
Abstract
A number of factors more or less unique to multiple sclerosis have suggested that this disease may be particularly amenable to cell-based reparative therapies. The relatively focussed damage to oligodendrocytes and myelin at least in early disease implies that only a single population of cells need be replaced—and that the daunting problem of re-establishing connectivity does not apply. The presence of significant though partial spontaneous myelin repair in multiple sclerosis proves there to be no insurmountable barrier to remyelination intrinsic to the CNS: the therapeutic challenge becomes that of supplementing this spontaneous process, rather than creating repair de novo. Finally, the large body of available knowledge concerning the biology of oligodendrocytes, and the success of experimental myelin repair, have allowed cautious optimism that future prospects for such therapies are not unrealistic. Nonetheless, particular and significant problems are not hard to list: the occurrence of innumerable lesions scattered throughout the CNS, axon loss, astrocytosis, and a continuing inflammatory process, to name but a few. Here we review the progress and the areas where difficulties have yet to be resolved in efforts to develop remyelinating therapies for multiple sclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Following the publication of a number of studies testing adult stem cell therapy in patients with cardiac disease, the past year or so has seen the beginning of a comparable therapeutic effort in neurological disease: small numbers of patients with stroke have received autologous bone marrow cells [6], as have comparable numbers of individuals with amyotrophic lateral sclerosis [82]. At least one (unpublished) study of patients with multiple sclerosis has been completed, wherein the effects of intracerebral implants of autologous Schwann cells and fibroblasts were studied, apparently with no demonstrable effect (http://www.myelin.org/12082003.htm).
What is the future of cell therapy in multiple sclerosis? If there be one, does it lie with Schwann cells, with oligodendrocyte progenitors, olfactory glia, or with stem cells? Is the time ripe to try further clinical studies? If not, what are the remaining hurdles? Here we propose that remyelination treatments by cell-based therapy represent an approachable challenge offering a realistic prospect of successful implementation for the current generation of patients with multiple sclerosis.
Multiple sclerosis is particularly suitable for cell therapy
A primary demyelinating disease—with substantial secondary axon loss
A combination of factors suggests that multiple sclerosis provides an attractive and tempting testing ground for neurological cell therapy.
The first is its nature: primarily a demyelinating disease. But axon loss of course represents the principal pathophysiological cause of disability in chronic progressive disease: does this undermine the rationale of a cell-based therapy?
Rather the opposite—for two reasons. First, none of the recent experimental, imaging or neuropathological studies re-confirming the importance of axon damage have challenged the concept that disease processes in MS are primarily directed against oligodendrocytes and/or myelin, and that axons are relatively spared until late disease [12, 115]. Therefore, in the main, axon pathways remain intact. Cell therapies therefore aim ‘only’ to reinvest axons with myelin, rather than addressing the almost overwhelming challenge presented by most other neurological diseases: that of re-establishing connectivity in highly complex but fragmented axonal circuitry.
Secondly, the mechanism of axon loss is important to consider. The course of secondary progression—and by implication, of axon loss—is significantly neither related to early inflammatory disease activity [10, 31, 64] nor impeded by even the most profound immune suppressant treatments. These and other observations have fuelled the hypothesis that progressive axonal damage is (at least in part) a consequence of persistent myelin loss [12, 112, 128]. Pathological studies have indicated that chronic axon loss does not correlate with either inflammatory cell infiltrate, tumour necrosis factor expression, nitric oxide expression, or demyelinating activity, but does correlate with the overall extent of established myelin loss [10, 64]. It is seen in lesions which are demyelinated but which exhibit sparse or no inflammation, but is rare in remyelinated lesions [64]. Demyelination-induced axon loss might occur by several possible mechanisms: directly, through the loss of oligodendrocyte-derived trophic support [47, 85, 128], or sustained demyelination-induced conduction block and electrical silence [78], or indirectly through increased vulnerability of the exposed axon to injurious agents [99]. A further important driver for early cell therapy thus emerges: the restoration of a normal oligodendroglial environment in order to sustain (previously demyelinated) axons.
Supplementing spontaneous myelin repair
The second positive feature of MS, in terms of developing cell therapies, is found in the clear evidence of spontaneous if partial myelin repair in multiple sclerosis [71, 79, 96, 101]. The nature of relentlessly progressive disability in many patients implies bears witness to the insufficiency of this process, but supplementing spontaneous remyelination must appear a less fanciful proposition than imposing repair in a fundamentally non-reparative environment.
Therapeutic remyelination works experimentally
The third reason to be cheerful is the now large body of experimental evidence proving that cell therapy can achieve successful remyelination. Schwann cells, oligodendrocyte lineage cells or cell lines, olfactory ensheathing cells (OEC), and rodent embryonic, adult neural and adult bone marrow stem cells all have been shown successfully to remyelinate the rodent CNS [2, 8, 9, 18, 46, 48, 50, 54, 93, 105, 109, 120, 134], and variously demonstrated to restore normal conduction [52, 122] and/or function [56].
