. Dopamine replacement therapy was calculated as daily levodopa equivalents (LED) for each patient based on theoretical equivalence to levodopa [20].
The study was approved by the institutional review boards and the local research ethics committee. Written informed consent was obtained from all study participants in accordance with the Declaration of Helsinki.
Data acquisition and processing
All participants were scanned on a 3-T Philips Intera whole-body MRI scanner. A T1-weighted three-dimensional magnetization-prepared rapid acquisition gradient-echo (MPRAGE; time echo: 4.6 ms; time repetition: 9.7 ms; field of view: 240 ms; voxel size = 1 × 1 × 1 mm) was acquired. All planar sequence acquisitions were acquired along the Anterior Commissure–Posterior Commissure (AC-PC) line. Particular care was taken to center the subject’s head in the head coil and to restrain his/her movements with cushions and pads. Further, a medical tape going from end-to-end of the coil was set, such that the adhesive parts guarantee grip to the coil itself, while the non-adhesive part softly touches subject’s forehead. In this way if the subject (even modestly) moves his/her head, he/she will immediately feel the tape touch, thus preventing him/her from moving further [21].
FreeSurfer’s image analysis suite (version 5.3.0 http://surfer.nmr.mgh.harvard.edu) processing pipeline was used to derive measures of cortical thickness (CTh) and deep gray matter (GM) nuclei volume. This process consists of several stages, detailed elsewhere [22, 23]. Briefly, the whole-brain T1-weighted images underwent a correction for intensity homogeneity, skull strip, and segmentation into GM and white matter (WM) with intensity gradient and connectivity among voxels. CTh was measured as the distance from the gray/white matter boundary to the corresponding pial surface. Reconstructed data sets were visually inspected for accuracy, and segmentation errors were corrected. Subcortical structure volumes were derived by automated procedures, which automatically assign a neuroanatomical label to each voxel in an MRI volume based on probabilistic information automatically estimated from a manually labeled training set [23]. The accuracy of FreeSurfer estimations has been validated against histologic measurements [24] and expert manual delineation [25].
We considered CTh measures of 10 selected cortical regions belonging to the mesocorticolimbic network. Specifically, we included from the frontal lobe: medial and lateral orbitofrontal cortices; frontal pole; superior frontal cortex; and rostral middle frontal cortex. From the temporal lobe: parahippocampal cortex; entorhinal cortex; and fusiform cortex. From cingulate areas: caudal anterior cingulate cortex and rostral anterior cingulate cortex. We included the following subcortical GM nuclei: caudate, putamen, globus pallidum, hippocampus, nucleus accumbens and amygdala. All individual volumes were normalized for intracranial volume (ICV) automatically generated by FreeSurfer [26]. Since no laterality was observed and for minimizing the number of comparisons, average hemispheric CTh and deep GM values were processed in the statistical analysis.
Statistical analysis
Statistical analyses were performed using Statistical Package for Social Science version 21.0. (SPSS, Inc. Chicago, IL, USA). For all variables, homogeneity and Gaussianity were tested with Bartlett and Kolmogorov–Smirnov tests. Demographic and clinical characteristics among groups were assessed by univariate analysis of variance with post hoc Bonferroni’s correction for continuous variables and by Chi-square test for categorical variables. Differences in clinical characteristics between PD patients with and without ICDs were analyzed using unpaired t test and Chi-square test where appropriate. First, we run a one-way multivariate analysis of variance (MANOVA) with a single independent variable (i.e., group) with three levels to investigate differences in CTh and subcortical GM nuclei volume among groups. The omnibus level of significance for MANOVA was set at P < 0.05. If the MANOVA was significant, we conducted a series of one-way ANOVAs, followed by Bonferroni’s multiple-comparisons post hoc tests (P < 0.05 Bonferroni corrected), to compare means among groups. The level of significance was set at P < 0.05 for the ANOVA comparative measurements. In addition, even though age was not significantly different among groups, the multivariate analysis was repeated adding age as covariate. Subsequently, to clarify our main research question, MRI data between the groups of PD with ICDs and PD without ICDs were compared for F and P values with multivariate analysis of co-variance (MANCOVA) using disease duration, and UPDRS-III score as covariates. If MANCOVA P values were significant (P < 0.05), we calculated a series of one-way ANOVAs to compare means between-group. The level of significance was set at P < 0.05 for the ANOVA comparative measurements. Although we did not find a significant difference between the groups of PD with ICDs and PD without ICDs in total daily LED, we run an additional MANCOVA between PD groups using total daily LED, disease duration and UPDRS-III as covariates. In addition, even though age was not significantly different among groups, all the multivariate analyses were repeated adding age as covariate. We investigated differences in clinical and morphometrics data between PD patients with one ICDs and PD patients with multiple ICDs using MANOVA. Further, we ran linear regression analysis split by group (i.e., PD with ICDs; PD without ICDs) between total daily LED and those brain structures showing significant differences between PD groups by the results of MANCOVA. All data are presented as mean ± SD, and the level α was set for all comparisons at P < 0.05, corrected.
