
Abstract
The kinetochore, the proteinaceous structure on the mitotic centromere, functions as a mechanical latch that hooks onto microtubules to support directional movement of chromosomes. The structure also brings in a number of signaling molecules, such as kinases and phosphatases, which regulate microtubule dynamics and cell cycle progression. Erroneous microtubule attachment is destabilized by Aurora B-mediated phosphorylation of multiple microtubule-binding protein complexes at the kinetochore, such as the KMN network proteins and the Ska/Dam1 complex, while Plk-dependent phosphorylation of BubR1 stabilizes kinetochore–microtubule attachment by recruiting PP2A-B56. Spindle assembly checkpoint (SAC) signaling, which is activated by unattached kinetochores and inhibits the metaphase-to-anaphase transition, depends on kinetochore recruitment of the kinase Bub1 through Mps1-mediated phosphorylation of the kinetochore protein KNL1 (also known as Blinkin in mammals, Spc105 in budding yeast, and Spc7 in fission yeast). Recruitment of protein phosphatase 1 to KNL1 is necessary to silence the SAC upon bioriented microtubule attachment. One of the key unsolved questions in the mitosis field is how a mechanical change at the kinetochore upon microtubule attachment is converted to these and other chemical signals that control microtubule attachment and the SAC. Rapid progress in the field is revealing the existence of an intricate signaling network created right on the kinetochore. Here we review the current understanding of phosphorylation-mediated regulation of kinetochore functions and discuss how this signaling network generates an accurate switch that turns on and off the signaling output in response to kinetochore–microtubule attachment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Equal distribution of genetic material to dividing cells relies on the segregation of each replicated pair of sister chromatids. This task is carried out by the bipolar attachment of spindle microtubules to each pair of kinetochores assembled on the centromeric DNA of sister chromatids that are topologically linked. Each kinetochore is composed of a variety of conserved multi-protein complexes that form multiple connections between centromeric DNA on the “inner” side to microtubules at the “outer” edge of the kinetochore (Fig. 1) (Cheeseman and Desai 2008; Przewloka and Glover 2009; Takeuchi and Fukagawa 2012). Resolution of sister chromatids or cell division prior to proper bipolar attachment leads to random segregation of chromosomes and causes aneuploidy, which is closely associated with cancers and birth defects (Nagaoka et al. 2012; Weaver and Cleveland 2006). To prevent this, the spindle assembly checkpoint (SAC) signaling pathway is activated on unattached kinetochores to inhibit the metaphase-to-anaphase transition (Kops and Shah 2012; Musacchio and Salmon 2007), while erroneous kinetochore–microtubule attachments are actively corrected (Lampson et al. 2004; Tanaka 2010; Tanaka et al. 2002). Once sister chromatids are separated upon SAC silencing, they can never re-establish cohesion to repeat the chromosome segregation process. Thus, SAC silencing is a critical decision-making step, which must be triggered only after all chromosomes accomplish bioriented attachment. Several protein kinases and phosphatases play essential roles in these processes by controlling the phosphorylation of a number of kinetochore proteins (Supplementary Table S1 and Fig. 2). Discovery of the kinetochore phosphoepitope “3F3/2,” whose presence depends on a lack of tension at the kinetochore, hinted at the existence of a mechanosensory mechanism to monitor the status of kinetochore–microtubule attachments and convert that status into a chemical signal (Gorbsky and Ricketts 1993; Nicklas et al. 1995).

Schematic architecture of the vertebrate kinetochore. Kinetochore components are arranged to highlight the overall geometry of the kinetochore, with more chromosome-proximal components considered inner and the microtubule-proximal components considered outer

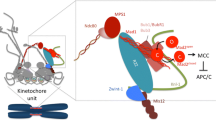
Phosphorylation sites at the kinetochore. A comparison of known phosphorylations present at the kinetochore of a chromosome before (top panel) and after (bottom panel) achieving bioriented microtubule attachments. The unattached kinetochore is generating the spindle assembly checkpoint (SAC) cell cycle arrest via formation of the mitotic checkpoint complex (MCC). The color and letter within each phosphorylation (star shapes) correspond to the kinase responsible (see legend, top). Question marks in the lower panel denote phosphorylations that may be retained upon biorientation but for which more information is required
Although pairs of sister kinetochores in mitosis are arranged in opposing directions, providing an intrinsic geometric preference for bipolar attachment (Indjeian and Murray 2007; Sakuno et al. 2009), erroneous attachments, such as syntelic attachment (both sister kinetochores attached to the same pole) and merotelic attachment (one kinetochore attached to both poles), are also formed. Correction of these erroneous configurations is mediated by destabilization of kinetochore–microtubule attachment, which is facilitated by kinases such as Aurora B (Cimini et al. 2006; Lampson et al. 2004; Tanaka et al. 2002), a subunit of the chromosomal passenger complex (CPC), which also contains INCENP, Survivin, and Borealin (also known as Dasra) (Carmena et al. 2012b; van der Waal et al. 2012). The CPC is localized to the inner centromere from prophase to metaphase where it controls kinetochore–microtubule attachment and the SAC. Aurora B-dependent phosphorylation of substrates at the kinetochore is sensitive to kinetochore–microtubule attachment status, with levels of phosphorylation greatest on kinetochores without microtubule attachment and reduced on those with bipolar attachment (Fig. 2) (DeLuca et al. 2011; Liu et al. 2009; Welburn et al. 2010). Therefore, it has been predicted that a mechanism exists to switch from phosphorylation to dephosphorylation of Aurora B substrates upon bipolar attachment (Kelly and Funabiki 2009; Lampson and Cheeseman 2011; Maresca and Salmon 2010; Tanaka 2010). Inactivation of Aurora B stabilizes syntelic and merotelic attachments (Cimini et al. 2006; Ditchfield et al. 2003; Hauf et al. 2003), indicating that this phospho-switch is important to selectively stabilize proper bipolar attachments. Aurora B-dependent destabilization of erroneous attachments helps correct these attachments but also creates unattached kinetochores, which would activate the SAC (Biggins and Murray 2001; Pinsky et al. 2006b; Tanaka et al. 2002).
The SAC generates a cell cycle arrest by inhibiting the Anaphase Promoting Complex/Cyclosome (APC/C), an E3 ubiquitin ligase complex that ubiquitylates mitotic cyclins and securin to target them for proteosomal degradation to promote cell cycle progression and sister chromatid separation (Peters 2006). Correlated with the fact that SAC activity is induced by unattached kinetochores, critical components of the SAC, such as Mad1, Mad2, Bub1, BubR1 (Mad3 in yeasts), Bub3, and Mps1, are recruited to kinetochores (Abrieu et al. 2001; Chan et al. 1998, 1999; Chen et al. 1996, 1998; Fisk and Winey 2001; Kops and Shah 2012; Li and Benezra 1996; Taylor et al. 1998; Taylor and McKeon 1997; Waters et al. 1998). Binding of Mad2 to Mad1 at the kinetochore triggers a structural conversion of Mad2, which stimulates Mad2 binding to the APC/C activator Cdc20 (Luo and Yu 2008; Musacchio and Salmon 2007; Yang et al. 2008), resulting in inhibition of Cdc20’s stimulatory activity toward the APC/C (Chao et al. 2012). The Mad2–Cdc20 complex further assembles into the mitotic checkpoint protein complex (MCC) with Bub3 and BubR1, which acts as a pseudosubstrate to inhibit the APC/C (Burton and Solomon 2007; Chao et al. 2012; King et al. 2007b; Lara-Gonzalez et al. 2011; Sczaniecka et al. 2008; Sudakin et al. 2001). While Mad1 and Bub1 stably associate with unattached kinetochores, the MCC proteins all show dynamic kinetochore localization, indicating that the MCC formed at an unattached kinetochore diffuses to inhibit cellular APC/C (Howell et al. 2004; Shah et al. 2004). Upon microtubule attachment, the levels of all these SAC proteins at the kinetochore decrease, with Mad1 and Mad2 showing the most robust reduction while a substantial fraction of Bub1 remains (Howell et al. 2004; Liu et al. 2003; Martinez-Exposito et al. 1999; Shah et al. 2004; Taylor et al. 1998). One mechanism for removal of checkpoint proteins from kinetochores is dynein-mediated transport along microtubules (Gassmann et al. 2010; Howell et al. 2001; Kasuboski et al. 2011). However, other dynein-independent mechanisms exist as Mad1 and Mad2 are removed from kinetochores after microtubule attachment in mammalian cells depleted of the protein Spindly, which is required for kinetochore recruitment of dynein (Gassmann et al. 2010), and in yeasts, dynein-dependent removal of kinetochore components does not appear to be conserved (Mayer et al. 2006; Yeh et al. 1995). It has also been shown in yeasts that protein phosphatase 1 (PP1) at the kinetochore is essential for SAC silencing, indicating that dephosphorylation at the kinetochore is required for SAC silencing upon microtubule binding (Pinsky et al. 2009; Rosenberg et al. 2011; Vanoosthuyse and Hardwick 2009).
Robust coupling between microtubule attachment status and SAC signaling is critical. If the SAC is erroneously silenced prior to bipolar attachment, it triggers a series of irreversible events, including sister chromatid separation and cell division. The SAC system is tuned so that a single unattached kinetochore can effectively generate the “wait anaphase” signal, but that signal is swiftly silenced upon attachment of the last unattached kinetochore (Clute and Pines 1999; Rieder et al. 1995). This decision-making process depends on phosphorylation and dephosphorylation of substrates present in small numbers at each kinetochore. If these kinetochore targets are “good” substrates for mitotic kinases and phosphatases, i.e., there are low kinetic barriers for the phosphorylation and dephosphorylation reactions, then they would be vulnerable to the stochastic actions of kinases/phosphatases that exist abundantly in the cytoplasm. How might the system insulate kinetochores from this type of inappropriate signaling while still maintaining the ability to rapidly respond to proper bioriented attachment? To address this question, we will first review the current understanding of phospho-regulation at the kinetochore critical for controlling microtubule attachment and SAC signaling. Then, we will discuss hypothetical mechanisms that could produce this tight coupling between microtubule attachment status and signaling outputs.