What problems remain?
Several! Perhaps the most pressing are readily summarised as “where?”, “when?”, “with what?” and “then what?”
Where?
Multiple inoculations of cells into widely distributed lesions in the brain and spinal cord of patients with MS is neither attractive nor realistic. Two possibilities are here relevant.
First, many plaques are clinically silent. A disproportionately large degree of disability frequently emanates from a few critical lesions in eloquent areas. Thus implantation into a very small number of carefully selected lesions—for example, the optic nerves, the spinal cord, or the superior cerebellar peduncle—could yield a commensurately disproportionate therapeutic dividend [30].
Alternatively, some experimental evidence using adult bone marrow- and brain-derived stem cells suggests that the tropism of certain reparative cells for diseased tissue can be exploited, and disseminated (or perhaps also diffuse) disease may be addressed by intravenous delivery of cells [2, 93].
When?
On the basis of “first, do no harm”, late MS seems safest: progressive disability is established, with sadly negligible hope of spontaneous recovery, and the possibility of doing damage or compromising spontaneous repair is remote.
However, arguments can be offered in favour of the early intervention—although at this stage, little disability is present and therefore there is much to lose, and the natural history is such that some patients will never develop significant disability. In addition, implanting cells into early lesions exposes remyelinating glia, and their new myelin, to ongoing inflammatory activity.
But spontaneous remyelination appears to occur maximally in acute inflammatory lesions [95, 101], suggesting (paradoxically) that these offer an optimal reparative environment. Some evidence suggests that anti-inflammatory drugs [116], or the suppression of inflammation in general [33], may impair myelin regeneration.
The clinical impact and irreversibility of accumulating axon loss in secondary progressive disease [11, 12] provide a more potent reason for earlier remyelinating intervention. Quite apart from the futility of attempting to remyelinate absent axons, it has become clear that changes in the cell surface expression of various molecules (e.g. PSA-NCAM) in chronically demyelinated axons actively inhibit myelination [23]. Accumulated myelin debris also inhibits myelination [65], and chronic astrocytosis offers a profound inhibitory effect on the migration of remyelinating glia [41].
With what?
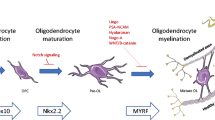
A number of cell types may be considered. (see Fig. 1)

cell-based therapies for myelin repair in MS: potential sources
Cells of the oligodendrocyte lineage
Oligodendrocytes are the most obvious candidates: they are the cells lost, and their normal function is to myelinate the CNS. Immature oligodendrocytes and oligodendrocyte precursors are found in fresh lesions [22, 80, 97, 100, 114, 131] and are generally considered responsible for the great majority of spontaneous remyelination [21, 40, 113, 130].
Despite their motility in vitro, oligodendrocyte progenitors however show poor survival and migration when implanted into normal white matter, although they are able to populate and remyelinate when injected into, or very close to, lesioned tissue [44]. A further difficulty is that investigations of human CNS glia have consistently demonstrated significant biological differences from rodent cells, so that data concerning rodent OPCs cannot be directly extrapolated to human glia. Thus, notwithstanding the success of rodent glial progenitors in experimental myelin repair, initial studies of adult human oligodendrocyte progenitors suggested a very limited capacity for remyelination (in the irradiated rodent spinal cord) [119]. However, oligodendrocyte lineage cells can successfully be derived from ES-cells, and can repair myelin, with demonstrable functional recovery [59, 89]—but human embryonic stem cells as a source of therapeutic material bring a number of problems (see below). Improved methods for isolating adult human oligodendrocyte progenitors (by genetically labelling) have also emerged [106]. Interestingly, comparative studies suggest that adult human CNS-derived oligodendrocyte progenitors have a significantly greater remyelinating capacity than their foetal counterparts [129].
Schwann cells
Schwann cells make a small but significant contribution to endogenous myelin repair in multiple sclerosis, perhaps particularly in the spinal cord [55, 79, 90]. Experimental methods for preparing, from adult peripheral nerve biopsies, cultures of Schwann cells, and for purifying and expanding the cells in vitro to generate large populations of Schwann cells have been established [88, 107]. When so purified, human Schwann cells successfully lay down new myelin in the mouse [75] and the rat spinal cord [17, 62]; autologous, expanded Schwann cells successfully repair relatively large areas of demyelination in the macaque demyelinated spinal cord [5].