Results
Structural changes: comparisons to normality
We found significant differences in MRI measures among the three groups (F 2,57 = 2.62, P = 0.001, \(\eta_{p}^{2}\) = 0.494). Bonferroni’s post hoc tests showed volume loss in the nucleus accumbens of both PD patient groups: with ICDs (P = 0.001) and PD patients without ICDs (P = 0.001) when compared to healthy controls. PD patients with ICDs showed further significant atrophy in caudate (P = 0.02), hippocampus (P = 0.04) and amygdala (P = 0.03) compared to healthy controls. Cortical thickness in rostral anterior cingulate and frontal pole was increased in PD patients with ICDs compared to the control group, but the differences failed to reach corrected levels of statistical significance (Table 3). Covarying for age did not influence results.
Structural changes: comparisons within Parkinson’s patients
Since PD patients with ICDs had significant longer disease duration (P = 0.02) and higher UPDRS-III scores (P < 0.01) than PD patients without ICDs, we investigated differences in MRI measures between PD patients with and without ICDs using disease duration and UPDRS-III score as covariates (F 1,32 = 2.97, P = 0.02, \(\eta_{p}^{2}\) = 0.737). We found that Cth of rostral anterior cingulate cortex (P = 0.002) and frontal pole (P = 0.001) were thicker in PD patients with ICDs compared to the group of PD patients without ICDs (Fig. 1; Table 4). The difference between the PD groups in total daily LED was not significant although it was considerable (Table 1), therefore, we further calculated an additional MANCOVA including total daily LED, disease duration and UPDRS-III score as covariates (F 1,31 = 2.81, P = 0.02, \(\eta_{p}^{2}\) = 0.74). This analysis confirmed our previous findings. Cth of rostral anterior cingulate cortex (P = 0.002) and frontal pole (P = 0.001) were thicker in PD patients with ICDs compared to the group of PD patients without ICDs. Further, covarying for age did not influence results. The linear regression analyses split by group between Cth of rostral anterior cingulate cortex and total daily LED and Cth of frontal pole and total daily LED were not significant in both groups.

Cortical thickness differences between Parkinson’s disease patients with and without impulse control disorders. For display purpose only, we have visualized our results using the whole-brain GLM analysis provided by qdec. Given the aim of the figure, FDR and Monte Carlo correction were not applied. The figure shows sagittal section of cortical areas with significant increased cortical thickness (i.e., rostral anterior cingulate, left side; frontal pole, right side) between PD patients with and without ICDs corrected for disease duration and UPDRS-III score. Difference maps are shown via a pseudo-color statistical overlay where black represents P 0.1 and yellow P 0.001. Red and yellow indicate thicker cortex in PD patients with ICDs with respect to patients without ICDs
No significant differences were found in clinical and morphometric data between PD patients with one ICDs and PD patients with multiple ICDs.
Discussion
ICDs are relatively common, impairing and distressing complications of dopamine replacement therapy in patients with PD. Our findings demonstrate structural alterations in mesocortical and limbic reward-related areas including volume loss in nucleus accumbens, caudate, hippocampus and amygdala, and increased thickening of anterior cingulate (Brodmann Area-BA 32) and frontal pole (part of BA 10) cortices in PD patients with ICDs.
The hub of the reward network is the cortico-ventral basal ganglia circuit including the orbitofrontal cortex and anterior cingulate cortex, the ventral striatum, comprising the nucleus accumbens, the amygdala, the hippocampus and the midbrain dopamine neurons (i.e., ventral tegmental area A10) [27, 28]. Dopaminergic signals originating in the midbrain and which propagate throughout reward circuitry reflect salience attribution, imprinting an incentive value on rewards and motivating appetitive behavior towards associated stimuli. The incentive sensitization theory propounded by Berridge and Robinson proposes that, as a result of chronic dopaminergic overstimulation, elevation of mesolimbic dopamine reactivity via neural sensitization magnifies “wanting” or craving for rewarding stimuli independently of reward “liking” [29]. Excess dopaminergic stimulation may also contribute to abnormally strong or aberrant forms of reward learning, altered coding of risk and impaired decision making [30].
