
Abstract
Genetically transmitted diseases are an important cause of juvenile sudden cardiac death (SCD). In a considerable proportion of individuals in which a medicolegal investigation is performed, structural heart disease is absent, and the medical examiner fails to discover an adequate cause of death. In such cases, an inherited arrhythmogenic disease should be considered, which manifests with life-threatening ventricular tachycardia or SCD. Molecular diagnosis is progressively becoming an important tool for these questions. Therefore, postmortem genetic testing (“molecular autopsy”) should be considered as a part of the comprehensive medicolegal investigation in SCD cases without apparent structural heart disease. It will have implications not only for the deceased individual but also for living family members in preventing (further) cardiac events by expert counseling, appropriate lifestyle adjustment, and adequate treatment, if available.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The frequency of sudden death in children and adolescents has been investigated in retrospective reviews by several groups. It accounts for approximately 5% of all deaths in children and adolescents, with an incidence of 1.5–8 per 100,000 patient-years [1]. About 5,000–7,000 “asymptomatic” children die suddenly in the United States annually, compared to 300,000–400,000 sudden cardiac deaths (SCDs) per year in adults.
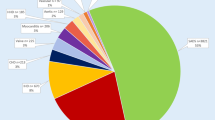
Whereas SCD in the adult is mostly due to coronary artery disease often associated with previous myocardial infarction [2], a large spectrum of cardiac disorders underlies SCD in the young [1]. The majority of deaths in childhood are due to previously diagnosed diseases. The most frequent causes include infection, asthma, epilepsy, as well as myocarditis and other cardiovascular abnormalities. In a study on juvenile sudden death in the Veneto region (Italy), cardiac causes accounted for more than 80% of the cases, and about one third of sudden deaths were due to congenital heart defects [3]. Wren et al. [4] documented the incidence and causes of sudden death in a region in England over a 10-year period. Eleven percent were sudden deaths (270 of 2,523), and more than half of these (53%) were attributed to previously diagnosed conditions (e.g., epilepsy, asthma, cardiovascular disease, etc.). One third (32%) were attributed to causes discovered at necropsy (e.g., respiratory tract infection, cardiovascular abnormalities), and 15% remained unexplained after a thorough examination (Fig. 1). Driscoll and Edwards [1] reviewed 13 studies with 61 children and adolescents with unexpected sudden death. The most common structural abnormalities identified were hypertrophic cardiomyopathy (HCM), coronary abnormalities, and aortic stenosis. Corrado et al. [5] reported the results of 273 SCD victims not more than 35 years of age in the Veneto region of Italy and found 76 patients (28%) with apparently normal hearts and 197 (72%) patients with macroscopic structural changes such as cardiomyopathy, obstructive coronary atherosclerosis, valve disease, non-atherosclerotic coronary artery disease, aortic rupture, and postoperative congenital heart disease (Fig. 2).

Incidence and causes of more than 2,500 sudden deaths in children and young adolescents in an English region over a 10-year period from 1985 to 1994 (modified from Wren et al. [4])

Prevalence of subtle morphologic substrates and causes of sudden cardiac death in 237 patients not more than 35 years of age over a time interval from 1979 to 1998 in the Veneto region of Italy (modified from Corrado et al. [5]). SCD Sudden cardiac death, ARVCM arrhythmogenic right ventricular cardiomyopathy, CAD coronary artery disease
However, in a certain percentage, no structural abnormalities can be identified at autopsy, which suggests the presence of primary electrical diseases, i.e., molecular changes that lead to subcellular changes without gross structural abnormalities. These can be suspected by possible premonitory symptoms like (recurrent) syncope, a family history of sudden death at young age, or electrocardiographic abnormalities identified before death. The suspicion can be confirmed by “molecular autopsy.” Therefore, the knowledge of inherited arrhythmias should be of special interest for all those who perform autopsies in these victims.
Spectrum of cardiac abnormalities associated with SCD
Cardiac abnormalities associated with SCD include not only known and recognizable (congenital and acquired) heart diseases [e.g., primary cardiomyopathies, arrhythmogenic right ventricular cardiomyopathy (ARVCM), and primary pulmonary hypertension] but also known but unrecognizable (electrical) heart diseases [e.g., long-QT syndrome (LQTS), Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia (CPVT)].
Inherited ventricular arrhythmias
Ventricular tachycardia and SCD are not common in children and young adults. However, whenever such an event occurs, it has a great impact on the family as well as on the physician who took care of the patient. In the previous two decades, a number of inherited ventricular arrhythmias were identified, which account for the majority of SCD in young otherwise healthy children. The spectrum includes so-called primary electrical diseases, in which an organic heart disease is not detectable, and so-called arrhythmogenic cardiomyopathies, in which an inherited myocardial disease may primarily manifest with ventricular tachycardia and/or SCD. This may happen in a very early stage of the disease when structural abnormalities are absent and autopsy fails to discover a morphologic abnormality.
Arrhythmias without structural heart disease
By definition, in primary electrical heart disease, there is no morphologic substrate at autopsy. The changes occur on the molecular level, which leads to electrophysiological abnormalities that form the electrical substrate.
The prevalence of primary electrical heart diseases in a population is difficult to estimate due to the lack of easily applicable tools to discover underlying morphologic or genetic abnormalities. Most of these diseases have a variable expression and incomplete penetrance. For ventricular tachycardia or ventricular fibrillation, there may be minor structural abnormalities that may remain undetected if not specifically searched for.
The long-QT syndrome
The long-QT syndrome (LQTS) is associated with SCD in children [6, 7] and has been well documented by the International LQTS Registry [8, 9].