Autologous Schwann cell harvesting from peripheral nerve biopsy, expansion in vitro, then transplantation into patients offers the considerable attractions of relative ease of availability, and the avoidance of rejection. Furthermore, Schwann cells (and their myelin) should be resistant to continuing MS-related immunological attack. Firm evidence is required however, that expanded human Schwann cells do not form tumours in vivo, a hazard described when rodent Schwann cells immortalised by growth factor expansion were transplanted [70]; unpurified preparations of human peripheral nerve cells result in substantial fibroblast overgrowth with axon destruction [17]. The apparent inhibitory effect of astrocytes on Schwann cell mediated CNS remyelination [45, 49, 133] represents another potential problem—though genetic modification of Schwann cells to show increased and sustained surface expression of PSA on NCAM significantly improves migration [72].
Olfactory glia
Olfactory ensheathing cells, found in the olfactory bulb, nerves and epithelium, ensheath axons emanating from olfactory epithelial neurons that penetrate the olfactory bulb of the CNS. Rodent OEC’s assume a myelinating phenotype closely resembling that of Schwann cells when transplanted into lesions containing demyelinated axons [46, 54]. Human OEC’s, like rodent OEC’s, are also capable of remyelination following transplantation into the demyelinated rodent spinal cord [7, 58]. This, and the OEC’s ability to migrate in an astrocytic environment [46, 69], in stark contrast to Schwann cells, has helped generate much interest in olfactory glia in the field of CNS repair [42].
Stem cells
Stem cells have enormous therapeutic potential, not least for treating neurodegenerative disease [13, 20, 91, 127]: a consequence of their proliferative and multipotent capacities. Most studies have concentrated on using embryonic tissue as a source of stem cells [18], but to develop therapies would obviously require the use of human embryos as the stem cell source, and this raises significant practical, immunological, and insurmountable ethical concerns [15, 111]. One serious risk is that of teratoma formation (Bjorklund et al. 2002): removing this capacity from embryonic stem cells with absolute success in order to develop safe cell therapies may pose considerable problems. In addition, the emergence of significant chromosomal abnormalities in cultured human embryonic stem cells raises further concerns about their safe use [39]. The problem of rejection would also have to be circumvented. Early, optimistic suggestions were that using stem cells from embryos cloned (by cell nuclear transfer) from individual putative recipients (recently legalised uniquely in the UK) would solve this, the implication that every patient requiring a transplant would first have to be cloned seems quite unrealistic, would not bypass the major ethical difficulties associated with the use of human embryonic material—and may in any case not prove possible, as recent events in Korea have shown.
These problems have helped stimulate the largely successful search for alternative sources of stem cells [103, 104]. There is increasing evidence that adult stem cells have a greater capacity to differentiate into a wider range of cell types than previously anticipated, and the use of adult stem cells—particularly autologous in origin—would avoid many of the difficulties associated with embryonic stem cells [27, 94, 98, 110].
Neural stem cells are present in the adult rodent brain [126]; large numbers of oligodendrocyte lineage cells can be generated using neurosphere/oligosphere techniques, which, upon transplantation, successfully remyelinate axons. Neural stem cells are also present in the adult human brain [68]. Adult rodent CNS-derived stem cells repair multifocal demyelinating lesions (in EAE-affected rodents) even after intravenous delivery [39, 93].
It is now beyond doubt that adult bone marrow does indeed harbour a sub-population of potentially proliferative stem cells [25, 29, 57, 61, 66, 67, 86, 92, 94, 102], whose differentiation capacity includes glial cells and neurones [37, 60, 108, 132]. Cell fusion of bone marrow-derived cells can provide an alternative explanation for apparent trans-differentiation, and this is particularly important in some organs—the liver, for example [123, 124]. However, cell fusion cannot explain the extensive in vitro data indicating multipotent differentiation, and in vivo studies confirm transdifferentiation without fusion in a variety of tissues [53, 81, 121]. Furthermore, from a pragmatic perspective, fusion may simply be part of the means by which bone marrow-derived stem cells stimulate successful regeneration [83, 104]. Recent studies indicate that polyploidy is in fact far commoner phenomenon that previously realised; the possible occurrence of fusion does not necessarily imply diminished regenerative capacity in a putative reparative cell [14].
Bone marrow derived stem cells migrate into (presumably) normal adult human brain and transdifferentiate into highly complex, apparently functionally integrate neural cell types [28, 32, 125]. Experimentally, they show an ability to home from the circulation to various damaged tissue(s) [38, 63], including tropism in regard to areas of CNS injury [1, 25]. Directly or peripherally injected bone marrow-derived cells will repair damage, often with demonstrable functional as well as anatomical recovery, in rodent models of traumatic, degenerative and ischaemic CNS damage [4, 16, 24–26, 74, 77, 87]. Remyelination is reported not only after intra-lesional injection [3, 109], but also following peripheral injection [2].