Our results show significant volumetric differences in several regions within the reward pathways, which reached corrected levels of statistical significance in PD ICD patients compared to healthy controls. We speculate that subcortical alterations in PD patients with ICDs may constitute a predisposing neural trait linked to impulsivity. Volumetric reductions in the accumbens and amygdala have previously been described in individuals at increased genetic risk for addiction [31] or with high levels of trait impulsivity [32] and a recent study of rats bred to be highly impulsive found volume reductions in the nucleus accumbens [10]. Alternatively, our findings might reflect the expression of secondary rearrangements of mesocorticolimbic system including derangement of deep gray matter nuclei and subsequent cortical remodeling. Indeed, a recent study in mice found that neural sensitization following repeated exposure to cocaine resulted in volume reductions in striatum and amygdala [11].
It is of interest that we found increased cortical thickness in specific frontal cortical areas, in particular rostral anterior cingulate and frontal polar cortex. The rostral anterior cingulate cortex and frontal polar region form part of a medial frontal neural network implicated in the control of various autonomic and neuroendocrine functions [33], mediating subjective reward experience [34], and are believed to be important for action selection, error and conflict monitoring and value-based decision making [35].
While these increases in thickness may be related to a pre-existing neurobiological trait making subjects with thicker cortex more impulsive [36], they may also be secondary to neuroplastic adaptations associated with non-physiological dopaminergic stimulation occurring only in subjects with increased susceptibility to neural sensitization. Analogous levodopa-induced increases in dorsal striatal dopamine release found in PD patients with ICDs are observed in PD patients with levodopa-induced dyskinesia (LIDs) [37]. Although LIDs and ICDs may occur under different clinical conditions (i.e., at different time from disease onset, levodopa vs. dopamine agonist intake, different prevalence), abnormal behaviors in ICDs might parallel involuntary movements of LIDs as part of a pathological spectrum influenced by chronic non-physiological dopaminergic transmission and as results of similar form of neural sensitization operating on different brain systems [38].
Recent structural MRI studies have demonstrated increased thickness in the right inferior frontal sulcus of PD patients with LIDs compared to PD patients without LIDs [39, 40]. The combined effects of dopamine denervation and chronic dopamine replacement therapy might determine morphological alterations on cortico-striatal networks. Increased synaptic levels of dopamine and post-synaptic modifications such as changes in proteins and genes might determine an aberrant increase in the dendritic arborization, neuronal sprouting and neuropil and result in maladaptive synaptic plasticity [41–45]. Therefore, given the critical role of inferior frontal sulcus in the regulation of action, abnormal cytoarchitectonic modifications of this specific cortical region determine a dysfunction in a motor inhibition network. In our study, we found increased thickness in medial frontal cortical areas involved in reward valuation and decision making. We speculate that analogous maladaptive synaptic plasticity could occur in these cortical areas resulting in the behavioral manifestations of ICDs. Notably, frontal CTh is similarly increased in cocaine-sensitized animals [11] and these macrostructural alterations may result from increase in dendritic length and spine density [46].
In a recent study, Biundo et al. [13] also found morphometric alterations in fronto-striatal-limbic circuitry of PD patients with ICDs. These researchers applied 1.5 T MRI scans and FreeSurfer automated reconstruction in a group of PD patients with and without ICDs and healthy controls. They found thinning of several areas within prefrontal cortex and relative volume increase of left amygdala in PD patients with ICDs. It is possible that the divergences observed with our study might be in part due to the different magnet strength employed and to the dissimilar analysis approach chosen: a whole-brain vertex-wise versus region of interest (ROI)-based analysis driven by a robust a priori hypothesis. Furthermore, one cannot exclude that the inclusion of patients with significant cognitive impairment in Biundo’s study might account for discrepancies between the two sets of findings.
The PD patients with ICDs in our study had more severe parkinsonian symptoms as expressed by higher UPDRS-III scores and longer disease duration. However, we reduced the effect of these differences in our analysis using both parameters as covariates thus minimizing disease severity as an explanation for our findings. Our results held when accounting for the effect of daily total dopaminergic dosage. Moreover, the regression analysis between brain structures and total daily LED was not significant. These findings indicate that morphometric changes might be independent of Parkinson’s disease severity and/or dopaminergic dosage per se.