Congenital LQTS is a hereditary disease. There is a more common form, the autosomal dominant Romano–Ward syndrome, with a frequency of approximately 1:7,000 newborns. The Jervell and Lange–Nielsen syndrome (JLNS) is the more severe but rarer homozygous form which is associated with sensorineural deafness. It occurs with a frequency of about 1–5% of that of the Romano–Ward syndrome. Both syndromes are caused predominantly by mutations of genes that encode for cardiac ion channels. At present, over 200 mutations in five potassium channel genes (KCNQ1, HERG, KCNE1, KCNE2, KCNJ2) [10–14], one sodium channel gene (SCN5A) [15], and one non-ion channel protein (Ankyrin-B) [16] that is involved in the structural integrity of ion channels have been identified. Recently, mutations in the L-type calcium channel (CaV1.2) gene were found to be responsible for QT prolongation in Timothy syndrome, a multisystem disorder that causes syncope and sudden death [17]. Thus, the L-type calcium channel gene has to be considered as a further LQT-causing channel, if mutated.
The LQTS accounts for 3,000–4,000 sudden deaths annually in childhood in the USA. The leading premonitoring symptoms are syncope and subsequent SCD due to ventricular arrhythmias, typically of the torsade de pointes type (Fig. 3a,b), or SCD may be the first manifestation of the disease. In most cases, torsade de pointes is self-terminating, producing a syncopal episode, but occasionally, it degenerates into ventricular fibrillation and causes SCD. Symptoms can manifest at any time from the neonatal period to young adulthood and later. However, more than 50% of patients have experienced their first episode of syncope or cardiac arrest by the age of 15 years [8]. Therefore, syncope in the history of otherwise healthy children should always raise the suspicion of an undiagnosed LQTS.

a Surface ECG of a patient with congenital long-QT syndrome, QT interval corrected for heart rate of 650 ms1/2. b Torsade de pointes tachycardia (not from the same patient) which is typical for patients with long-QT syndrome. Note the typical twisting axis of the QRS vector that gave this particular form of ventricular tachyarrhythmia the name
Zareba et al. [18] reported the clinical characteristics and risk of cardiac events in 844 LQTS children below 15 years and in 1,123 affected family members. The children were followed in three age groups (0–5 years, n=844; 6–10 years, n=812; and 11–15 years, n=748) over a period of 4–5 years, and the frequency of cardiac events (syncope, aborted cardiac arrest, or death related to LQTS) was documented. Cardiac events occurred in 14, 25, and 34% in the respective age groups compared with a frequency of cardiac events in the affected family members of 5, 8, and 11%. The gender differences showed a significant higher risk for males [16] having cardiac events in the age groups of 0–5 years (7 vs 3%) and 6–10 years (11 vs 6%) but not in the age group of 11–15 years (11 vs 11%). The authors concluded that children not more than 5 years of age have a lower risk of cardiac events than children at age 6–15 years. In 1998, Zareba et al. [19] also reported the clinical and genetic data and the prognosis in 112 LQT1, 72 LQT2, and 62 LQT3 subjects of the International LQTS Registry. LQT5 and LQT6 as sporadic causes for LQTS and their function as β-subunits in the potassium channel were not included. The median age at first cardiac event was 9 years for LQT1 and 14 and 16 years for LQT2 and LQT3, respectively. By the age of 15 years, the cumulative probability of cardiac events was 53% in LQT1 patients, 29% in LQT2 patients, and only 6% in LQT3 patients [17]. Another important observation was that death as the first cardiac event in these patients occurred in 1–3% without preceding symptoms. In 2001, the same authors [20] reported the influence of the genotype on the clinical course of 504 LQTS children (224 LQT1, 202 LQT2, and 78 LQT3). They documented the first cardiac event (syncope, aborted cardiac arrest, or LQTS-related death) at specific age intervals of 0–5, 6–10, and 11–15 years and the cardiac event rate per patient-year. In LQT1 children, the cardiac event rate was the highest between 6 and 10 years (0.28), whereas in LQT2 children, it was the highest at age 11–15 years (0.13). In LQT3 children, it was infrequent. First cardiac events were found more often in LQT1 patients in all age intervals. Aborted cardiac arrest or death as the first event was observed in 3 of 103 LQT1, 0 of 42 LQT2, and 2 of 5 LQT3 carriers, respectively.
In 1993, Garson et al. [6] defined children at low risk for SCD when they were asymptomatic with normal QTc and high risk when the QTc exceeded 600 ms1/2. This observation could be confirmed by others who observed several children in whom an extensively prolonged QT interval was associated with an increased risk of fatal arrhythmias [21, 22]. Priori et al. [23] analyzed three variables (gender, QTc, and mutated gene) that were sufficient to identify low-, intermediate-, or high-risk patients for developing LQTS-related symptoms until the age of 40 years. The initiation and association of arrhythmic events by specific circumstances were investigated by Schwartz et al. [9] and Chiang and Roden [24] in LQTS patients. Patients with mutations in the KCNQ1 gene (LQT1 subtype) developed early and frequent symptoms during exercise or periods of physical stress, whereas patients with a sodium channel defect (SCN5A gene) developed symptoms more often during sleep [8].
Overall, LQTS plays an important role as a cause of SCD in young otherwise healthy children. Due to large numbers of well-characterized LQTS index patients and their families, genotype/phenotype correlations and risk stratification have been established on a solid basis.
Thus, inquiries in cases of sudden unexpected death should include a search for prior ECG tracings which naturally exist less frequently in children of any age than in adults. However, from our own experience, children may have presented with syncope leading to an ECG recording with marginally or even markedly prolonged QT interval, which had not been accounted for in the subsequent management of these patients. However, even if no ECG is available from the deceased patient, ECG from the parents and other relatives may point to a genetic background if some of them have prolonged QT intervals.