These properties, their easy accessibility, and not least the absence lack of ethical problems, and also their significant track history in the treatment of haematological disease makes bone marrow cells strong candidates for use in cellular therapies for CNS disease. Further elucidation of the mechanisms involved may allow for mobilisation of endogenous cells, perhaps even obviating the need for transplantation.
The advantages and disadvantages of the different sources of stem cells and progenitor cells to achieve demyelination in MS are summarised in Table 1.
Then what?
How can the effects of a trial cell therapy in MS be assessed? At present, MRI detection of new myelin is not reliably feasible, but new techniques continue to emerge, of which magnetisation transfer contrast is one promising candidate [36], 3-dimensional MRI using multiple contrasts [84], and radial diffusivity [117, 118] likewise. Magnetic resonance spectroscopy measurement of N-acetyl aspartate levels might offer means of assessing any impact on local neuron/axon survival [34, 35]. Cells can be labelled to render them MRI-visible [19, 43, 76], but from a safety perspective, even trivial manipulation of cells prior to implantation is best avoided. Furthermore, graft survival cannot be inferred from migration, since dead cells remain visible [19], and this method not only fails to show new myelin formation but may also impair the ability of other MR modalities to do so.
Serial neurophysiology may prove valuable, and monitoring conduction times may provide evidence of returning saltatory conduction in the targetted pathway(s). The optic nerve has particular advantages in this respect, but various approaches to more generalised neurophysiological assessment have been described and may prove useful for any intervention aimed at multifocal or more diffuse myelin repair [73].
Finally, robust and reproducible methods of clinical assessment need to be applied. Specific clinical outcomes measures of function, disability, and handicap must be adopted; considerable advances in clinical scale design have improved physical and functional measurement in multiple sclerosis [51], so that the tools for assessing clinical outcome, on which remyelination therapies must stand or fall, are becoming available.
References
Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, Breakefield XO, Snyder EY (2000) Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci USA 97:12846–12851
Akiyama Y, Radtke C, Honmou O, Kocsis JD (2002) Remyelination of the spinal cord following intravenous delivery of bone marrow cells. Glia 39:229–236
Akiyama Y, Radtke C, Kocsis JD (2002) Remyelination of the rat spinal cord by transplantation of identified bone marrow stromal cells. J Neurosci 22:6623–6630
Azizi SA, Stokes D, Augelli BJ, Digirolamo C, Prockop DJ (1998) Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats – similarities to astrocyte grafts. Proc Natl Acad Sci USA 95:3908–3913
Bachelin C, Lachapelle F, Girard C, Moissonnier P, Serguera-Lagache C, Mallet J, Fontaine D, Chojnowski A, Le GE, Nait-Oumesmar B, Baron-Van EA (2005) Efficient myelin repair in the macaque spinal cord by autologous grafts of Schwann cells. Brain 128:540–549
Bang OY, Lee JS, Lee PH, Lee G (2005) Autologous mesenchymal stem cell transplantation in stroke patients. Ann Neurol 57:874–882
Barnett SC, Alexander CL, Iwashita Y, Gilson JM, Crowther J, Clark L, Dunn LT, Papanastassiou V, Kennedy PG, Franklin RJ (2000) Identification of a human olfactory ensheathing cell that can effect transplant-mediated remyelination of demyelinated CNS axons [see comments]. Brain 123:1581–1588
Barnett SC, Franklin RJM, Blakemore WF (1993) In vitro and in vivo analysis of a rat bipotential O-2A progenitor cell line containing the temperature-sensitive mutant gene of the SV40 large T antigen. Eur J Neurosci 5:1247–1260
Baron-Van Evercooren A, Avellana-Adalid V, Lachapelle F, Liblau R (1997) Schwann cell transplantation and myelin repair of the CNS. Mult Scler 3:157–161
Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W (2000) Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 123:1174–1183
Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD (2000) Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol 48:893–901
Bjartmar C, Trapp BD (2001) Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol 14:271–278
Bjorklund A (2000) Cell replacement strategies for neurodegenerative disorders. Novartis Found Symp 231:7–15; discussion 16–20
Blau HM (2002) A twist of fate. Nature 419:437
Braude P, Minger SL, Warwick RM (2005) Stem cell therapy: hope or hype? BMJ 330:1159–1160