We also acknowledge the limitations of our study. The relative small sample size did not allow specific analysis of different variants of ICDs and a comprehensive neuropsychological and psychiatric evaluation was not performed on every patient. Moreover, we did not perform a scale providing severity score for each ICDs and this has hampered correlation analysis between morphometric data and ICDs severity. Further studies on larger series investigating a more homogeneous group of PD patients with a longitudinal design and using multimodal imaging analysis will be needed to confirm our results.
In conclusion, this study provides evidence of morphological abnormalities in PD patients with ICDs. We found increased cortical thickness in cortical regions involved in reward valuation and decision making, as well as reduced volumes of subcortical components of reward circuitry. Our results suggest similarities between ICDs and LIDs as analogous side effects of non-physiologic chronic dopaminergic stimulation.
References
Wu K, Politis M, Piccini P (2009) Parkinson disease and impulse control disorders: a review of clinical features, pathophysiology and management. Postgrad Med J 85:590–596
Weintraub D, Koester J, Potenza MN et al (2010) Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 67:589–595
Leeman RF, Billingsley BE, Potenza MN (2012) Impulse control disorders in Parkinson’s disease: background and update on prevention and management. Neurodegener Dis Manag 2:389–400
O’Sullivan SS, Wu K, Politis M et al (2011) Cue-induced striatal dopamine release in Parkinson’s disease-associated impulsive–compulsive behaviours. Brain 134:969–978
Steeves TD, Miyasaki J, Zurowski M et al (2009) Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a [11C] raclopride PET study. Brain 132:1376–1385
Cilia R, Cho SS, van Eimeren T et al (2011) Pathological gambling in patients with Parkinson’s disease is associated with fronto-striatal disconnection: a path modeling analysis. Mov Disord 26:225–233
vanEimeren T, Pellecchia G, Cilia R et al (2010) Drug-induced deactivation of inhibitory networks predicts pathological gambling in PD. Neurology 75:1711–1716
Voon V, Pessiglione M, Brezing C et al (2010) Mechanisms underlying dopamine-mediated reward bias in compulsive behaviors. Neuron 65:135–142
Politis M, Loane C, Wu K et al (2013) Neural response to visual sexual cues in dopamine treatment-linked hypersexuality in Parkinson’s disease. Brain 136:400–411
Caprioli D, Sawiak SJ, Merlo E et al (2014) Gamma aminobutyric acidergic and neuronal structural markers in the nucleus accumbens core underlie trait-like impulsive behavior. Biol Psychiatry 75:115–123
Wheeler AL, Lerch JP, Chakravarty MM et al (2013) Adolescent cocaine exposure causes enduring macroscale changes in mouse brain structure. J Neurosci 33:1797–1803a
Cerasa A, Salsone M, Nigro S et al (2014) Cortical volume and folding abnormalities in Parkinson’s disease patients with pathological gambling. Parkinsonism Relat Disord. pii: S1353-8020(14)00323-X [Epub ahead of print]
Biundo R, Weis L, Facchini S et al (2015) Patterns of cortical thickness associated with impulse control disorders in Parkinson’s disease. Mov Disord. doi:10.1002/mds.26154 [Epub ahead of print]
Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56:33–39
Weintraub D, Hoops S, Shea JA et al (2009) Validation of the questionnaire for impulsive–compulsive disorders in Parkinson’s disease. Mov Disord 24:1461–1467
American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders, 4th edn. American Psychiatric Association, Washington, DC
Movement Disorder Society Task Force on Rating Scales for Parkinson’s Disease (2013) The Unified Parkinson’s Disease Rating Scale (UPDRS): status and recommendations. Mov Disord 18:738–750
Goetz CG, Poewe W, Rascol O et al (2004) Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord 19:1020–1028
Folstein MF, Folstein SE, McHugh PR (1975) “Mini Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiat Res 12:189–198
Politis M, Wu K, Loane C et al (2010) Staging of serotonergic dysfunction in Parkinson’s disease: an in vivo 11C-DASB PET study. Neurobiol Dis 40:216–221
Reuter M, Tisdall MD, Qureshi A, Buckner RL, van der Kouwe AJ, Fischl B (2015) Head motion during MRI acquisition reduces gray matter volume and thickness estimates. Neuroimage 107:107–115