The long-QT syndrome and the sudden infant death syndrome
The sudden infant death syndrome (SIDS) is a frequent cause of death among infants. The etiology of SIDS is unknown, and several theories, including fatal ventricular arrhythmias, have been suggested. Therefore, several studies were conducted in the past, with different results of an association between QT prolongation and SIDS [25, 26]. In 2000, Schwartz et al. [27] demonstrated a relation between LQTS and SIDS in performing a postmortem analysis and thereby identifying a de novo mutation in the cardiac sodium channel gene SCN5A. A more comprehensive genetic analysis of the LQT genes in SIDS victims was reported by Ackerman et al. [28]. They investigated the LQT3 gene (SCN5A) in 93 consecutive SIDS cases. Two heterozygous mutations have been identified, and the frequency of SCN5A mutations in SIDS was estimated at about 2%. In the further extended investigation of the other LQTS genes, the data indicate a prevalence of 5.1% in white infants and 2.9% in black infants [29]. Our group performed a comprehensive gene mutation screening in 41 consecutively SIDS victims and identified in a single SIDS case a LQTS mutation (LQT1) without a yet identified functional effect [30]. Taking the various reports of the associative and genetic analyses together, the data confirm that in some SIDS victims, mutations in cardiac ion channels may provide an arrhythmogenic substrate for malignant tachyarrhythmias, although the frequency is yet unclear and has to be investigated in larger SIDS collectives.
Short-QT syndrome
Recently, a new syndrome associated with SCD has been described, the short-QT syndrome (SQTS) [31]. Six patients from two European families with histories of SCD were extensively tested by noninvasive and invasive methods. QT intervals never exceeded 280 ms or a QTc of 300 ms1/2. In each family, three living members had short-QT intervals and histories of syncope, palpitations, atrial fibrillation, and, in one case, resuscitated cardiac arrest at the age of 8 months after a loud noise (adrenergic stress). One relative died suddenly at the age of 3 months and was diagnosed as SIDS. On electrophysiological evaluation, all investigated individuals had short atrial and ventricular refractory periods, and three out of four tested had inducible ventricular fibrillation. Four patients received an implantable cardioverter-defibrillator.
Genetic analysis revealed mutations in the HERG gene (SQT1 gene). The gene product I Kr (rapidly acting component of the outward-rectifying potassium current) is largely responsible for repolarization and, thus, the QT interval duration. The mutation increases I Kr (gain of function), leading to an increase of the repolarizing current and, thus, to shortening of action potential duration and refractoriness. In contrast, patients with LQT2 have mutations in the HERG gene that decrease I Kr (loss of function). Meanwhile, two other groups demonstrated that SQTS is like other primary electrical diseases that are genetically heterogeneous and can also be caused by mutation in the KCNQ1 (=SQT2) and KCNJ2 (=SQT3) genes [32, 33]. ECG analysis of patients with SQT3 show a special ECG phenotype characterized by asymmetrical T waves [32].
The Brugada syndrome
In 1992, Brugada and Brugada [34] described eight patients with a history of aborted sudden death and a characteristic electrocardiogram with a right bundle branch block-like pattern with ST segment elevation in leads V1 to V2 and normal QT intervals in the absence of any structural heart disease (Fig. 4). The prognosis of symptomatic patients with this syndrome is poor if they do not receive an implantable defibrillator [35, 36]. In 1998, the genetic nature of the disease as an ion channelopathy (mutation of the gene SCN5A) was identified [37]. The electrical heterogeneity within right ventricular epicardium leads to the development of phase 2 and circus movement reentry, which then precipitates ventricular tachycardia or ventricular fibrillation [38].

ECG features of Brugada syndrome. The 12-lead surface ECG demonstrates a typical coved-type (so-called type 1) ST segment elevation with atypical right bundle branch block in V1–V3 as a hallmark of the syndrome
The prevalence of a Brugada syndrome-like disease is highest in men of Asian origin. Sudden and unexpected death of young adults during sleep is endemic in Japan and Southeast Asia, where it is a leading cause of death among young adults. In Thailand, the incidence of ECGs compatible with the syndrome is 4:10,000. SCD due to Brugada syndrome is the most common cause of death after traffic accidents in these regions [39]. Studies of the adult Japanese population showed prevalences of ECGs compatible with the syndrome between 0.05% [40] and 0.6% [40]. However, in children from Japan, the incidence of ECG changes was only 0.0006% [40]. Oe et al. [41] determined the prevalence and clinical course in children exhibiting Brugada-type ECG in a community-based population in Osaka, Japan. Of the 21,944 subjects (11,282 boys and 10,662 girls) who underwent ECG during their first year elementary school health examinations between 1992 and 2001, four subjects (0.02%) showed a Brugada-type ECG (two boys and two girls). No history of structural heart disease was documented in these four subjects, and no episode of unexpected sudden death, syncopal attack, and fatal arrhythmia occurred during a follow-up of about 7 years. These data suggest that SCD due to the Brugada syndrome occurs predominantly in adulthood. However, in a case report, a family is described in which five children died after unexplained cardiac arrest. Brugada syndrome was suspected because of a transient manifestation of the typical ECG pattern in one of them and further confirmed by mutation analysis [42]. In another report, Todd et al. [43] identified a Brugada syndrome in a 15-month-old girl who presented after cardiac arrest and who later died. Further investigation of the proband revealed a mutation in the SCN5A gene. This mutation was also present in two deceased maternal relatives. Both of them were attributed to the SIDS because of death at 8 and 6 months without identifying an adequate cause of death.
Catecholaminergic polymorphic ventricular tachycardia
In 1978, Coumel et al. [44] described four cases of severe ventricular tachycardia in children with normal QT intervals. Today, catecholaminergic polymorphic ventricular tachycardia (CPVT) is a clearly defined entity that occurs preferably in children and young adults. The terms CPVT and familial polymorphic ventricular tachycardia both apply to the same clinical entity. Affected individuals present with a distinct pattern of stress-related bidirectional or polymorphic ventricular tachycardia, syncope, and SCD (Fig. 5) [45, 46]. In one third of the cases, a family history of juvenile sudden death and stress-related syncope is present. Mortality is high and reaches up to 30–50% by the age of 30 years [47].