Brazelton TR, Rossi FM, Keshet GI, Blau HM (2000) From marrow to brain: expression of neuronal phenotypes in adult mice. Science 290:1775–1779
Brierley CM, Crang AJ, Iwashita Y, Gilson JM, Scolding NJ, Compston DA, Blakemore WF (2001) Remyelination of demyelinated CNS axons by transplanted human schwann cells: the deleterious effect of contaminating fibroblasts. Cell Transplant 10:305–315
Brustle O, Jones KN, Learish RD, Karram K, Choudhary K, Wiestler OD, Duncan ID, McKay RD (1999) Embryonic stem cell-derived glial precursors: a source of myelinating transplants. Science 285:754–756
Bulte JW, Zhang S, van Gelderen P, Herynek V, Jordan EK, Duncan ID, Frank JA (1999) Neurotransplantation of magnetically labeled oligodendrocyte progenitors: magnetic resonance tracking of cell migration and myelination. Proc Natl Acad Sci USA 96:15256–15261
Cao Q, Benton RL, Whittemore SR (2002) Stem cell repair of central nervous system injury. J Neurosci Res 68:501–510
Carroll WM, Jennings AR (1994) Early recruitment of oligodendrocyte precursors in CNS demyelination. Brain 117:563–578
Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD (2000) NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 20:6404–6412
Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C (2000) Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc Natl Acad Sci USA 97:7585–7590
Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M (2001) Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 32:1005–1011
Chopp M, Li Y (2002) Treatment of neural injury with marrow stromal cells. Lancet Neurology 1:92–99
Chopp M, Zhang XH, Li Y, Wang L, Chen J, Lu D, Lu M, Rosenblum M (2000) Spinal cord injury in rat: treatment with bone marrow stromal cell transplantation. Neuroreport 11:3001–3005
Clarke D, Frisen J (2001) Differentiation potential of adult stem cells. Curr Opin Genet Dev 11:575–580
Cogle CR, Yachnis AT, Laywell ED, Zander DS, Wingard JR, Steindler DA, Scott EW (2004) Bone marrow transdifferentiation in brain after transplantation: a retrospective study. Lancet 363:1432–1437
Colter DC, Sekiya I, Prockop DJ (2001) Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc Natl Acad Sci USA 98:7841–7845
Compston DAS (1996) Remyelination of the central nervous system. Mult Scler 1:388–392
Confavreux C, Vukusic S, Moreau T, Adeleine P (2000) Relapses and progression of disability in multiple sclerosis. N Engl J Med 343:1430–1438
Crain BJ, Tran SD, Mezey E (2005) Transplanted human bone marrow cells generate new brain cells. J Neurol Sci 233:121–123
Cuzner ML, Loughlin AJ, Mosley K, Woodroofe MN (1994) The role of microglia macrophages in the processes of inflammatory demyelination and remyelination. Neuropathol Appl Neurobiol 20:200–201
Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, Harding AE, McDonald WI, Miller DH (1995) Persistent functional deficit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axon loss. Brain 118:1583–1592
De Stefano N, Matthews PM, Antel JP, Preul M, Francis G, Arnold DL (1995) Chemical pathology of acute demyelinating lesions and its correlation with disability. Ann Neurol 38:901–909
Deloire-Grassin MS, Brochet B, Quesson B, Delalande C, Dousset V, Canioni P, Petry KG (2000) In vivo evaluation of remyelination in rat brain by magnetization transfer imaging. J Neurol Sci 178:10–16
Deng W, Obrocka M, Fischer I, Prockop DJ (2001) In vitro differentiation of human marrow stromal cells into early progenitors of neural cells by conditions that increase intracellular cyclic AMP. Biochem Biophys Res Commun 282:148–152
Devine SM, Cobbs C, Jennings M, Batholomew A, Hoffman R (2003) Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into non-human primates. Blood 101:2999–3001
Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, Meisner L, Zwaka TP, Thomson JA, Andrews PW (2004) Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol 22:53–54
Duncan ID, Grever WE, Zhang SC (1997) Repair of myelin disease: strategies and progress in animal models. Mol Med Today 3:554–561
Fawcett JW, Asher RA (1999) The glial scar and central nervous system repair. Brain Res Bull 49:377–391
Franklin RJ, Barnett SC (2000) Olfactory ensheathing cells and CNS regeneration: the sweet smell of success? Neuron 28:15–18
Franklin RJ, Blaschuk KL, Bearchell MC, Prestoz LL, Setzu A, Brindle KM, Ffrench-Constant C (1999) Magnetic resonance imaging of transplanted oligodendrocyte precursors in the rat brain. Neuroreport 10:3961–3965
Franklin RJM, Bayley SA, Blakemore WF (1996) Transplanted CG4 cells (an oligodendrocyte progenitor cell line) survive, migrate, and contribute to repair of areas of demyelination in X-irradiated and damaged spinal cord but not in normal spinal cord. Exp Neurol 137:263–276