Phosphorylations that control kinetochore–microtubule attachment
It is widely believed that proper anaphase chromosome movements rely on attachment between the dynamic plus ends of microtubules and kinetochores (end-on attachments). However, detailed live analysis of kinetochore motions corroborated with electron microscopy revealed that stable end-on attachments are not immediately accomplished upon entry into mitosis or meiosis (Kitajima et al. 2011; Magidson et al. 2011). Instead, a majority of kinetochores in early prometaphase form transient, unstable attachments to the sides of microtubule filaments (Fig. 3). Conversion from initial lateral attachments to end-on attachments is also observed in budding yeast (Tanaka et al. 2005). Although these lateral attachments can assist bioriented attachment, support positioning of chromosomes at the spindle equator, and facilitate partial removal of SAC proteins Mad1 and Mad2 from kinetochores (DeLuca et al. 2003), more stable attachments rely on forming end-on attachments, which occurs during late prometaphase and metaphase.

Lateral attachments. Diagram of the kinetochore components important for establishing initial, lateral attachments to microtubules early in prometaphase. The dual roles of Aurora B-dependent phosphorylation in recruiting the microtubule-interacting motor proteins dynein and CENP-E to kinetochores and destabilizing the interactions between microtubules and the Ndc80 complex are shown
Phospho-regulation of lateral attachment
At least two motor proteins are known to contribute to lateral attachments in vertebrates, the minus-end-directed cytoplasmic dynein and the plus-end-directed kinesin-7 motor, CENP-E (Kapoor et al. 2006; Rieder and Alexander 1990; Vorozhko et al. 2008; Wood et al. 1997). In the absence of dynein activity or CENP-E, congression of chromosomes to the metaphase plate is severely inhibited (Firestone et al. 2012; Kapoor et al. 2006; Schaar et al. 1997; Varma et al. 2008; Vorozhko et al. 2008; Wood et al. 1997), supporting the importance of these lateral interactions for establishing end-on attachments. Both motors are preferentially localized to unattached kinetochores in an Aurora B-dependent manner (Ditchfield et al. 2003; Emanuele et al. 2008; Kasuboski et al. 2011), which suggests that Aurora B plays a role in establishing attachments in addition to its role in destabilizing attachments (Fig. 3).
It has been proposed that kinetochore recruitment of dynein is mediated by Aurora B-dependent phosphorylation of Zwint-1, which associates with the kinetochore component KNL1 and recruits the dynein–dynactin complex through interaction with the RZZ complex, composed of Rod, Zwilch, and ZW10 (Fig. 3) (Kasuboski et al. 2011; Kiyomitsu et al. 2011; Lin et al. 2006; Petrovic et al. 2010). There are multiple consequences of Aurora B inhibition that are all rescued by expressing a phosphomimetic mutant of Zwint-1, including failure in kinetochore recruitment of dynein and ZW10, dynamic chromosome movement, and metaphase plate formation (Kasuboski et al. 2011), indicating the functional significance of this Zwint-1 phosphorylation. However, it remains to be established whether Zwint-1 residues are phosphorylated directly by Aurora B in vivo, as the sites studied are only remotely related to the consensus target motif for Aurora B (R-X-[S/T]-Φ, where Φ is any hydrophobic amino acid except Pro) (Alexander et al. 2011).
Kinetochore recruitment of CENP-E is also facilitated by Aurora B (Ditchfield et al. 2003; Emanuele et al. 2008; Vigneron et al. 2004), perhaps through enriching BubR1, which is important for CENP-E recruitment (Chen 2002; Johnson et al. 2004; Mao et al. 2003). CENP-E is phosphorylated by Aurora B (and Aurora A, which concentrates at the spindle poles) at its neck domain (Kim et al. 2010), which decreases its affinity for microtubules and its motor processivity. This phosphorylation is essential for congression of chromosomes from the poles to the spindle equator (Kim et al. 2010). The high levels of Aurora B-dependent phosphorylation at the kinetochore specifically on unattached kinetochores may facilitate rapid lateral attachments through recruitment of dynein and CENP-E, prior to the establishment of end-on attachment.
Aurora B-dependent destabilization of kinetochore–microtubule attachments
Aurora B is required for destabilizing erroneous attachments (Cimini et al. 2006; Lampson et al. 2004; Tanaka et al. 2002). The major targets of this phospho-regulation are the components of the KMN network, composed of KNL1 (also known as Blinkin, Spc105 [budding yeast and Drosophila] and Spc7 [fission yeast]), the Mis12 complex (also known as MIND), and the Ndc80 complex, which is critical for load-bearing end-on attachment (Cheeseman et al. 2004, 2006; Cheeseman and Desai 2008; Ciferri et al. 2008; DeLuca et al. 2006; Foley and Kapoor 2012; Kiyomitsu et al. 2007; Perpelescu and Fukagawa 2011; Petrovic et al. 2010; Wei et al. 2007). Microtubule binding of the KMN network is supported by Ndc80 (also known as Hec1 in human cells) and KNL1 in a cooperative manner (Cheeseman et al. 2006), while the Mis12 complex is important for kinetochore recruitment of the KNM network and other outer kinetochore components (De Wulf et al. 2003; Kline et al. 2006; Obuse et al. 2004).
Ndc80 has two distinct microtubule-binding modules, the calponin homology domain (CHD) and the unstructured, positively charged N-terminal tail (Alushin et al. 2010; Ciferri et al. 2008; Tooley et al. 2011), the latter of which is subject to Aurora B-dependent regulation. Binding of the CHD to microtubules is sensitive to structural changes in microtubules, showing substantial binding to taxol-stabilized microtubules, which have straight ends, but not with vinblastin-induced tubulin spirals, which mimic peel structures seen at depolymerizing microtubule ends. In contrast, the N-terminal tail interacts with the negatively charged C-terminal tail of tubulin and thus can bind microtubules laterally or at microtubule ends regardless of polymerization status (Ciferri et al. 2008; DeLuca et al. 2006; Miller et al. 2008; Tooley et al. 2011; Wei et al. 2007). The Ndc80 tail also promotes clustering of the Ndc80 complex on microtubules, further stabilizing its microtubule-binding capacity (Alushin et al. 2010; Ciferri et al. 2008). Aurora B phosphorylates multiple sites on the N-terminal tail of Ndc80 and weakens both the microtubule-induced clustering and the microtubule-binding affinity in vitro (Alushin et al. 2010; Cheeseman et al. 2006; Ciferri et al. 2008; DeLuca et al. 2006, 2011). Aurora B-dependent phosphorylation of Ndc80 is important for kinetochore–microtubule attachment in human cells, as phosphomimetic Ndc80 mutants are unable to support stable K-MT attachments (Guimaraes et al. 2008), while unphosphorylatable mutants are sufficient to cause the accumulation of syntelic and merotelic attachments in tissue culture cells (DeLuca et al. 2006, 2011; Kasuboski et al. 2011). Aurora B-dependent phosphorylation of other kinetochore proteins that interact with the Ndc80 complex, such as KNL1, Dsn1 (a member of the Mis12 complex), and CENP-U (a member of the constitutive centromere-associated network (CCAN) complex), also weakens microtubule attachment (Hua et al. 2011; Welburn et al. 2010). In addition, Nek2-dependent phosphorylation of Ser165 at the CHD of Ndc80 has been implicated in destabilization of microtubule attachment and recruitment of Mad1 and Mad2 (Wei et al. 2011). Thus, it is possible that the microtubule-binding modules of Ndc80 are negatively regulated by Aurora B and Nek2 in a cooperative manner (Fig. 4a).

Phospho-regulation of kinetochore functions. a–e Schematic illustrations of kinase and phosphatase signaling networks controlling kinetochore function. Colored arrows and inhibition symbols show the activity of kinases and phosphatases of the corresponding color. Large black arrows and inhibition symbols denote the functional consequences and feedback loops generated by this phospho-signaling, with small black arrows representing subprocesses within each signaling network
The function of the Ndc80 complex to support end-on attachment is augmented by additional microtubule-binding proteins that are also subject to phospho-regulation. In Saccharomyces cerevisiae, whereas the contribution of Aurora B-dependent phosphorylation of Ndc80 is subtle (Akiyoshi et al. 2009a; Kemmler et al. 2009), the Dam1 complex is the major functional target of Aurora B for microtubule attachment control because the temperature-sensitive growth of Aurora B mutant cells (ipl1-2) can be partially suppressed by phosphomimetic mutations of Dam1 (Cheeseman et al. 2002). Like the Ndc80 complex, the Dam1 complex can accomplish load-bearing attachment to dynamic microtubules and phosphorylation weakens this attachment (Gestaut et al. 2008). The Dam1 complex forms a ring-shaped oligomer that encircles microtubules and also directly interacts with the Ndc80 complex, which enhances the processivity of microtubule binding. Both of these Dam1 functions are opposed by Aurora B (Lampert et al. 2010; Tien et al. 2010). Phosphorylation of the Ser20 residue of Dam1 disrupts its binding to the Ndc80 complex (Tien et al. 2010), while phosphorylation of C-terminal residues compromises Dam1 oligomerization (Wang et al. 2007). Although phosphorylation-mediated regulation of Dam1 does not seem to be conserved in fission yeast (Buttrick et al. 2012), in higher eukaryotes, Aurora B-mediated phosphorylation negatively regulates kinetochore localization and microtubule binding of the Ska complex, which has very similar functions as the Dam1 complex despite no apparent structural similarity (Jeyaprakash et al. 2012). The Ska complex binds to the Ndc80 complex and supports kinetochore–microtubule attachment by providing affinity for curved protofilaments, which facilitates binding to depolymerizing microtubules (Chan et al. 2012; Schmidt et al. 2012).