Series of ECG tracings from an 11-year-old patient with CPVT before and during exercise testing. The family history revealed an SCD of the 14-year-old brother while playing soccer. a At rest, a normal sinus rhythm with a heart rate of 72 bpm is documented. With increasing exercise level, first a bigeminus (b, 80 W) and then later on (c, 105 W) polymorphic ventricular beats occurred, which finally led to discontinuation of exercise test
CPVT is a genetically diverse disease, involving several loci: one form of familial polymorphic ventricular tachycardia is inherited in an autosomal dominant pattern that was initially linked to chromosome 1q42–q43 [48]. Subsequent studies identified mutations in the gene for the human cardiac ryanodine receptor (RyR2), which is also called the cardiac sarcoplasmic calcium release channel. The ryanodine receptor is localized across the membrane of the sarcoplasmic reticulum, and it releases Ca2+ from the sarcoplasmic reticulum in response to the calcium influx through the L-type channels, during the phase 2 of the action potential. Mutations found in RyR2 gene linked to a CPVT phenotype are all missense mutations located in functionally important regions of the protein: Functional characterization of the mutants has shown that they all produce abnormal calcium release “leaky RyR2 channels” in response to adrenergic stimulation (e.g., exercise, beta-adrenergic stimulation). This mechanism supports the hypothesis that life-threatening ventricular tachycardia is induced by triggered activity.
The clinical features of this form of familial polymorphic ventricular tachycardia were evaluated in two unrelated families with 24 members who had experienced exercise-induced ventricular tachycardia or syncope or had an episode of cardiac arrest. Some of the family members had delayed onset of clinical manifestations, which necessitated continued observation and repeated evaluation. The cumulative incidence of SCD by the age of 30 was 31%. Only one heterozygous carrier was clinically unaffected, suggesting high disease penetrance by adulthood in these families. The frequency with which RyR2 mutations occur in patients with familial catecholaminergic ventricular tachycardia was assessed in a series of 30 probands and 118 family members [46]. RyR2 mutations were detected in 14 of the 30 probands (47%) and in nine family members, four of whom were silent carriers. The patients with RyR2 mutations, compared to those without such mutations, had events at a younger age (age at first syncope, 8 vs 20 years). Male sex was an important risk factor for syncope. In contrast, 90% of the subjects without an RyR2 mutation were female. The distribution of arrhythmias in these 14 patients was polymorphic ventricular tachycardia in seven, bidirectional ventricular tachycardia in five, and catecholaminergic idiopathic ventricular fibrillation (IVF) in two. Tester et al. [49] performed targeted molecular analysis at autopsy in 49 victims of sudden unexplained death, 13 with a family history of syncope, cardiac arrest, or SCD. The mean age at death was 14.2 years (SD ±10.9 years). They investigated 18 of the 105 exons and identified six RyR2 missense mutations in seven cases. They concluded that analysis of the cardiac ryanodine receptor revealed a potential CPVT-causing RyR2 mutations in one of every seven cases (14%). Denjoy et al. [50] reported on the genetic and clinical data from 25 children (19 with syncope and 6 with resuscitated sudden death during exercise) with CPVT. Analysis of the RyR2 gene showed mutations in 13 of the 25 cases. Ninety-six percent of patients remained asymptomatic over an average follow-up of about 7.5 years under beta-blocker therapy, although some of them continued to display polymorphic ventricular extrasystoles on exercise. Twelve percent of the cases died suddenly or suffered further syncope during follow-up.
A second genetic form of familial polymorphic ventricular tachycardia, with autosomal recessive inheritance, has been identified in seven related Bedouin families [51]. These families included nine children who died suddenly at an average age of 7 years and 12 others with a history of recurrent syncope or seizures beginning at 6 years of age. The 12 symptomatic patients had a relative resting bradycardia and polymorphic ventricular tachycardia induced by exercise or isoproterenol infusion. Genetic studies of these families have identified a mutation in the calsequestrin 2 gene (CASQ2) that is responsible for the disease [52]. The calsequestrin 2 protein has an ability to bind extremely large amounts of calcium. While the mechanism by which the reported mutation causes ventricular arrhythmias is not clearly established, the mutated protein may directly increase the calcium content within the sarcoplasmic reticulum, alter the function of the ryanodine receptor to which it is connected, or impair the calcium release process. In conclusion, CPVT is rare but clearly, in young age, occurring as inherited arrhythmia, and children with syncope or sudden death during exercise may suffer from this disease.
Idiopathic right ventricular outflow tract tachycardia
Idiopathic ventricular tachycardia (RVOVT) in the absence of structural heart disease may arise from the right or left ventricle causing either a left bundle branch block-like pattern (right ventricular origin) or a right bundle branch block-like pattern (left ventricular origin) in the ECG during tachycardia. The majority of cases occur sporadic rather than familial. Idiopathic RVOVT is the most frequent form of idiopathic ventricular tachycardia and is usually catecholamine sensitive, believed to be secondary to cAMP-mediated triggered activity. The VT may be repetitive and occur at rest, or it may be provoked by exercise. It is sometimes impossible to differentiate this entity from mild or subclinical forms of ARVCM. Although long-term prognosis of idiopathic RVOVT is good, syncope, cardiac arrest due to life-threatening arrhythmias, or SCD have been reported in rare cases. However, RVOVT or SCD is uncommon in children and young adults, and episodes of ventricular tachycardia are well tolerated in most cases due to the preserved global ventricular function.
Lerman et al. [53] identified in a patient with RVOVT and insensitivity to adenosine therapy a mutation in the GTP-binding domain of the inhibitory G protein Gαi2. This mutation was shown to increase intracellular cAMP concentration and inhibit suppression of cAMP by adenosine. The mutation was detected in biopsy samples from the arrhythmic focus but not in myocardial tissue sampled from regions remote from the origin of tachycardia or from peripheral lymphocytes. The authors concluded that somatic cell mutations in the cAMP-dependent signal transduction pathway occurring during myocardial development may be responsible for some forms of idiopathic ventricular tachycardia.