Franklin RJM, Blakemore WF (1993) Requirements for Schwann cell migration within CNS environments: A viewpoint. Int J Dev Neurosci 11:641–649
Franklin RJM, Gilson JM, Franceschini IA, Barnett SC (1996) Schwann cell-like myelination following transplantation of an olfactory bulb-ensheathing cell-line into areas of demyelination in the adult CNS. Glia 17:217–224
Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA (1998) Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280:1610–1613
Groves AK, Barnett SC, Franklin RJM, Crang AJ, Mayer M, Blakemore WF, Noble M (1993) Repair of demyelinated lesions by transplantation of purified O-2A progenitor cells. Nature 362:453–455
Harrison B (1985) Schwann cell and oligodendrocyte remyelination in lysolecithin-induced lesions in irradiated rat spinal cord. J Neurol Sci 67:143–159
Harrison BM (1980) Remyelination by cells introduced into a stable demyelinating lesion in the central nervous system. J Neurol Sci 46:63–81
Hobart J, Lamping D, Fitzpatrick R, Riazi A, Thompson A (2001) The Multiple Sclerosis Impact Scale (MSIS-29): a new patient-based outcome measure. Brain 124:962–973
Honmou O, Felts PA, Waxman SG, Kocsis JD (1996) Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. J Neurosci 16:3199–3208
Ianus A, Holz GG, Theise ND, Hussain MA (2003) In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J Clin Invest 111:843–850
Imaizumi T, Lankford KL, Waxman SG, Greer CA, Kocsis JD (1998) Transplanted olfactory ensheathing cells remyelinate and enhance axonal conduction in the demyelinated dorsal columns of the rat spinal cord. J Neurosci 18:6176–6185
Itoyama Y, Webster HD, Richardson EP Jr, Trapp BD (1983) Schwann cell remyelination of demyelinated axons in spinal cord multiple sclerosis lesions. Ann Neurol 14:339–346
Jeffery ND, Crang AJ, O’leary MT, Hodge SJ, Blakemore WF (1999) Behavioural consequences of oligodendrocyte progenitor cell transplantation into experimental demyelinating lesions in the rat spinal cord. Eur J Neurosci 11:1508–1514
Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418:41–49
Kato T, Honmou O, Uede T, Hashi K, Kocsis JD (2000) Transplantation of human olfactory ensheathing cells elicits remyelination of demyelinated rat spinal cord. Glia 30:209–218
Keirstead HS, Nistor G, Bernal G, Totoiu M, Cloutier F, Sharp K, Steward O (2005) Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci 25:4694–4705
Kim BJ, Seo JH, Bubien JK, Young SO (2002) Differentiation of adult bone marrow stem cells into neuroprogenitor cells in vitro. Neuroreport 13:1185–1188
Koc ON, Lazarus HM (2001) Mesenchymal stem cells: heading into the clinic. Bone Marrow Transplant 27:235–239
Kohama I, Lankford KL, Preiningerova J, White FA, Vollmer TL, Kocsis JD (2001) Transplantation of cryopreserved adult human Schwann cells enhances axonal conduction in demyelinated spinal cord. J Neurosci 21:944–950
Korbling M, Estrov Z (2003) Adult stem cells for tissue repair. N Engl J Med 349:570–582
Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 157:267–276
Kotter MR, Li WW, Zhao C, Franklin RJ (2006) Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 26:328–332
Krause DS (2002) Plasticity of marrow-derived stem cells. Gene Ther 9:754–758
Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ (2001) Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105:369–377
Kukekov VG, Laywell ED, Suslov O, Davies K, Scheffler B, Thomas LB, O’Brien TF, Kusakabe M, Steindler DA (1999) Multipotent stem/progenitor cells with similar properties arise from two neurogenic regions of adult human brain. Exp Neurol 156:333–344
Lakatos A, Franklin RJ, Barnett SC (2000) Olfactory ensheathing cells and Schwann cells differ in their in vitro interactions with astrocytes. Glia 32:214–225
Langford LA, Porter S, Bunge RP (1988) Immortalized rat Schwann cells produce tumours in vivo. J Neurocytol 17:521–529
Lassmann H, Bruck W, Lucchinetti CF, Rodriguez M (1997) Remyelination in multiple sclerosis. Mult Scler 3:133–136
Lavdas A, Franceschini I, Dubois-Dalcq M, Matsas R (2006) Schwann cells genetically engineered to express PSA show enhanced migratory potential without impairment of their myelinating ability in vitro. Glia in press
Leocani L, Medaglini S, Comi G (2000) Evoked potentials in monitoring multiple sclerosis. Neurol Sci 21:S889–S891
Lescaudron L, Unni D, Dunbar GL (2003) Autologous adult bone marrow stem cell transplantation in an animal model of Huntington’s disease: behavioral and morphological outcomes. Int J Neurosci 113:945–956