Kinetochore–microtubule attachment is also controlled by proteins affecting microtubule stability. The kinesin-13 family proteins MCAK and Kif2b, which catalyze microtubule depolymerization, are localized to the centromere and the kinetochore and contribute to the error correction mechanism. During prometaphase, phosphorylation by Plk1 promotes localization of Kif2b at the outer kinetochore through its interaction with CLASP1, and this destabilizes kinetochore–microtubule attachment, which is required for correction of erroneous attachments (Hood et al. 2012). When Aurora B-dependent phosphorylation at the kinetochore is suppressed upon bioriented attachment, the astrin–kinastrin/SKAP complex displaces Kif2b from CLASP1 to stabilize microtubule attachment (Dunsch et al. 2011; Manning et al. 2010; Schmidt et al. 2010). However, it is not clear how this potential role of Plk1 in kinetochore–microtubule destabilization is coordinated with its critical function for establishing and maintaining stable end-on microtubule attachments (see below) (Lenart et al. 2007).
MCAK is required for correcting merotelic attachments and is recruited to centromeric chromatin by Aurora B-dependent phosphorylation of MCAK itself and Sgo2 (Andrews et al. 2004; Lan et al. 2004; Tanno et al. 2010). A role for Aurora B in recruiting centromeric MCAK is at odds with its effect on MCAK function, as Aurora B-dependent phosphorylation of MCAK reduces its microtubule depolymerization activity (Andrews et al. 2004; Lan et al. 2004; Ohi et al. 2004). However, Aurora B-dependent inhibition of another kinesin-13 family protein Kif13a is neutralized by a centromeric protein ICIS in vitro (Knowlton et al. 2009), so centromeric MCAK activity may also be maintained by ICIS at the centromere despite high Aurora B activity.
The small Rho GTPase Cdc42 and its downstream effector mDia3 (mammalian diaphanous-related formin 3), which is localized to mitotic kinetochores, support stable kinetochore–microtubule attachment by a mechanism independent of mDia3’s capacity to nucleate actin (Cheng et al. 2011; Yasuda et al. 2004). The formin homology domain of mDia3 can directly bind and stabilize microtubules in vitro, but its phosphorylation by Aurora B neutralizes these activities. Since a phosphomimetic mutant of mDia3 cannot fully support stable bipolar attachment, phospho-regulation of mDia3 appears to control the stability of kinetochore–microtubule attachment (Cheng et al. 2011).
The kinase cascades regulating kinetochore–microtubule attachment
Aurora B’s role in destabilizing kinetochore–microtubule attachment is supported by Mps1. In yeast and human cells, Mps1 is required for correcting erroneous kinetochore–microtubule attachments (Jelluma et al. 2008; Maure et al. 2007; Sliedrecht et al. 2010). However, its underlying mechanism remains to be established. While it has been reported that Mps1-dependent phosphorylation of the CPC subunit Borealin stimulates Aurora B activity at the kinetochore in human cells (Jelluma et al. 2008), this was not seen in other studies (Maciejowski et al. 2010; Maure et al. 2007). This discrepancy may be due to the use of different cell lines, or the specific requirement of Mps1 for initial stimulation but not maintenance of Aurora B activity (Sliedrecht et al. 2010). Recently, it was demonstrated that Mps1-dependent phosphorylation at multiple residues of Spc105/Spc7/KNL1 is necessary and sufficient for its recruitment of the Bub1–Bub3 complex (Fig. 4b) (London et al. 2012; Shepperd et al. 2012; Yamagishi et al. 2012), which is required for proper kinetochore–microtubule attachment (Bernard et al. 2001; Logarinho et al. 2008; Meraldi and Sorger 2005; Warren et al. 2002). Phosphorylation site mutants of fission yeast spc7 show an increased level of chromosome missegregation, similar to deletion mutants of mph1 (fission yeast mps1) and bub1 (Shepperd et al. 2012; Yamagishi et al. 2012). Although a requirement for this pathway in the error correction mechanism remains to be established, these data indicate that one of the major functions of Mps1 is to target Bub1–Bub3 to KNL1.
The checkpoint protein Bub1 is a protein kinase whose major established substrate is histone H2A at Ser121 in fission yeast or at Thr120 in human (Kawashima et al. 2010). Phosphorylation of H2A at Thr120 (in human residue notation) recruits shugoshin proteins (Sgo1 and Sgo2) to the inner centromere (Kawashima et al. 2010). Shugoshin proteins then recruit the CPC by interacting with Survivin (in fission yeast) or Borealin (in humans) that is phosphorylated by Cdk1 (Fig. 4c) (Tsukahara et al. 2010). In fission yeast, a Bub1 kinase-dead mutant, a histone H2A S121A mutant, deletion of Sgo2 (the sole mitotic form of shugoshin in fission yeast), and Cdk1 phosphorylation site mutants of Survivin defective in Sgo2 binding all show comparable defects in chromosome segregation (Kawashima et al. 2010; Tsukahara et al. 2010). Artificial targeting of INCENP to the centromere bypassed the requirement of Bub1 kinase activity for chromosome alignment in mouse cells (Ricke et al. 2012), confirming the model that Bub1 promotes proper kinetochore attachment by regulating centromeric localization of the CPC.
The clear role of Aurora B-dependent phosphorylation of Ndc80 and its interacting proteins in destabilizing microtubule attachment makes the observation that the highest levels of phosphorylation on these substrates are present on unattached kinetochores enigmatic, because these kinetochores must prepare to capture microtubules (DeLuca et al. 2011; Welburn et al. 2010). This enigma may be explained if dynein and CENP-E, whose kinetochore recruitment are facilitated by Aurora B, and perhaps other microtubule binding proteins, not the Ndc80 complex, support initial kinetochore–microtubule attachment during prophase. Consistent with this hypothesis, initial kinetochore–microtubule attachments during early prometaphase are unstable and they are independent of the Ndc80 complex (Cai et al. 2009; Magidson et al. 2011). In addition, it was recently proposed that another kinase, Plk1, promotes kinetochore–microtubule attachment in part through counteracting the action of Aurora B (Liu et al. 2012; Suijkerbuijk et al. 2012b). The tension-sensitive 3F3/2 epitope depends on Plk1 (Ahonen et al. 2005; Wong and Fang 2007), and the reduction of Plk1-dependent phosphorylation at the kinetochore upon bipolar attachment has been confirmed using a FRET-based sensor (Liu et al. 2012). One of the critical kinetochore substrates for Plk1 is BubR1 (Fig. 4d) (Elowe et al. 2007; Matsumura et al. 2007; Suijkerbuijk et al. 2012b). Plk1-dependent phosphorylation of BubR1 at the Kinetochore Attachment Regulatory Domain is important for recruitment of PP2A-B56α to counteract the action of Aurora B and thus to stabilize kinetochore–microtubule attachment (Suijkerbuijk et al. 2012b). In addition, Plk1 is likely to phosphorylate other substrates, such as CLIP-170, which can bind microtubule plus ends (Li et al. 2010). Plk1 also promotes kinetochore recruitment of CENP-E (Ahonen et al. 2005; Nishino et al. 2006), which drives chromosome congression to the metaphase plate through supporting lateral attachment (Kapoor et al. 2006). During chromosome congression, microtubules bind laterally to the side of the leading kinetochore, while the trailing kinetochore attaches the ends of microtubules (Kapoor et al. 2006). It has been shown that the Plk1-dependent phosphoepitope 3F3/2 is high at the leading kinetochore but is low at the trailing kinetochores (Gorbsky and Ricketts 1993). Thus, high Plk1 activity on the laterally attached kinetochore may recruit PP2A, which can promote dephosphorylation of Aurora B substrates to help the conversion from lateral attachment to end-on attachment.
Phosphorylations that support the SAC
The metaphase-to-anaphase transition is triggered by the APC/C-mediated ubiquitylation of cyclin B and securin (Peters 2006). Recognition of these substrates by the APC/C is mediated by Cdc20, which is the primary target of the SAC. The APC/C is inhibited by the MCC, composed of Mad2, BubR1, Bub3, and Cdc20, whose assembly is controlled by kinetochore-dependent and kinetochore-independent mechanisms (Kulukian et al. 2009; Maciejowski et al. 2010; Malureanu et al. 2009; Meraldi et al. 2004; Sudakin et al. 2001; Tang et al. 2001). The kinetochore-independent MCC is constitutively required for inhibition of the APC/C during interphase and early M phase to set a basal duration from entry into mitosis to anaphase onset. The kinetochore-dependent generation of the MCC responds to unattached kinetochores to activate and maintain the SAC until all kinetochores accomplish bioriented microtubule attachments (Meraldi et al. 2004). Laser ablation of a single unattached kinetochore triggers anaphase (Rieder et al. 1994), supporting the idea that the kinetochore is critical for generation of SAC signaling. This view was confirmed by the fact that kinetochores assembled on magnetic beads can activate the SAC in Xenopus egg extracts (Guse et al. 2011). Unattached kinetochores recruit Mad1, which converts the cytoplasmic open form of Mad2 (O-Mad2) into the closed form of Mad2 (C-Mad2), which binds and inhibits Cdc20 (Musacchio and Salmon 2007). This amplification mechanism to generate C-Mad2 explains how a single unattached kinetochore can produce the robust signal to inhibit cell cycle progression. However, artificial recruitment of Mad1–Mad2 to the kinetochore (by a Mis12–Mad1 fusion protein) but not to bulk chromatin (by a H2B–Mad1 fusion) is able to activate the SAC (Maldonado and Kapoor 2011), indicating that generation of C-Mad2 and/or formation of the MCC must require co-localization of Mad1 with other kinetochore proteins, such as Bub3, BubR1, Bub1, and Mps1.