Idiopathic ventricular fibrillation
The remaining group of sudden deaths with neither an identifiable structural heart disease nor an inherited genetic background, myocardial ischemia, drug effects, or electrolyte or metabolic abnormalities is classified as having idiopathic ventricular fibrillation (IVF). IVF accounts for approximately 6–12% of all SCD. Although it occurs more often in the younger age group below 40 years, children are infrequently affected. In a review from Viskin and Belhassen [54], the mean age of patients belonging to the group of IVF was 36, with a male to female ratio of 2.5:1. A combined Task Force of the Unexplained Cardiac Arrest Registry of Europe (UCARE) and Idiopathic Ventricular Fibrillation Registry of the USA (IVF-US) recently summarized a variety of minor abnormalities that may be compatible with the diagnosis of IVF [55]. Since the mechanism of initiating ventricular fibrillation has been discovered in the Brugada syndrome, this entity has not been considered as a distinct subgroup of IVF anymore.
Arrhythmias with structural heart disease
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by left and/or right ventricular hypertrophy and is familial in a large proportion of cases with an autosomal dominant trait. The results of molecular genetic studies have shown that HCM is a disease of the sarcomere involving mutations in 11 different genes encoding proteins of the myofibrillar apparatus. In a recent analysis of 744 consecutively enrolled patients with HCM, Maron et al. [56] identified 86 HCM-related deaths over 8±7 years. Fifty-one percent of these occurred suddenly and unexpectedly and were most common in young patients (45±20 years), whereas heart failure- and stroke-related deaths were observed more frequently in midlife and beyond. Although data on the incidence of HCM in sudden death in children and young adults are lacking, HCM was identified in nearly all series ranging from sporadic cases to the most predominate finding [57]. According to these data, it is appropriate to investigate any child with exercise-related death, a family history of HCM, or sudden death for the presence of HCM.
The phenotype of HCM is first influenced by factors varying the penetrance like age and gender. Second, the phenotype depends on the responsible mutation. Especially in HCM, several mutations have been identified which are associated with a high risk of SCD, e.g., Arg403Gln in the beta-myosin heavy chain (MYH7) gene, whereas others (e.g., Val606Met) in the same gene have a good prognosis as regards arrhythmic events. Furthermore, the propensity to ventricular fibrillation does not always correlate to the extent of hypertrophy; for example, some mutations in the cardiac troponin T (TNNT2) gene lead to subclinical hypertrophy but are associated with a high risk of SCD [58], whereas others are completely penetrant but without a high risk of arrhythmic events [59]. This underscores the importance of identifying malignant mutations during medicolegal workup when considering death related to HCM and when counseling living family members.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is a genetically and clinically heterogeneous disease. It is an important cause of chronic congestive heart failure and cause of heart transplantation in infants and children. However, whereas SCD accounts for approximately 30% of deaths in adult DCM patients, it was found only in sporadic cases in several postmortem analyses in children. The prevalence of DCM is about 2.6 patients in 100,000 inhabitants [60]. Manifestation age is often the first years of life with nonspecific symptoms. If symptoms are present, they usually develop gradually. Some patients are asymptomatic and have left ventricular dilatation for months or years, and others rapidly develop symptoms of heart failure including ventricular tachycardia. Death in DCM patients occurs not only due to progressive congestive heart failure or as a complication of thromboembolism but also abruptly due to an arrhythmic cardiac arrest when the disease is still in its early stage. Idiopathic DCM seems to represent a common expression of myocardial damage produced by a variety of myocardial insults. The three most possible basic mechanisms of damage could be (1) familial and genetic factors, (2) viral myocarditis and other cytotoxic insults, and (3) immunological abnormalities. In 20–30% of patients, a first-degree relative also shows evidence of DCM, suggesting that familial transmission is frequent and at least 25% of the DCM cases are familial [61].
Although DCM is clinically and genetically heterogeneous, some clinical presentations lead to the suspicion of a familial form of DCM especially in children [62]. If the patient is male, has a short stature, and presents with granulocytopenia, Barth syndrome should be suspected, and the methylglutaconic acid should be measured in the urine. Barth syndrome [63] is a rare X-linked disease for which the gene tafazzin has recently been identified. If the patient presents with hypoglycemia without ketonuria and chronic muscle weakness, carnitine deficiency should be suspected [64]. Carnitine plays a key role in the metabolism of fatty acids since it is essential for their transfer into the mitochondria. Disorders in the oxidation of fatty acids are associated with ventricular tachycardia and conduction defects in children [65]. If the patient has early contractures of skeletal muscles and associated atrioventricular block, the Emery–Dreifuss disease with defects in the X-linked emerin has to be ruled out [66].
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVCM) is characterized by regional atrophy of right ventricular myocardium and subsequent replacement by fatty and fibrous tissues. The disease may often be overlooked in the presence of an early stage of the disease (“concealed disease”). ARVCM manifests in young adulthood with an approximately 2:1 predominance in males. The most frequent clinical features of ARVCM consist of ventricular arrhythmias with left bundle branch block morphology ranging from isolated ventricular beats to sustained VT or, rarely, ventricular fibrillation [67]. Typical symptoms include palpitations due to ventricular extrasystoles, dizziness, syncope, and SCD due to ventricular tachyarrhythmias. On the surface ECG at rest, a somewhat broader than normal QRS complex (0.11 s) in V1–V3 with an epsilon potential (low voltage potentials at the end of the QRS due to delayed conduction in the right ventricular myocardium) might be visible.
Since ARVCM has a familial occurrence, an inherited disorder was assumed early on [68]. Meanwhile, several gene loci and at least four genes which are associated with ARVCM have been identified (plakoglobin, desmoglobin, ryanodine receptor, plakophilin-2). In 2000, McKoy et al. [69] identified the first gene for autosomal recessive ARVCM in Naxos disease (autosomal recessive ARVCM, palmoplantar keratoderma, and wooly hair) on chromosome 17, occurring on the island of Naxos in Greece. The gene product is a key protein component of the desmosomes and adherens junctions and is responsible for the tight cell-to-cell adhesion in the heart. In the recent years, several other genes have been identified, all of them playing a crucial role as cytoskeletal proteins to form and stabilize cell adhesion structures at the cytoplasmic membrane and the connection of these contact sites to corresponding sites of adjacent cells. The plakoglobin and desmoglobin genes encode for proteins that maintain desmosomal cell junction integrity [69, 70]. Very recently, Gerull et al. [71] have been identified in 32 of 120 unrelated individuals with ARVCM heterozygous mutations in the desmosomal protein plakophilin-2 (PKP2). PKP2 are armadillo repeat-containing proteins, localized in the desmosomal plaque and cell nucleus. According to these findings, the authors concluded that mutations in this gene are common in patients with ARVCM.