Levi ADO, Bunge RP (1994) Studies of myelin formation after transplantation of human SchwannX X cells into the severe combined immunodeficient mouse. Exp Neurol 130:41–52
Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R (2000) Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol 18:410–414
Li Y, Chen J, Wang L, Zhang L, Lu M, Chopp M (2001) Intracerebral transplantation of bone marrow stromal cells in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neurosci Lett 316:67–70
Lipton SA (1986) Blockade of electrical-activity promotes the death of mammalian retinal ganglion-cells in culture. Proc Natl Acad Sci USA 83:9774–9778
Ludwin S (1988) Remyelination in the central nervous system and in the peripheral nervous system. Adv Neurol 47:215–254
Maeda Y, Solanky M, Menonna J, Chapin J, Li W, Dowling P (2001) Platelet-derived growth factor-alpha receptor-positive oligodendroglia are frequent in multiple sclerosis lesions. Ann Neurol 49:776–785
Masuya M, Drake CJ, Fleming PA, Reilly CM, Zeng H, Hill WD, Martin-Studdard A, Hess DC, Ogawa M (2003) Hematopoietic origin of glomerular mesangial cells. Blood 101:2215–2218
Mazzini L, Fagioli F, Boccaletti R, Mareschi K, Oliveri G, Olivieri C, Pastore I, Marasso R, Madon E (2003) Stem cell therapy in amyotrophic lateral sclerosis: a methodological approach in humans. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the world federation of neurology, Research group on motor neuron diseases 4:158–161
Medvinsky A, Smith A (2003) Stem cells: fusion brings down barriers. Nature 422:823–825
Merkler D, Boretius S, Stadelmann C, Ernsting T, Michaelis T, Frahm J, Bruck W (2005) Multicontrast MRI of remyelination in the central nervous system. NMR Biomed 23:7710–7718
MeyerFranke A, Kaplan MR, Pfrieger FW, Barres BA (1995) Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron 15:805–819
Mezey E, Chandross KJ (2000) Bone marrow: a possible alternative source of cells in the adult nervous system. Eur J Pharmacol 405:297–302
Mezey E, Chandross KJ, Harta G, Maki RA, McKercher SR (2000) Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science 290:1779–1782
Morrissey TK, Levi AD, Nuijens A, Sliwkowski MX, Bunge RP (1995) Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbB2. Proc Natl Acad Sci USA 92:1431–1435
Nistor GI, Totoiu MO, Haque N, Carpenter MK, Keirstead HS (2005) Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia 49:385–396
Ogata J, Feigin I (1975) Schwann cells and regenerated peripheral myelin in multiple sclerosis: an ultrastructural study. Neurology 25:713–716
Park KI, Ourednik J, Ourednik V, Taylor RM, Aboody KS, Auguste KI, Lachyankar MB, Redmond DE, Snyder EY (2002) Global gene and cell replacement strategies via stem cells. Gene Ther 9:613–624
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, Furlan R, Comi G, Vescovi AL, Martino G (2003) Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature 422:688–694
Poulsom R, Alison MR, Forbes SJ, Wright NA (2002) Adult stem cell plasticity. J Pathol 197:441–456
Prineas JW, Barnard RO, Kwon EE, Sharer LR, Cho ES (1993) Multiple sclerosis: remyelination of nascent lesions. Ann Neurol 33:137–151
Prineas JW, Connell F (1979) Remyelination in multiple sclerosis. Ann Neurol 5:22–31
Prineas JW, Kwon EE, Goldenberg PZ (1989) Multiple sclerosis: oligodendrocyte proliferation and differentiation in fresh lesions. Lab Inv 61:489–503
Prockop DJ (2002) Adult stem cells gradually come of age. Nat Biotechnol 20:791–792
Raine CS, Cross AH (1989) Axonal dystrophy as a consequence of long-term demyelination. Lab Invest 60:714–725
Raine CS, Scheinberg L, Waltz JM (1981) Multiple sclerosis. Oligodendrocyte survival and proliferation in an active established lesion. Lab Invest 45:534–546
Raine CS, Wu E (1993) Multiple sclerosis: remyelination in acute lesions. J Neuropathol Exp Neurol 52:199–204
Reyes M, Lund T, Lenvik T, Aguiar D, Koodie L, Verfaille CM (2001) Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood 98:2615–2625
Rice CM, Halfpenny C, Scolding NJ (2004) Cell therapy in demyelinating diseases. NeuroRx 1:415–423
Rice CM, Scolding NJ (2004) Adult stem cells – reprogramming neurological repair? Lancet 364:193–199
Rogister B, Ben Hur T, Dubois-Dalcq M (1999) From neural stem cells to myelinating oligodendrocytes. Mol Cell Neurosci 14:287–300
Roy NS, Wang S, Harrison-Restelli C, Benraiss A, Fraser RA, Gravel M, Braun PE, Goldman SA (1999) Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J Neurosci 19:9986–9995
Rutkowski JL, Kirk CJ, Lerner MA, Tennekoon GI (1995) Purification and expansion of human schwann cells in vitro. Nat Med 1:80–83