Bub1 and BubR1
Although Bub1 has kinase activity and it has been reported that Bub1 phosphorylates Cdc20 to inhibit the catalytic activity of the APC/C (Tang et al. 2004), its kinase activity is not directly required for SAC activation in yeasts (Fernius and Hardwick 2007; Kawashima et al. 2010; Warren et al. 2002), Xenopus egg extracts (Sharp-Baker and Chen 2001), and mammalian cells (Klebig et al. 2009; Perera and Taylor 2010; Ricke et al. 2012). Instead, the N-terminal non-kinase domain supports the SAC by recruiting proteins critical for SAC activation, such as Mad1–Mad2, Bub3, BubR1/Mad3, and CENP-E (Klebig et al. 2009; Rischitor et al. 2007). However, Bub1 kinase activity contributes to SAC activation through H2A phosphorylation, which indirectly recruits Aurora B (Kawashima et al. 2010; Ricke et al. 2012) (discussed above). This may explain why the SAC is partially compromised in Bub1-depleted Xenopus egg extracts supplemented with kinase-deficient Bub1 (Chen 2004).
BubR1, a paralog of Bub1, plays a critical role in inhibiting the APC/C through formation of the MCC. In addition to assisting the kinetochore recruitment of Mad1–Mad2 at the kinetochore, its N-terminal KEN-box acts as a pseudosubstrate of Cdc20–APC/C (Lara-Gonzalez et al. 2011). BubR1 has a kinase domain, though the functional significance of the kinase domain has been a subject of debate. Several motifs critical for canonical kinases are mutated or absent in BubR1, while Mad3, the yeast BubR1 homolog, lacks the entire kinase domain. Kops and colleagues recently proposed that the kinase domain of BubR1 does not function as an enzyme but as a structural stabilizer (Suijkerbuijk et al. 2012a). A study using mouse BubR1 knockout cells indicates that the kinase domain is dispensable for the kinetochore-independent inhibition of the APC/C, further confirming the kinase-independent function of BubR1 for SAC activity (Malureanu et al. 2009). However, in that same study, kinetochore recruitment of BubR1 including the kinase domain partially contributed to prolonged mitotic arrest in response to nocodazole or taxol. In addition, it has been shown that BubR1 kinase activity depends on binding to the kinetochore motor protein CENP-E (Mao et al. 2003, 2005; Zhang et al. 2007) and is required for SAC activation and kinetochore recruitment of other SAC components, such as Mad2, though other studies conflict with this observation (Chen 2002). Very recently, Mao’s team demonstrated that CENP-E-dependent BubR1 phosphorylation supports the SAC and chromosome alignment in human cells (Guo et al. 2012). The lack of this phosphorylation resulted in reduced phosphorylation of Ndc80 by Aurora B and reduced levels of Mad2 on unattached kinetochores. Based on the observation that a phosphomimetic BubR1 mutant bypasses the requirement for CENP-E in metaphase chromosome alignment, Mao and colleagues proposed that the major function of CENP-E for the SAC and kinetochore–microtubule attachment is to activate BubR1 (Guo et al. 2012). The apparent conflict regarding BubR1 kinase activity is highlighted by an observation that a D911N mutation in human BubR1, which would nullify conventional kinases, can still support the SAC and chromosome alignment (Suijkerbuijk et al. 2012a). Testing whether this mutation indeed inactivates CENP-E-dependent BubR1 kinase activity will help reconcile this issue.
Mps1
One of the key upstream regulators of the SAC is Mps1, a kinase that is recruited to kinetochores by an unclear mechanism dependent on the Ndc80 complex (Abrieu et al. 2001; Martin-Lluesma et al. 2002; Stucke et al. 2004; Stucke et al. 2002). Inhibition of Mps1 causes displacement of SAC proteins from mitotic kinetochores and also dissociates Cdc20 from Mad2 and BubR1 during interphase and mitosis, indicating that Mps1 activity is constitutively required for both kinetochore-independent and kinetochore-dependent mechanisms of MCC assembly (Maciejowski et al. 2010; Sliedrecht et al. 2010; Tighe et al. 2008). In fission yeast, Mps1-dependent phosphorylation of Mad2 at Ser92 contributes to maintenance of the SAC by supporting the interaction of the MCC with the APC/C (Zich et al. 2012). For the kinetochore-dependent mechanism, Mps1 contributes to SAC activation by phosphorylating Spc105 and recruiting the Bub1–Bub3 complex (London et al. 2012; Shepperd et al. 2012; Yamagishi et al. 2012), which further recruits BubR1 and the Mad1–Mad2 complex (Chen 2002; Hewitt et al. 2010; Johnson et al. 2004; Meraldi et al. 2004; Yamagishi et al. 2012). However, recruitment of Bub1–Bub3 to the kinetochore is insufficient to recruit Mad1 and Mad2 (Ito et al. 2011; Yamagishi et al. 2012), indicating an additional requirement for Mad1–Mad2 recruitment. In budding yeast, Mps1-dependent phosphorylation of Ndc80 contributes to SAC maintenance without affecting bioriented kinetochore–microtubule attachment (Kemmler et al. 2009).
The kinetochore recruitment of Mps1 depends on an N-terminal region containing the tetratricopeptide repeat, which also exists in Bub1 and BubR1 (Lee et al. 2012; Maciejowski et al. 2010). This domain is required for kinetochore localization of Bub1 and long-lasting SAC maintenance, but not for SAC activation in human tissue culture cells (Maciejowski et al. 2010). Therefore, kinetochore localization of Mps1 can contribute to, but is normally dispensable for SAC function. However, Mps1 kinetochore localization is critical for SAC activation and chromosome alignment in female mouse meiosis I (Hached et al. 2011), indicating a context-dependent requirement of Mps1 at the kinetochore.
Aurora B
Aurora B contributes to activation of the SAC by at least three distinct mechanisms: destabilizing kinetochore–microtubule attachment, which leads to generation of unattached kinetochores; kinetochore recruitment of SAC components; and an unknown process after the kinetochore recruitment of Mad1 and Mad2. While the CPC is clearly important for SAC activation in response to lack of microtubule attachment in Xenopus egg extracts and in fission yeast (Kallio et al. 2002; Petersen and Hagan 2003; Vanoosthuyse and Hardwick 2009; Vigneron et al. 2004), its contribution to the SAC in budding yeast appears to be limited to creating unattached kinetochores prior to bipolar attachment (Pinsky et al. 2006b). Similarly, in human cells, the effect of Aurora B inhibition is more pronounced in cells treated with taxol, which stabilizes microtubules and reduces tension at the kinetochore, than those treated with nocodazole, which inhibits microtubule polymerization and creates unattached kinetochores, supporting the idea that Aurora B plays a limited role in activating the SAC in response to unattached kinetochores (Ditchfield et al. 2003). This result is in line with the finding that in budding yeast Mad3 phosphorylation by Aurora B is important for SAC signaling induced by a lack of tension but not by a lack of attachment (King et al. 2007a).
In vertebrates, Aurora B promotes kinetochore recruitment of SAC proteins (Mad1, Mad2, Bub1, BubR1, Mps1, and CENP-E) and outer kinetochore proteins such as the RZZ complex and dynein (Ditchfield et al. 2003; Emanuele et al. 2008; Kasuboski et al. 2011; Saurin et al. 2011; Vigneron et al. 2004). It was proposed that Aurora B-dependent phosphorylation of Zwint-1 recruits the RZZ complex (Kasuboski et al. 2011), which is important for recruitment of the dynein–dynactin complex and the Mad1–Mad2 complex, but it remains to be tested whether Zwint-1 is an in vivo target of Aurora B as discussed above. Intriguingly, the requirement of Aurora B for Mad2 recruitment is most evident during prophase and early prometaphase, but not in late prometaphase (Saurin et al. 2011). This correlates with the observation that Aurora B inhibition delays rapid activation of the SAC upon entry into mitosis, but that the SAC eventually engages. Since this early requirement of Aurora B is bypassed by a forced targeting of Mps1 to the kinetochore, Aurora B appears to promote SAC activation by recruiting Mps1 to the kinetochore (Saurin et al. 2011). Consistent with the auxiliary role of Aurora B for SAC activation in response to unattached kinetochores, Aurora B becomes indispensable for SAC activation upon partial inhibition of Mps1, depleting Ndc80 (Hec1), which is critical for kinetochore recruitment of Mps1 (Santaguida et al. 2011; Saurin et al. 2011), or depleting members of the CCAN, CENP-H, CENP-I, and CENP-N, which also contribute to kinetochore recruitment of Mad1 and Mad2 (Matson et al. 2012). Mps1 and Aurora B are not only required for Mad1–Mad2 recruitment but also for conducting the SAC signal after Mad1–Mad2 recruitment (Maldonado and Kapoor 2011), likely through controlling MCC formation (Maciejowski et al. 2010) and the MCC–APC/C interaction (Morrow et al. 2005; Vanoosthuyse and Hardwick 2009).