Despite several reports on the familial occurrence of ARVCM, its prevalence in childhood and young adulthood can only be guessed. Zardini et al. [72] reported the clinical course and the onset of first symptoms of 80 patients with ARVCM (mean age at onset of symptoms: 33±14 years). In a study from Dalal et al. [73], only 1 out of 100 ARVCM patients was below 13 years of age. Most of the patients presented between the second and fifth decades of life either with symptoms of palpitations (27%) and syncope associated with ventricular tachycardia (26%) or with SCD (23%). Risk factors for SCD in the adult age group are a markedly dilated right ventricle and progressive fibrofatty replacement of right ventricular myocardium. However, at present, no similar data are available in the young age group. Thiene et al. [67] conducted a postmortem study of 60 cases under 35 years of age who had died suddenly. At autopsy, they found 12 subjects (7 males and 5 females, 20%) at the age of 13–30 with the morphologic features of right ventricular cardiomyopathy. In five cases, sudden death was the first sign of the disease.
Wolff–Parkinson–White syndrome
The Wolff–Parkinson–White (WPW) syndrome is characterized by accessory pathways that, beside the AV node/His bundle system, represent additional pathway conduction from the atria to the ventricles or vice versa. The conduction properties of the accessory pathway determine the rate of ventricular response (the shorter the refractory period of the pathway, the higher is the ventricular rate, e.g., during atrial fibrillation).
Population studies have detected WPW syndrome in up to 2:1,000 apparently normal teenagers and young adults [74]. A minority suffers from arrhythmias, suggesting that a considerable proportion of WPW syndrome is asymptomatic. The spectrum of symptoms includes palpitations, dizziness, and syncope or SCD. The latter two occur when atrial fibrillation complicates WPW syndrome with a rapid ventricular response over the accessory pathway, which degenerates into ventricular fibrillation. In patients with WPW syndrome, natural history of SCD rate has been reported, 0.15% per year. Patients who survived cardiac arrest tend to be symptomatic and have short cycle (RR) intervals (<250 ms) during atrial fibrillation. The clinical course of asymptomatic children with WPW syndrome has recently been investigated [75]. The authors investigated 165 children (5–12 years) in which prophylactic radiofrequency catheter ablation of accessory pathways was compared with no ablation in asymptomatic children who were at high risk for arrhythmias (i.e., reproducible induction of atrioventricular reciprocating tachycardia or atrial fibrillation). During follow-up (median 34 months), one child in the ablation group (5%) and 12 in the control group (44%) had arrhythmic events. Two children in the control group had ventricular fibrillation, and one died suddenly. Thus, even in asymptomatic children, WPW syndrome is associated with a risk of SCD and may be first detected during postmortem investigation. According to these results, high-risk children with WPW syndrome may benefit from prophylactic catheter ablation by reducing the risk of life-threatening arrhythmias. Furthermore, in those cases, siblings and first-degree relatives should be screened for preexcitation.
Five to ten percent of HCM patients have ventricular preexcitation. An association between WPW and familial HCM had been noted in earliest descriptions of the latter condition. It is proposed that abnormal ventricular activation might result in regional myocardial hypertrophy or that localized hypertrophy might disrupt normal cardiac electrical discontinuity at the atrial ventricular ring.
In 2001, Gollob et al. [76] noted that the PRKAG2 gene is located in the critical genomic region of the WPW syndrome locus identified by linkage on chromosome 7q34–q36. In affected members of two families with WPW syndrome, they identified a mutation in the PRKAG2 gene. An additional sequence variation in the 3’ untranslated region of PRKAG2 was present in all affected members of family 2 but not family 1, indicating that the two families are unrelated. The same author reported a third, unrelated family in which affected members manifested ventricular preexcitation, atrial fibrillation, and conduction defects from childhood. The molecular defect in this family was a novel PRKAG2 mutation.
Conclusion
Although sudden unexpected death in children and young adolescents is relatively uncommon, it has a devastating psychosocial impact on the families. Many patients who die suddenly have identifiable cardiac disease and are known to have been at risk. In the past, the etiology of many of these deaths was unknown and deemed “idiopathic.” However, subsequent discoveries have identified the cause of death in more and more of these patients. During recent years, the genetic basis of several ion channel disorders has been identified. In these diseases, structural abnormalities are usually absent, and SCD may be the first symptom (Table 1). Especially the pathologist or the forensic pathologist who is involved in such cases should be aware of these diseases. Analysis of published reports on children with syncope, cardiac arrest, and sudden death shows that very different populations have been included. This may be partly explained by the fact that postmortem diagnosis of previously unidentified arrhythmias is almost impossible. However, the following could help us further elaborate on these categories:
-
Morphological investigation
-
Very detailed autopsy which must contain extended histological examination of all organs, immunohistochemistry, microbiology, virology, toxicological screening, and x-ray examination of the skeleton to exclude morphologically defined natural and unnatural causes of death. Highly specialized morphological investigation of the heart including histology, immunohistochemistry, and investigation of the cardiac conduction system to exclude myocarditis and other morphologically defined causes of death.
-
-
Molecular autopsy
-
Genetic postmortem investigations in cases without morphologically defined cause of death. We strongly suggest the construction of a multilocus/multigenic kit carrying all known disorders of genetic origin.
-
-
Heart history
-
Evaluation of the “heart history” of the child and in family members including available cardiac documents (Table 2).
-
Moreover, such process will not only elucidate the cause of death in these groups of cases but also will have implications for the remaining family members in preventing (further) cardiac events due to expert counseling, appropriate lifestyle adjustment, and adequate prophylactic treatment.