Sanchez-Ramos J, Song S, Cardozo-Pelaez F, Hazzi C, Stedeford T, Willing A, Freeman TB, Saporta S, Janssen W, Patel N, Cooper DR, Sanberg PR (2000) Adult bone marrow stromal cells differentiate into neural cells in vitro. Exp Neurol 164:247–256
Sasaki M, Honmou O, Akiyama Y, Uede T, Hashi K, Kocsis JD (2001) Transplantation of an acutely isolated bone marrow fraction repairs demyelinated adult rat spinal cord axons. Glia 35:26–34
Scolding N (2001) New cells from old. Lancet 357:329–330
Scolding N (2005) Stem-cell therapy: hope and hype. Lancet 365:2073–2075
Scolding N, Franklin R (1998) Axon loss in multiple sclerosis. Lancet 352:340–341
Scolding NJ, Franklin RJM (1999) Remyelination in demyelinating disease. Clin Neurol Int Pract Res 6:525–548
Scolding NJ, Franklin RJM, Stevens S, Heldin CH, Compston DAS, Newcombe J (1998) Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain 121:2221–2228
Smith KJ, McDonald WI (1999) The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Philos Trans R Soc Lond B Biol Sci 354:1649–1673
Smith PM, Franklin RJ (2001) The effect of immunosuppressive protocols on spontaneous CNS remyelination following toxin-induced demyelination. J Neuroimmunol 119:261–268
Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH (2002) Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage 17:1429–1436
Song SK, Yoshino J, Le TQ, Lin SJ, Sun SW, Cross AH, Armstrong RC (2005) Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage 26:132–140
Targett MP, Sussman J, Scolding N, OLeary MT, Compston DAS, Blakemore WF (1996) Failure to achieve remyelination of demyelinated rat axons following transplantation of glial cells obtained from the adult human brain. Neuropath App Neurobiol 22:199–206
Tontsch U, Archer DR, Dubois-Dalcq M, Duncan ID (1994) Transplantation of an oligodendrocyte cell line leading to extensive myelination. Proc Natl Acad Sci USA 91:11616–11620
Tran SD, Pillemer SR, Dutra A, Barrett AJ, Brownstein MJ, Key S, Pak E, Leakan RA, Kingman A, Yamada KM, Baum BJ, Mezey E (2003) Differentiation of human bone marrow-derived cells into buccal epithelial cells in vivo: a molecular analytical study. Lancet 361:1084–1088
Utzschneider DA, Archer DR, Kocsis JD, Waxman SG, Duncan ID (1994) Transplantation of glial cells enhances action potential conduction of amyelinated spinal cord axons in the myelin-deficient rat. Proc Natl Acad Sci USA 91:53–57
Vassilopoulos G, Wang PR, Russell DW (2003) Transplanted bone marrow regenerates liver by cell fusion. Nature 422:901–904
Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al Dhalimy M, Lagasse E, Finegold M, Olson S, Grompe M (2003) Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 422:897–901
Weimann JM, Charlton CA, Brazelton TR, Hackman RC, Blau HM (2003) Contribution of transplanted bone marrow cells to Purkinje neurons in human adult brains. Proc Natl Acad Sci USA 100:2088–2093
Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson AC, Reynolds BA (1996) Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J Neurosci 16:7599–7609
Weissman IL (2000) Translating stem and progenitor cell biology to the clinic: barriers and opportunities. Science 287:1442–1446
Wilkins A, Majed H, Layfield R, Compston A, Chandran S (2003) Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: a novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J Neurosci 23:4967–4974
Windrem MS, Nunes MC, Rashbaum WK, Schwartz TH, Goodman RA, McKhann G, Roy NS, Goldman SA (2004) Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat Med 10:93–97
Wolswijk G (2000) Oligodendrocyte survival, loss and birth in lesions of chronic-stage multiple sclerosis. Brain 123(Pt 1):105–115
Wolswijk G (1998) Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci 18:601–609
Woodbury D, Schwarz EJ, Prockop DJ, Black IB (2000) Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res 61:364–370
Woodruff RH, Franklin RJ (1999) Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: a comparative study. Glia 25:216–228
Zhang SC, Ge B, Duncan ID (1999) Adult brain retains the potential to generate oligodendroglial progenitors with extensive myelination capacity. Proc Natl Acad Sci USA 96:4089–4094
Acknowledgements
The authors thank the UK Multiple Sclerosis Society and the Ipsen Trust for support. The Burden Chair Clinical Neurosciences is supported by the Burden Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rice, C., Scolding, N. Strategies for achieving and monitoring myelin repair. J Neurol 254, 275–283 (2007). https://doi.org/10.1007/s00415-006-0455-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-006-0455-x