Cdk1 and MAPK
Cdk1 is the major cell cycle driver, whose activity is controlled by cyclins. The primary Cdk1 substrate recognition motif is [S/T]Px[R/K], where the [R/K] residue at the +2 position relative to the phosphorylation site facilitates but is dispensable for phosphorylation (Alexander et al. 2011; Ubersax et al. 2003). Since Cdk1-cyclin B is the major downstream target for the SAC, it has been difficult to demonstrate a specific role for Cdk1 in SAC regulation. The role of phosphorylation at [S/T]P motifs in SAC components can be examined as possible Cdk1 targets; however, it has been shown that mitogen-activated protein kinase (MAPK), which also phosphosphorylates [S/T]P motifs, is required for SAC activation in Xenopus egg extracts and in somatic vertebrate cells (Minshull et al. 1994; Takenaka et al. 1997; Wang et al. 1997). Studies in Xenopus egg extracts elegantly demonstrated the importance of phosphorylation at multiple residues of the N-terminal region of Cdc20 for SAC activation by promoting association with Mad2 and inhibiting association with the APC/C (Chung and Chen 2003; D'Angiolella et al. 2003; Labit et al. 2012). Among these residues, phosphorylation of Thr64 and Thr68 depends on MAPK activity while Ser50 and Thr79 are potential Cdk1 target sites and all are all required for SAC activation (Chung and Chen 2003). Interestingly, phosphatases for Thr64, Thr68, and Thr79, but not for Ser50, are active in mitotic extracts (Labit et al. 2012). How the balance between phosphorylation and dephosphorylation of these sites is controlled in response to SAC activation and silencing is an important future question.
In addition, MAPK contributes to SAC activation by phosphorylating at least two more substrates. MAPK-dependent phosphorylation at Ser844 of Xenopus Mps1 is critical for SAC activation by promoting kinetochore localization of Mps1 and other checkpoint components, such as Mad1, without affecting the kinase activity of Mps1 (Zhao and Chen 2006). It was also reported that five residues of Bub1 are targets of MAPK, and this is important for Bub1 kinase activity, thereby partially contributing to the SAC (Chen 2004). Since cyclin B1 and the active phosphorylated form of MAPK are preferentially localized to unattached kinetochores during prometaphase (Bentley et al. 2007; Shapiro et al. 1998; Zecevic et al. 1998), it would be interesting to examine whether phosphorylation by Cdk1-cyclin B and MAPK at the kinetochore is regulated by microtubule attachment status.
Roles of phosphatases at the kinetochore and the centromere
PP1
The identification of PP1 mutants showing mitotic arrest phenotypes revealed a specific role for PP1 in mitotic progression (Booher and Beach 1989; Doonan and Morris 1989; Ohkura et al. 1989). More recently, it has become clear that these mitotic arrests are largely due to a failure in silencing the SAC (Espeut et al. 2012; Meadows et al. 2011; Pinsky et al. 2009; Rosenberg et al. 2011; Vanoosthuyse and Hardwick 2009). A variety of PP1 functions can be modulated by its regulatory subunits, which often contain PP1-docking segments called SILK and RVxF motifs (Hendrickx et al. 2009). The essential role of PP1 during mitosis is mediated by recruitment of PP1 to the highly conserved RVxF motif of the kinetochore protein KNL1 (Fig. 4e) (Espeut et al. 2012; Liu et al. 2010; Meadows et al. 2011; Rosenberg et al. 2011). Mutation of this RVxF motif is lethal, and this lethality can be rescued by deleting Mad2 in budding yeast (Rosenberg et al. 2011), indicating that the essential role of PP1 bound to KNL1 (Spc105) is silencing the SAC in that organism, not regulating kinetochore–microtubule attachment. In contrast, the RVxF motif mutant of KNL1 in Caenorhabditis elegans shows delayed formation of load-bearing kinetochore–microtubule attachments and enhanced lethality upon Mad2 depletion (Espeut et al. 2012), indicating a more important role in regulating kinetochore–microtubule attachment in that system. In vertebrates, the RVxF motif mutant of KNL1 (Blinkin) is lethal and mildly defective in stabilization of kinetochore–microtubule attachment (Liu et al. 2010), but its impact on SAC silencing remains to be established. Vertebrates express three isotypes of PP1 (α, β, γ) and their localization is most prominent on unattached kinetochores and is reduced upon microtubule attachment (Posch et al. 2010; Trinkle-Mulcahy et al. 2006; Trinkle-Mulcahy et al. 2003). Phosphorylation of KNL1 by Aurora B can weaken the PP1–KNL1 interaction, raising the possibility that PP1 localization at the kinetochore is negatively regulated by Aurora B (Fig. 4e) (Liu et al. 2010; Rosenberg et al. 2011). Furthermore, the catalytic activity of PP1 is suppressed by Cdk1-cyclin B (Dohadwala et al. 1994; Yamano et al. 1994). Since cyclin B1 is preferentially localized to unattached kinetochores but is dissociated from kinetochores at the metaphase plate (Bentley et al. 2007), the catalytic activity of PP1 may also be regulated by microtubule attachment.
A series of genetic, biochemical, and cell biological experiments strongly supports the idea that PP1 acts to oppose the function of Aurora B (Francisco et al. 1994; Hsu et al. 2000; Pinsky et al. 2006a, 2009; Vanoosthuyse and Hardwick 2009). Consistently, lethality of the spc105 RVxF mutant and temperature sensitivity of the ipl1 (Aurora B) mutant partially complement each other (Rosenberg et al. 2011). It may be surprising that recruitment of PP1 to Spc105 (KNL1) is dispensable for kinetochore–microtubule attachment in budding yeast since dephosphorylation of Aurora B substrates is important for accomplishing stable kinetochore–microtubule attachments. One possible explanation is that different levels of dephosphorylation may be required for stable kinetochore–microtubule attachment and SAC silencing. For example, since the multisite phosphorylation of the KMN network has synergistic effects on microtubule binding (Welburn et al. 2010), partial dephosphorylation by PP1 may provide the kinetochore with substantial affinity to microtubules to achieve load-bearing attachment but more robust dephosphorylation may be required for SAC silencing and thus require the high local concentration of kinetochore-bound PP1.
Alternatively, specific substrates relevant for SAC signaling, but not for microtubule attachment, may require PP1 recruitment to KNL1. One candidate for a SAC-specific substrate is KNL1 itself, whose Mps1-dependent phosphorylation recruits Bub1. However, it is unlikely that Bub1 dissociation from the kinetochore triggers SAC silencing since Bub1 remains on bioriented kinetochores (Howell et al. 2004; Shah et al. 2004). Other candidates include Mad3 (BubR1), which is phosphorylated by Ipl1 (Aurora B) (King et al. 2007a) and Ndc80, which is phosphorylated by Mps1 in budding yeast (Kemmler et al. 2009). In higher eukaryotes, it has been suggested that dephosphorylation of Zwint-1 is necessary for SAC silencing, as a phosphomimetic mutant of Zwint-1 delays metaphase with accumulated SAC components even on kinetochores aligned at the metaphase plate (Kasuboski et al. 2011).
Consistent with the idea that PP1 bound to KNL1 can dephosphorylate only a subset of kinetochore substrates, other kinetochore proteins also interact with PP1 through their RVxF motifs. These PP1-binding proteins include Fin1 in budding yeast (Akiyoshi et al. 2009b), kinesin-8 (Klp5 and Klp6) in fission yeast (Meadows et al. 2011), and CENP-E in human cells (Kim et al. 2010). The PP1-binding motifs in Klp5 and Klp6 contribute to both SAC silencing and kinetochore–microtubule attachment. Like KNL1, Aurora-dependent phosphorylation of an RVxF motif in CENP-E causes it to dissociate from PP1. This dissociation has been implicated in establishment of stable kinetochore–microtubule attachment (Kim et al. 2010), though the specific role of PP1-binding to CENP-E remains to be established.
Direct fusion of PP1 to the N-terminus of Spc105 (KNL1) bypasses the requirement for Spc105’s RVxF motif in budding yeast (Rosenberg et al. 2011), indicating that association of the RVxF motif with PP1 does not affect PP1’s catalytic activity. However, PP1 activity is likely to be regulated by Sds22, which binds PP1 through its leucine-rich repeats (Ceulemans et al. 2002; Stone et al. 1993), and plays an essential role for mitotic progression (Ohkura and Yanagida 1991; Stone et al. 1993). Cells with reduced Sds22 show a metaphase delay with mild chromosome misalignment (Ohkura and Yanagida 1991; Posch et al. 2010; Stone et al. 1993). Consistent with its augmenting PP1 function, reduction of Sds22 in mammalian cells causes enhanced levels of Aurora B autophosphorylation and Aurora B-dependent phosphorylation of a member of the Mis12 complex, Dsn1 (Wurzenberger et al. 2012), though it was also reported that phosphorylation of Ndc80 and MCAK was rather decreased (Posch et al. 2010). This difference may reflect the role of Sds22 as an inhibitor for specific substrates but not others (Stone et al. 1993). Although Sds22 does not contribute strongly to kinetochore–microtubule attachment in metaphase, it does do so during anaphase chromosome movement, along with Repo-Man, which recruits PP1 to anaphase chromosomes (Wurzenberger et al. 2012). Altogether, the major role of PP1–Sds22 at the kinetochore appears to be silencing the SAC with an auxiliary function for kinetochore–microtubule attachment.