References
Driscoll DJ, Edwards WD (1985) Sudden unexpected death in children and adolescents. J Am Coll Cardiol 5(Suppl):118B–121B
Davies MJ, Thomas A (1984) Thrombosis and acute coronary artery lesions in sudden cardiac ischemic death. N Engl J Med 310:1137–1140
Basso C, Frescura C, Corrado D, Muriago M, Angelini A, Daliento L, Thiene G (1995) Congenital heart disease and sudden death in the young. Hum Pathol 26:1065–1072
Wren C, O’Sullivan JJ, Wright C (2000) Sudden death in children and adolescents. Heart 83:410–413
Corrado D, Basso C, Thiene G (2001) Sudden cardiac death in young people with apparently normal heart. Cardiovasc Res 50:399–408
Garson A Jr, Dick M II, Fournier A, Gillette PC, Hamilton R, Kugler JD, van Hare GF III, Vetter V, Vick GW III (1993) The long QT syndrome in children. An international study of 287 patients. Circulation 87:1866–1872
Weintraub RG, Gow RM, Wilkinson JL (1990) The congenital long-QT syndromes in childhood. J Am Coll Cardiol 16:674–680
Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH, Hall WJ (1998) Influence of the genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med 339:960–965
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R (2001) Genotype–phenotype correlation in the long-QT syndrome. Circulation 103:89–95
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT (1995) A molecular basis for cardiac arrhythmias: HERG mutations cause long QT syndrome. Cell 80:795–803
Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, Van Raay TJ, Shen J, Towbin JA, Mon AJ, Atkinson DL, Lander GM, Conners TD, Keating MT (1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12:17–23
Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SAN (1999) MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97:175–187
Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hördt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H (1997) KCNE1 mutations cause Jervell and Lange–Nielsen syndrome. Nat Genet 17:267–268
Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R (2002) Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 110:381–388
Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT (1995) SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80:805–811
Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosin S, duBell WH et al (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421:634–639
Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT (2005) Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A 102:8089–8096
Zareba W, Moss AJ, Robinson JL, Andrews ML, for the International LQTS Research Group (2001) Clinical course of LQTS in children. PACE 24(Suppl):571 (abstract)
Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH, Hall WJ (1998) Influence of the genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med 339:960–965
Zareba W, Moss AJ, Robinson JL, Andrews ML, for the International LQTS Research Group (2001) Influence of genotype on clinical course of LQTS children. PACE 24(Suppl):571 (abstract)
Schulze-Bahr E, Fenge H, Etzrodt D, Haverkamp W, Mönnig G, Wedekind H, Breithardt G, Kehl HG (2004) Long QT syndrome and life threatening arrhythmia in a newborn: molecular diagnosis and treatment response. Heart 90:13–16
Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, Borggrefe M, Brinkmann B, Warnecke I, Funke H, Bhuiyan ZA, Wilde AA, Breithardt G, Haverkamp W (2001) De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation 104:1158–1164
Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D (2003) Risk stratification in the long-QT syndrome. N Engl J Med 348:1866–1874
Chiang CE, Roden D (2000) The long QT syndromes: genetic basis and clinical implications. J Am Coll Cardiol 36:1–12
Southall DP (1983) Identification of infants destined to die unexpectedly during infancy: evaluation of predictive importance of prolonged apnoea and disorders of cardiac rhythm or conduction. Br Med J 286:1092–1096
Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, Grancini F, Marni ED, Perticone F, Rosti D, Salice P (1998) Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 338:1709–1714
Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, Richard TA, Berti MR, Bloise R (2000) A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med 343:262–267
Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, Towbin JA (2001) Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA 286:2264–2269
Tester DJ, Ackerman MJ (2005) Sudden infant death syndrome: How significant are the cardiac channelopathies? Cardiovasc Res (in press)
Wedekind H, Bajanowski T, Friederich P, Breithardt G, Wülfing T, Siebrands C, Engeland B, Mönnig G, Haverkamp W, Brinkmann B, Schulze-Bahr E (2005) Sudden infant death syndrome and long QT syndrome: an epidemiological and genetic study. Int J Legal Med (in press)
Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M (2003) Short QT syndrome: a familial cause of sudden death. Circulation 108:965–970
Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J (2005) A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96:800–807
Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, Baro I, Wilde AA (2004) Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109:2394–2397
Brugada P, Brugada J (1992) Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. J Am Coll Cardiol 20:1391–1396
Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ (2000) Clinical and genetic heterogeneity of right bundle branch block and ST segment elevation syndrome. Circulation 102:2509–2515
Brugada J, Brugada R, Brugada P (1998) Right bundle brunch block and ST segment elevation in leads V1–V3: a marker for sudden death in patients with no demonstrable structural heart disease. Circulation 97:457–460
Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q (1998) Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392:293–296
Gussak I, Antzelevitch C, Bjerregaard P, Towbin JA, Chaitman BR (1999) The Brugada syndrome: clinical, electrophysiologic and genetic aspects. J Am Coll Cardiol 33:5–15
Nademanee K, Veerakul G, Immannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, Tunsanga K, Kuasirikul S, Malasit P, Tansupasawadikul S, Tatsanavivat P (1997) Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 96:2277–2285
Brugada J, Brugada P, Brugada R (1999) The syndrome of right bundle branch block, ST segment elevation in V1 to V3 and sudden death. Are asymptomatic patients at high risk for sudden death? In: Raviele A (ed) Cardiac arrhythmias. Springer, Berlin Heidelberg New York, pp 255–261
Oe H, Takagi M, Tanaka A, Namba M, Nishibori Y, Nishida Y, Kawarabayashi T, Yoshiyama M, Nishimoto M, Tanaka K, Yoshikawa J (2005) Prevalence and clinical course of the juveniles with Brugada-type ECG in Japanese population. Pacing Clin Electrophysiol 28:549–554