Fischl B, Salat DH, van der Kouwe AJ et al (2004) Sequence-independent segmentation of magnetic resonance images. Neuroimage 23(Suppl 1):S69–S84
Fischl B, Salat DH, Busa E et al (2002) Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 33:341–355
Cardinale F, Chinnici G, Bramerio M et al (2014) Validation of FreeSurfer-estimated brain cortical thickness: comparison with histologic measurements. Neuroinformatics 12:535–542
Mulder ER, de Jong RA, Knol DL (2014) Hippocampal volume change measurement: quantitative assessment of the reproducibility of expert manual outlining and the automated methods FreeSurfer and FIRST. Neuroimage 92:169–181
Malone IB, Leung KK, Clegg S et al (2015) Accurate automatic estimation of total intracranial volume: a nuisance variable with less nuisance. Neuroimage 104:366–372
Robinson TE, Berridge KC (2003) Addiction. Annu Rev Psychol 54:25–53
Haber SN (2014) The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 282:248–257
Berridge KC (2007) The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacology 191:391–431
Schultz W (2011) Potential vulnerabilities of neuronal reward, risk, and decision mechanisms to addictive drugs. Neuron 69:603–617
Hill SY, De Bellis MD, Keshavan MS et al (2001) Right amygdala volume in adolescent and young adult offspring from families at high risk for developing alcoholism. Biol Psychiatry 49:894–905
Muhlert N, Lawrence AD (2015) Brain structure correlates of emotion-based rash impulsivity. Neuroimage 115:138–146
Price JL (2005) Free will versus survival: brain systems that underlie intrinsic constraints on behavior. Comp Neurol 493:132–139
Kringelbach ML (2005) The human orbitofrontal cortex: linking reward to hedonic experience. Nat Rev Neurosci 6:691–702
Rushworth MF, Kolling N, Sallet J, Mars RB (2012) Valuation and decision-making in frontal cortex: one or many serial or parallel systems? Curr Opin Neurobiol 22:946–955
Cho SS, Pellecchia G, Aminian K et al (2013) Morphometric correlation of impulsivity in medial prefrontal cortex. Brain Topogr 26:479–487
Politis M, Wu K, Loane C et al (2014) Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J Clin Invest 124:1340–1349
Voon V, Fernagut PO, Wickens J et al (2009) Chronic dopaminergic stimulation in Parkinson’s disease: from dyskinesias to impulse control disorders. Lancet Neurol 8:1140–1149
Cerasa A, Messina D, Pugliese P et al (2011) Increased prefrontal volume in PD with levodopa-induced dyskinesias: a voxel-based morphometry study. Mov Disord 26:807–812
Cerasa A, Morelli M, Augimeri A et al (2013) Prefrontal thickening in PD with levodopa-induced dyskinesias: new evidence from cortical thickness measurement. Parkinsonism Relat Disord 19:123–125
Morgante F, Espay AJ, Gunraj C, Lang AE, Chen R (2006) Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain 129:1059–1069
Cerasa A, Fasano A, Morgante F et al (2014) Maladaptive plasticity in levodopa-induced dyskinesias and tardive dyskinesias: old and new insights on the effects of dopamine receptor pharmacology. Front Neurol 9(5):49
Vernon AC, Modo M (2012) Do levodopa treatments modify the morphology of the parkinsonian brain? Mov Disord 27:166–167
Finlay CJ, Duty S, Vernon AC (2014) Brain morphometry and the neurobiology of levodopa-induced dyskinesias: current knowledge and future potential for translational pre-clinical neuroimaging studies. Front Neurol 5:95
Salgado-Pineda P, Delaveau P, Falcon C, Blin O (2006) Brain T1 intensity changes after levodopa administration in healthy subjects: a voxel-based morphometry study. Br J Clin Pharmacol 62:546–551
Kolb B, Gibb R (2015) Plasticity in the prefrontal cortex of adult rats. Front Cell Neurosci 9:15
Acknowledgments
We thank all participants and their families. Marios Politis research is supported by Parkinson’s UK, Lily and Edmond J. Safra Foundation, Michael J. Fox Foundation (MJFF), and NIHR BRC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standard
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional review boards and the local research ethics committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Pellicano, C., Niccolini, F., Wu, K. et al. Morphometric changes in the reward system of Parkinson’s disease patients with impulse control disorders. J Neurol 262, 2653–2661 (2015). https://doi.org/10.1007/s00415-015-7892-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7892-3