PP2A
Phosphorylation of kinetochore proteins is also controlled by type 2A phosphatase (PP2A), which shows a stronger contribution to kinetochore–microtubule attachment than to the SAC (Foley et al. 2011). The PP2A holoenzyme is composed of a catalytic subunit, a scaffolding A subunit and a regulatory B subunit. Among a variety of B subunits, the B56 (B′) family (α, β, γ, δ, and ε) members localize to kinetochores during mitosis and seem to be the major regulatory subunits controlling PP2A’s role in microtubule attachment. The localization of the B56 subunits is most prominent on unattached kinetochores, and their levels are reduced (α, ε) or undetectable (β, γ, δ) on kinetochores with bioriented attachment. Upon depletion of B56 subunits, the levels of KNL1 and Dsn1 phosphorylation by Aurora B and BubR1 phosphorylation by Plk1 increase, and kinetochore–microtubule attachment is compromised. This reduced kinetochore–microtubule attachment is rescued by inhibiting Aurora B, indicating that PP2A-B56 antagonizes Aurora B to support stable association of microtubules at the kinetochore (Foley et al. 2011). Although PP2A-B56 has been implicated in the maintenance of sister chromatid cohesion through interaction with Shugoshin proteins (Kitajima et al. 2006; Riedel et al. 2006; Tang et al. 2006), its role in kinetochore–microtubule attachment appears to be distinct from its regulation of sister chromatid cohesion. As mentioned above, kinetochore recruitment of the B56 subunit is mediated by Plk1-dependent phosphorylation of BubR1 (Fig. 4d) (Suijkerbuijk et al. 2012b).
Taken together, studies to date suggest a division of labor between mitotic phosphatases in which PP2A-B56 promotes dephosphorylation of kinetochore substrates at unattached kinetochores to promote kinetochore–microtubule attachment while PP1 supports SAC silencing and maintenance of kinetochore–microtubule attachment on attached kinetochores. How the actions of these two phosphatases are coupled to kinetochore–microtubule attachment and how they can play distinct functions are subjects of future study.
Making an effective switch at the kinetochore in response to microtubule attachment
Kinetochore–microtubule attachments are stabilized when chromosomes achieve bipolar microtubule attachment, but erroneous attachments must be corrected by destabilizing them. The SAC can be activated by a sole unattached kinetochore in a cell, while bipolar attachment of the last unattached kinetochore swiftly silences the SAC (Clute and Pines 1999; Rieder et al. 1994). As discussed above, a number of protein kinases and phosphatases are required to control these processes, and so far Aurora B- and Plk1-dependent phosphorylations of kinetochore substrates are known to be sensitive to the status of microtubule attachments: high at unattached kinetochores and low at kinetochores with bipolar attachment (Ahonen et al. 2005; Cheng et al. 2011; DeLuca et al. 2011; Elowe et al. 2007; Lan et al. 2004; Liu et al. 2009, 2012; Nishino et al. 2006; Suijkerbuijk et al. 2012b; Welburn et al. 2010). Coincidentally, many of the kinases and phosphatases are also recruited to kinetochores and centromeres, and their local levels are affected by microtubule attachment status (Fig. 2). For example, more Mps1, Bub1, Plk1, Cdk1-cyclin B1, and PP2A-B56 are recruited to unattached/misaligned kinetochores than to kinetochores with bioriented attachment (Bentley et al. 2007; Foley et al. 2011; Hori et al. 2008; Howell et al. 2004; Liu et al. 2012; Saurin et al. 2011), while more PP1 is recruited to kinetochores upon bioriented attachment (Liu et al. 2010). Strikingly, either disruption of the PP1 (Glc7)–KNL1 (Spc105) interaction or recruitment of an extra copy of PP1 to KNL1 is lethal in budding yeast (Rosenberg et al. 2011), suggesting that the exact amount of PP1 residing at the kinetochore must be tightly tuned for proper phospho-regulation. Enrichment of these signaling components at the kinetochore may help couple regulation of kinetochore–microtubule dynamics and the SAC (Foley and Kapoor 2012). Furthermore, local regulation of phosphorylation/dephosphorylation allows the system to correct erroneous microtubule attachments at an individual kinetochore without affecting the established attachments at other kinetochores even if other kinetochores are very close by. It also allows each kinetochore to act as an independent sensor to detect its own microtubule attachment status and generate a diffusible SAC signal. But how can phosphorylation levels accurately respond to kinetochore–microtubule attachment status? Below we discuss several potential mechanisms.
Feedback mechanisms that control kinetochore localization of kinases and phosphatases
Cooperativity and feedback can contribute to formation of a switch-like response (Pomerening et al. 2003) and can be generated by enzymes whose activity affects their own localization either directly or indirectly. Plk1 and Aurora B are known to promote their own localization at the kinetochore and centromere. Plk1 is recruited to kinetochores through its C-terminal Polo-box domain (PBD), which recognizes a phosphorylated serine or threonine preceded by a serine residue (S-Sph/Tph) (Elia et al. 2003). Plk1 activity can thus create its own phospho-docking site on targets, such as NudC (Nishino et al. 2006; Zhou et al. 2003) and the CCAN component CENP-U (CENP-50, PBIP1) (Hori et al. 2008; Kang et al. 2006). Since binding ligands through its PBD stimulates the catalytic activity of Plk1, a cluster of CENP-U proteins may lead to the spreading of active Plk1 on a kinetochore as it phosphorylates adjacent CENP-U molecules and generates new Plk1-binding sites (Park et al. 2011). However, the importance of this mechanism of Plk1 recruitment to kinetochores in prometaphase is unclear since CENP-U is primarily required for kinetochore recruitment of Plk1 during late G2 phase (Hori et al. 2008; Kang et al. 2006), and its chicken homolog, CENP-50, is not essential in DT40 cells (Minoshima et al. 2005). Cdk1-dependent phosphorylation of Bub1, BubR1, and INCENP also generates Plk1 docking sites at the kinetochore (Goto et al. 2006; Qi et al. 2006; Wong and Fang 2007), but it remains to be established whether this docking merely represents substrate recognition or also contributes to enrich Plk1 at the kinetochore to facilitate phosphorylation of other neighboring substrates.
The mechanism of Plk1 dissociation from the kinetochore, which is coupled to bipolar microtubule attachment (Hori et al. 2008; Lenart et al. 2007) and SAC silencing (Liu et al. 2012), could be the simple reversal of the mechanism that generates its recruitment. Consistent with the PBD-dependent kinetochore recruitment of Plk1, PP1 and PP2A-B56 are both required for dissociation of Plk1 from metaphase kinetochores (Foley et al. 2011; Liu et al. 2012). Thus, dissociation of Plk1 from the kinetochore is likely to be caused by decreased Plk1 activity or increased dephosphorylation of the PBD recognition sites. Since the activation loop of Plk1 at the kinetochore is phosphorylated by Aurora B (Carmena et al. 2012a), downregulation of Aurora B-dependent phosphorylation upon bioriented attachment could contribute to dissociation of Plk1.
Centromeric enrichment of Aurora B is regulated by positive feedback involving two histone kinases: Haspin, which phosphorylates H3T3, and Bub1, which phosphorylates H2A T120. Phosphorylated H3T3 (H3T3ph), which is not restricted to the centromere at the entry into mitosis, recruits Aurora B to mitotic chromatin by directly binding the Survivin subunit of the CPC (Kelly et al. 2010; Wang et al. 2010; Yamagishi et al. 2010), while phosphorylated H2A, which is localized to the kinetochore-proximal region of the centromere, also recruits the CPC as described above. Local enrichment of Aurora B can facilitate phosphorylation at its activating loop and at the C-terminal TSS motif of INCENP (Kelly et al. 2007), both of which are critical for full activation of Aurora B. Activated Aurora B can in turn phosphorylate Haspin to stimulate H3T3 phosphorylation (Wang et al. 2011). In addition, Aurora B contributes to kinetochore accumulation of Bub1 (Boyarchuk et al. 2007; Morrow et al. 2005; Vigneron et al. 2004). These feedback mechanisms drive local enrichment of H3T3ph and Aurora B at the inner centromere (Fig. 4c) (Ricke et al. 2012; Wang et al. 2011; Yamagishi et al. 2010), and this enrichment is functionally important for Aurora B-dependent control of kinetochore–microtubule attachment and SAC activation. Artificial targeting of the CPC to the centromere and kinetochore by fusing INCENP to CENP-B and Mis12, respectively, destabilizes kinetochore–microtubule attachment (Liu et al. 2009), while CENP-B–INCENP fusion bypasses the requirement for H3T3ph in SAC activation (De Antoni et al. 2012; Wang et al. 2012). In addition, abrogating CPC targeting to the centromere causes severe chromosome misalignment defects (De Antoni et al. 2012; Lens et al. 2006; Wang et al. 2010, 2012) (except in chicken DT40 cells, where CPC mislocalization by Survivin depletion does not have strong impacts on chromosome alignment (Yue et al. 2008)). Although this positive feedback regulation of Aurora B is important for centromeric enrichment of Aurora B, turning off this positive feedback may not be the major trigger for SAC silencing. Aurora B shows preferential accumulation on centromeres of unattached/misaligned chromosomes by a Plk1-dependent mechanism in untransformed cell lines, but such a preference is not seen in HeLa cells (Salimian et al. 2011). Instead, removal of the CPC from the centromere depends on Cdk1 inactivation (Hummer and Mayer 2009; Parry et al. 2003), the process that requires SAC silencing.
In mammals, bioriented attachments facilitate kinetochore recruitment of PP1 (DeLuca et al. 2011; Liu et al. 2010), potentially supporting a mechanism that couples kinetochore–microtubule attachment and dephosphorylation. Aurora B-dependent phosphorylation of KNL1 dissociates the PP1–KNL1 interaction (Liu et al. 2010; Rosenberg et al. 2011), creating a double negative feedback loop to potentially support the switch-like behavior of phosphorylation status (Fig. 4e). However, evidence in budding yeast indicates that PP1 recruitment is insufficient for this switch. Although the constitutive recruitment of PP1 to the kinetochore (via fusion of PP1 to the N-terminus of Spc105 [KNL1] with a mutation at the authentic RVxF motif) supports normal chromosome segregation, it does not cause a premature silencing of the SAC (Rosenberg et al. 2011). Therefore, PP1 recruitment to Spc105 is necessary but not sufficient to turn off the SAC in the absence of microtubule attachment.