Priori SG, Napolitano C, Giordano U, Collisani G, Memmi M (2000) Brugada syndrome and sudden cardiac death in children. Lancet 355:808–809
Todd SJ, Campbell MJ, Roden DM, Kannankeril PJ (2005) Novel Brugada SCN5A mutation causing sudden death in children. Heart Rhythm 2(5):540–543
Coumel P, Fidelle J, Lucet V, Attuel P, Bouvrain Y (1978) Catecholaminergic-induced severe ventricular arrhythmias with Adams–Stokes syndrome in children: report of four cases. Br Heart J 40(Suppl):28–37
Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P (1995) Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 91:1512–1519
Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A (2002). Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106:69–74
Fisher JD, Krikler D, Hallidie-Smith KA (1999) Familial polymorphic ventricular arrhythmias: a quarter century of successful medical treatment based on serial exercise—pharmacologic testing. J Am Coll Cardiol 34:2015–2022
Swan H, Piippo K, Viitasalo M, Heikkila P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K, Toivonen L (1999) Arrhythmic disorder mapped to chromosome 1q42–q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol 34:2035–2042
Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ (2004) Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin Proc 79:1380–1384
Denjoy I, Postma A, Lupoglazoff JM, Vaksman G, Kamblock J, Leenhardt A, Wilde AA, Guicheney P (2005) Catecholinergic ventricular tachycardia in children. Arch Mal Coeur Vaiss 98:506–512
Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, Lorber A, Kastner DL, Goldman B, Pras E (2001) Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation 103:2822–2827
Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M (2001) A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 69:1378–1384
Lerman BB, Dong B, Stein KM, Markowitz SM, Linden J, Catanzaro DF (1998) Right ventricular outflow tract tachycardia due to a somatic cell mutation in G protein subunitalphai2. J Clin Invest 101:2862–2868
Viskin S, Belhassen B (1990) Idiopathic ventricular fibrillation. Am Heart J 120:661–671
Joint Steering Committees of the Unexplained Cardiac Arrest Registry of Europe and of the Idiopathic Ventricular Fibrillation Registry of the United States (1997) Consensus Statement of the Joint Steering Committees of the Unexplained Cardiac Arrest Registry of Europe and of the Idiopathic Ventricular Fibrillation Registry of the United States. Survivors of out-of-hospital cardiac arrest with apparently normal heart. Need for definition and standardized clinical evaluation. Circulation 95:265–267
Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohmann TE, Graham KJ, Burton DA, Cecchi F (2000) Epidemiology of hypertrophic cardiomyopathy-related death. Circulation 102:858–864
Niimura I, Maki T (1989) Sudden cardiac death in childhood. Jpn Circ J 53:1571–1580
Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F (2000) Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 102:858–864
Maron BJ (1990) Apical hypertrophic cardiomyopathy: the continuing saga. J Am Coll Cardiol 15(1):91–93
Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, Tikanoja T, Paavilainen T, Simell O (1997) Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol 146:385–393
Kokado H, Shimizu M, Yoshio H, Ino H, Okeie K, Emoto Y, Matsuyama T, Yamaguchi M, Yasuda T, Fujino N, Ito H, Mabuchi H (2000) Clinical features of hypertrophic cardiomyopathy caused by a Lys183 deletion mutation in the cardiac troponin I gene. Circulation 102:663–669
Arbustini E, Morbini P, Pilotto A, Gavazzi A, Tavazzi L (2000) Familial dilated cardiomyopathy: from clinical presentation to molecular genetics. Eur Heart J 21:1825–1832
D’Adamo P, Fassone L, Gedeon A (1997) The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 61:862–867
Marques JS (1998) Dilated cardiomyopathy caused by plasma membrane carnitine transport defect. J Inherit Metab Dis 21:428–429
Bonnet D, Martin D, de Lonlay P, Villain E, Jouvet P, Rabier D, Brivet M, Saudubray JM (1999) Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation 100:2248–2253
Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D (1994) Identification of a novel X-linked gene responsible for Emery–Dreifuss muscular dystrophy. Nat Genet 8:323–327
Thiene G, Nava A, Corrado D, Rossi L, Pennelli N (1988) Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 318:129–133
Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, Martini B, Stritoni P, Fasoli G (1988) Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol 12:1222–1228
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355:2119–2124
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP (2000) Recessive mutation in desmoplakin disrupts desmoplakin–intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 9:2761–2766
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L (2004) Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 36:1162–1164
Zardini MC, Lupo P, Titto M, Massacci E, De Ponti R, Storti C, Salerno JA (2001) Arrhythmogenic right ventricular cardiomyopathy: long term follow-up of 80 patients. PACE 24(Suppl):607 (abstract)
Dalal D, James C, Tichnell C, Calkins H (2005) Arrhythmogenic right ventricular dysplasia: a United States experience. Heart Rhythm 2(Suppl):115 (abstract)
Garson A Jr, Gillette PC, McNamara DG (1981) Supraventricular tachycardia in children: clinical features, response to treatment, and long-term follow-up in 217 patients. J Pediatr 98:875–882
Pappone C, Manguso F, Santinelli R, Vicedomini G, Sala S, Paglino G, Mazzone P, Lang CC, Gulletta S, Augello G, Santinelli O, Santinelli V (2004) Radiofrequency ablation in children with asymptomatic Wolff–Parkinson–White syndrome. N Engl J Med 351:1197–1205
Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Ali Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, Bachinski LL, Roberts R (2001) Identification of a gene responsible for familial Wolff–Parkinson–White syndrome. N Engl J Med 344:1823–1831
Acknowledgements
This work was partly supported by grants from the Dr. Adolf Schilling Foundation, Münster, Germany, the Deutsche Forschungsgemeinschaft (SFB 656-C1, Schu1082/3-1, Ki653/13-1 and FR 1625/1-1), Bonn, Germany, and the Fondation Leducq, Paris, France.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wedekind, H., Schulze-Bahr, E., Debus, V. et al. Cardiac arrhythmias and sudden death in infancy: implication for the medicolegal investigation. Int J Legal Med 121, 245–257 (2007). https://doi.org/10.1007/s00414-005-0069-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-005-0069-3