Budding yeast is so far unique in that the critical phosphorylation site for Cdk1-dependent inhibition on PP1 is not conserved (Dohadwala et al. 1994; Yamano et al. 1994). In other organisms, inhibition of PP1 catalytic activity by Cdk1-cyclin B could generate additional feedback regulation. Cyclin B1 is localized to the unattached kinetochore, but dissociates from the kinetochore upon bioriented attachment (Bentley et al. 2007). Since its kinetochore localization depends on Mad2, dissociation of cyclin B1 from the kinetochore may be caused by Mad2 dissociation. Lowering Cdk1-cyclin B1 could promote dephosphorylation of kinetochore substrates, including PP1 itself. This would remove Cdk1-dependent PP1 inhibition and further stimulate dephosphorylation of kinetochore substrates.
Plk1-dependent recruitment of PP2A potentially generates negative feedback. As mentioned above, Plk1-dependent phosphorylation of kinetochore substrates promotes its own kinetochore localization as well as the antagonistic phosphatase PP2A-B56 (Suijkerbuijk et al. 2012b). PP2A-B56 in turn dephosphorylates Plk1 substrates at the kinetochore, promoting dissociation of Plk1 at the kinetochore (Foley et al. 2011). Since microtubule attachment defects upon PP2A-B56 depletion can be rescued by inhibition of Plk1, dissociation of Plk1 stabilizes kinetochore–microtubule attachment. In addition, this correlates with dissociation of PP2A-B56 from kinetochores with bioriented attachment (Foley et al. 2011). Although it is not clear whether dissociation of PP2A-B56 has a functional consequence, it may help keep a low level of phosphorylation that is required for the integrity of kinetochore architecture, dynamic association of microtubules, or prevention of merotelic attachments. Indeed, the level of Aurora B-dependent phosphorylation decreases but does not necessarily vanish upon bipolar attachment (DeLuca et al. 2011; Kops and Shah 2012; Welburn et al. 2010). Similarly, saturated phosphorylation of kinetochore substrates may have to be avoided to support initial kinetochore–microtubule attachment. The negative feedback could be utilized for adaptation or oscillation (Pomerening et al. 2003), but more quantitative measurements of kinetochore phosphorylation levels during mitotic progression are needed in the presence or absence of microtubule attachment to understand the functional meaning of this negative feedback.
In summary, feedback mechanisms control the localization of critical enzymes at the kinetochore and the centromere, but it remains to be established whether modulations of these feedback mechanisms upon microtubule attachment act as a critical trigger for kinetochore–microtubule stabilization or SAC silencing.
Changing the catalytic activity of enzymes
Bipolar microtubule attachment at the kinetochore may directly affect the catalytic activity of enzymes. However, few examples of this type of regulation have been demonstrated. It was reported that the kinase activity of BubR1 is silenced by CENP-E-mediated microtubule attachment (Mao et al. 2005), though the presence of BubR1 kinase activity has been questioned (Suijkerbuijk et al. 2012a). Aurora B activity is stimulated by microtubule binding (Kelly et al. 2007; Rosasco-Nitcher et al. 2008; Tseng et al. 2010), and this activation was implicated in correcting merotelic attachments, a configuration in which Aurora B at the inner centromere may have better access to microtubules (Knowlton et al. 2006). However, this mechanism does not explain the high Aurora B-dependent phosphorylation of kinetochore substrates in nocodazole-treated cells.
Since Aurora B can be activated by binding to chromatin (Kelly et al. 2007, 2010), a change in chromatin structure at the centromere upon microtubule attachment could potentially alter Aurora B activity. Although the underlining mechanism remains unclear, it has been indicated that Aurora B-dependent phosphorylation of CENP-A or histone H3 is greatly reduced upon depletion or inhibition of DNA topoisomerase II (Topo 2). Since Topo 2 is important for DNA decatenation, Aurora B activity may be sensitive to the topological status of the centromeric DNA. Centromeric localization of Topo 2 depends on SUMOylation (Dawlaty et al. 2008), but Topo 2 SUMOylation also inhibits Topo 2 activity (Ryu et al. 2010). It would be interesting to learn how Topo 2 SUMOylation is regulated by kinetochore–microtubule attachments.
Spatial separation between enzymes and substrates
Microtubule attachment may alter the accessibility of substrates to kinases and phosphatases. It has been proposed that the change in the physical distance between Aurora B located at the inner centromere and PP1 at the outer kinetochore alters the relative kinetochore substrate accessibility (Kelly and Funabiki 2009; Lampson and Cheeseman 2011; Maresca and Salmon 2010; Tanaka et al. 2002). In addition, PP2A-B56, which is localized to the kinetochore, contributes to kinetochore–microtubule attachment regulation (Foley et al. 2011). In this spatial separation model, Aurora B, which is activated at the inner centromeric chromatin, diffuses to act on substrates at the kinetochore. Biorientation increases the distance between the kinetochore and the inner centromere and, thus, reduces Aurora B’s access to kinetochore substrates while phosphatases retain access. Supporting this idea, the levels of Aurora B-dependent phosphorylation at sites in the inner centromere are less sensitive to microtubule attachment status than the levels of phosphorylation at sites in the outer kinetochore (Liu et al. 2009; Welburn et al. 2010). However, it is not clear whether the dynamic range of this “Aurora B gradient” is large enough to support switch-like behavior over the small spatial change in substrate positioning within the kinetochore in response to bipolar attachment (at most ~50 nm) (Maresca and Salmon 2009; Uchida et al. 2009; Wan et al. 2009; Welburn et al. 2010). Furthermore, kinetochore structure fluctuates very rapidly in response to microtubule-dependent forces (Uchida et al. 2009). Thus, information about the separation between the centromere and the outer kinetochore has to be filtered by the SAC silencing machinery by, e.g., time-averaging or scoring incidents that the distance becomes larger than a threshold. In principle, enzyme kinetics could accomplish this type of filtering, but we currently have insufficient quantitative enzymological information for the relevant kinases and phosphatases to support that model.
Changing the stability of enzyme–substrate interactions
Microtubule-dependent force may directly alter the structure of binding modules that support substrate–enzyme interactions. In this model, stabilization of substrate–enzyme interactions is the major determinant for critical phosphorylation/dephosphorylation. Such a mechanism has an advantage when the system has to deal with small numbers of molecules. Proteins responsible for microtubule attachment (e.g., Ndc80) on each kinetochore are found in low numbers (~20 per kinetochore microtubule) (Coffman et al. 2011; Lawrimore et al. 2011). When an enzyme targets a small number of substrates, the reaction follows stochastic rather than deterministic (mass action) rate equations. The stochastic nature of substrate–enzyme recognition can be transformed into a deterministic signal if the substrate and enzyme interaction is stabilized (or the local concentration of substrates and enzymes is high). Conversion to a stable interaction can also drive the phosphorylation/dephosphorylation of an intrinsically “poor” but functionally critical substrate sequence. A stable interaction can be supported by a specific enzyme-binding module (such as the Cy/RxL motif in substrates of Cdk1-cyclin (Koivomagi et al. 2011; Takeda et al. 2001)), or a scaffold that can bring the substrate and the enzyme together (such as Ste5 in the MAP kinase cascades (Good et al. 2011)). This interaction stability hypothesis may explain why some enzymes can execute proper functions without full enrichment at the kinetochore, as long as their substrate interaction regulation remains intact. To test this hypothesis, identification of substrate recognition modules of kinases and phosphatases is critical. The PBD of Plk1 is responsible for this function, but retention of Plk1 at metaphase kinetochores by inhibiting PP1 or PP2A indicates that microtubule attachment does not directly interfere with substrate recognition (Foley et al. 2011; Liu et al. 2012). It will be important to investigate whether Aurora B and Mps1 possess similar substrate recognition modules and, if so, whether those modules are altered by microtubule attachment.
Requirement of phosho-independent mechanisms for SAC silencing
Although PP1 at the kinetochore is required for SAC silencing, a switch-like change in phosphorylation status may not be necessary as long as critical substrates are dephosphorylated below the threshold level upon microtubule attachment. In this case, the switch-like response of SAC silencing must be ensured by a phospho-independent mechanism, such as microtubule-dependent stripping of SAC components. Interestingly, KNL1 in C. elegans has a microtubule-binding domain that is required for SAC silencing independent of its role in PP1 recruitment (Espeut et al. 2012). Since KNL1 also recruits Bub1–Bub3, which generates the MCC with Mad1 and Cdc20, microtubule binding of KNL1 may influence the formation of the MCC. Furthermore, it has been suggested that deacetylation of BubR1 is required for silencing the SAC by promoting its own ubiquitylation and degradation through the APC/C (Choi et al. 2009). While BubR1 acetylation does affect BubR1 phosphorylation, it remains to be tested whether acetylation/deacetylation is controlled by phosphorylation and/or microtubule attachment. Disassembly of MCC by p31comet and APC-dependent Cdc20 autoubiquitylation also contribute to SAC silencing (Foster and Morgan 2012; Jia et al. 2011; Mansfeld et al. 2011; Reddy et al. 2007; Stegmeier et al. 2007; Teichner et al. 2011; Uzunova et al. 2012; Westhorpe et al. 2011). These phospho-independent mechanisms may create a strong AND gate with phospho-dependent mechanisms to support a switch-like response of SAC activation and inactivation.
Outstanding questions
In spite of the extensive investigation into kinetochore composition and function, a complete picture of how microtubule attachment is monitored and relayed into cell cycle progression feedback is still emerging. Below, we highlight some of the critical questions to be addressed and major challenges that must be overcome to obtain a more complete picture of kinetochore regulation.
Regulating composition and architecture of the kinetochore
Kinetochore recruitment of proteins that play direct roles in kinetochore–microtubule attachment regulation and/or in SAC signaling is mostly limited to mitosis. Little is known about the molecular basis for this cell cycle-dependent recruitment of proteins to the kinetochore. Recruitment of the KMN network by Cdk1-dependent phosphorylation on CENP-T is one mechanism (Gascoigne et al. 2011), but it is likely that phosphorylation and dephosphorylation of many other proteins also regulate assembly and disassembly of the kinetochore during the cell cycle.
In Xenopus egg extracts, Aurora B activity is critical for assembly of a wide variety of outer kinetochore proteins important for kinetochore–microtubule attachment (the KMN network) and SAC signaling (Emanuele et al. 2008), but almost nothing is known about the underlying molecular basis. The importance of Aurora B for kinetochore assembly raises the question of how some of these proteins dissociate (e.g., Mad1–Mad2) while others (e.g., the KMN network, Bub1) remain recruited to metaphase and anaphase chromosomes upon silencing of the Aurora B pathway. It is likely that residual phosphorylations on attached kinetochores can maintain a subset of protein recruitment, but how is the quantity or quality of this subset of phosphorylation determined? Dissecting the critical phosphorylation sites that contribute to kinetochore recruitment will help answer this question. Particularly, identification of the kinetochore protein that directly recruits the Mad1–Mad2 complex and Mps1 is critical.
Phosphorylation of subsets of kinetochore proteins may be differentially regulated by interaction with specific substrate recognition modules on each kinase and phosphatase (and their potential regulatory subunits). Among the kinases discussed here, Plk1 has an established substrate recognition module, the PBD, but other kinases may have intricate mechanisms to support robust and accurate substrate recognition that is sensitive to microtubule attachment status. Similarly, it will be important to understand how the substrate recognition of PP1 and PP2A-B56 is regulated. Specifically, Sds22 has been implicated in providing the substrate specificity for PP1 (Stone et al. 1993). Characterizing the role of Sds22 in PP1 substrate specificity on kinetochore proteins will be helpful to address this question.
It is also possible that accessibility of enzymes to their substrates may be governed by the physical proximity that is changed by microtubule-dependent force. Recent advancements in super-resolution microscopic techniques and immuno-electron microscopy have started to help reveal the dynamic architectural changes in the kinetochore upon microtubule attachment (Dumont et al. 2012; Maresca and Salmon 2009; Suzuki et al. 2011; Uchida et al. 2009; Wan et al. 2009). Further characterization of the physical and biochemical bases for substrate recognition by kinases and phosphatases is critical to understand how microtubule attachment status can be converted into chemical signals.
Microtubule attachment and error correction
Although we now know a great deal of molecular players involved in kinetochore–microtubule attachment and in regulating the dynamics of kinetochore microtubules, it is still far from clear how these activities are coordinated to regulate different modes of attachment. As we discussed above, it remains to be established how microtubules can make initial attachments to a kinetochore that is predicted to have the weakest capacity to establish end-on attachment due to high Aurora B-dependent phosphorylation of kinetochore proteins such as Ndc80. Although lateral attachments mediated by dynein and CENP-E likely contribute to these processes, little is known about how the conversion from lateral attachment to end-on attachment is controlled. It will be important to examine whether lateral attachment causes differential effects on substrates of Aurora B (e.g., Ndc80) and Plk1 (e.g., BubR1). The microtubule motors CENP-E and kinesin-8 can both bind to PP1 (Kim et al. 2010; Meadows et al. 2011). It was suggested that dephosphorylation of CENP-E’s PP1-binding module is important for conversion from lateral attachment to end-on attachment since microinjection of phospho-specific antibodies recognizing this site disrupt stable bipolar attachments (Kim et al. 2010). Human kinesin-8, Kif18A, which has a conserved PP1-binding module, is recruited to the kinetochore in a microtubule-dependent manner to suppress dynamic kinetochore movements (Stumpff et al. 2008). Therefore, it would be interesting to test whether CENP-E and/or Kif18A support(s) stable bipolar microtubule attachment by recruiting PP1 and dephosphorylating Ndc80 upon microtubule attachment.
Mps1 is also important for kinetochore–microtubule attachment control, and recent work indicates that KNL1 is an important substrate to recruit Bub1, which contributes to the centromeric localization of Aurora B. Does Mps1 then regulate kinetochore–microtubule attachment through Aurora B localization? Perplexingly, this is not the case: Mps1 is not required for Aurora B localization at the centromere (Jelluma et al. 2008; Maciejowski et al. 2010; Maure et al. 2007; Vigneron et al. 2004). Although the presence of Aurora B in the absence of the Mps1–Bub1 pathway may be explained by the redundant function of the Haspin pathway, it does not explain why Mps1 inhibition leads to chromosome missegregation. It is likely that Mps1 has additional substrates, such as Borealin, CENP-E, and BubR1 (Espeut et al. 2008; Huang et al. 2008; Jelluma et al. 2008) through which it regulates kinetochore–microtubule attachment. More interestingly, the lack of clear impacts of Mps1 inhibition on Aurora B localization begs the question of how the strength/importance of Mps1’s regulation of Aurora B is determined within the complex feedback loop of the kinase network.
SAC regulation
Identification of KNL1 as a critical Mps1-dependent phosphorylation for Bub1/BubR1 recruitment and SAC activation was a landmark discovery (London et al. 2012; Shepperd et al. 2012; Yamagishi et al. 2012). However, this phosphorylation is insufficient to recruit Mad1–Mad2 and to activate the SAC, indicating that there are other phospho-dependent mechanisms regulating SAC activation. In addition to their roles in kinetochore recruitment of proteins important for SAC activation, Aurora B and Mps1 support the SAC downstream of Mad1–Mad2 recruitment at the kinetochore (Maldonado and Kapoor 2011). Establishing the Aurora B- and Mps1-dependent phosphorylation sites required for SAC activation and maintenance will be necessary to understand how dephosphorylation of these sites is coupled to kinetochore–microtubule attachment status.
Prolonged mitotic arrest caused by microtubule poisons in human cells leads to various fates, such as apoptosis during mitosis, and mitotic escape, which is sometimes followed by apoptosis in interphase (Gascoigne and Taylor 2008). These differences are linked to the level of sustained cyclin B during mitotic arrest. Although the cancer cells studied in that work contain “intact” SAC activity, it is possible that the quality of the SAC is compromised. This variation can be due to subtle difference in the amount of SAC regulators at the kinetochore, as seen in the case of Mps1 (Maciejowski et al. 2010; Salimian et al. 2011). Thus, identifying the proteins whose kinetochore levels determine sensitivity to the effects of microtubule poisons may help predict the effectiveness of these drugs during chemotherapy.
Quantifying phosphorylation levels on the kinetochore
We have just begun accumulating knowledge of the phosphorylation sites responsible for kinetochore–microtubule attachment and SAC regulation. Quantitative analysis of these phosphorylation events has been very limited, despite its necessity for understanding how the phospho-switch works at the kinetochore. The current difficulty in quantitative characterization of kinetochore phosphorylation is due to technical limitations. Most current studies rely on indirect immunofluorescence using phospho-specific antibodies, but absolute quantitation by this method is difficult. Specifically, since the dynamic range and linearity of immunofluorescence signals are usually not addressed, it has been hard to validate the significance of observed differences in published data. Recent FRET-based phospho-sensor techniques have provided more quantitative data (Liu et al. 2009), but the dynamic range of these tools is a limitation and the method relies on artificial marker substrates. Thus, although this method has been useful to detect the spatial distribution of phosphorylation and dephosphorylation reactions by diffuse enzymes, it may not reflect reactions mediated by specific interactions governed by complex substrate recognition modules of enzymes. These technical issues must be resolved to obtain more quantitatively useful data amenable to simulation analysis. Quantitative analysis of isolated kinetochores from budding yeast or in Xenopus egg extracts (Akiyoshi et al. 2010; Guse et al. 2011) may help tackle this problem.
There are many hurdles to overcome before answering the question of how kinetochore–microtubule attachment status can be sensed and transduced into signaling pathways that control dynamic microtubule attachments and the SAC. However, with the list of critical enzymes and substrate complexes likely to be near complete, a growing list of critical phosphorylation sites being characterized, and the combined power of innovative in vivo imaging and new biochemical approaches currently being developed in diverse systems, we have powerful tools to dissect this complex, medically important biological mechanism.
References
Abrieu A, Magnaghi-Jaulin L, Kahana JA, Peter M, Castro A, Vigneron S, Lorca T, Cleveland DW, Labbe JC (2001) Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell 106:83–93
Ahonen LJ, Kallio MJ, Daum JR, Bolton M, Manke IA, Yaffe MB, Stukenberg PT, Gorbsky GJ (2005) Polo-like kinase 1 creates the tension-sensing 3F3/2 phosphoepitope and modulates the association of spindle-checkpoint proteins at kinetochores. Curr Biol 15:1078–1089
Akiyoshi B, Nelson CR, Ranish JA, Biggins S (2009a) Analysis of Ipl1-mediated phosphorylation of the Ndc80 kinetochore protein in Saccharomyces cerevisiae. Genetics 183:1591–1595
Akiyoshi B, Nelson CR, Ranish JA, Biggins S (2009b) Quantitative proteomic analysis of purified yeast kinetochores identifies a PP1 regulatory subunit. Genes Dev 23:2887–2899
Akiyoshi B, Sarangapani KK, Powers AF, Nelson CR, Reichow SL, Arellano-Santoyo H, Gonen T, Ranish JA, Asbury CL, Biggins S (2010) Tension directly stabilizes reconstituted kinetochore–microtubule attachments. Nature 468:576–579
Alexander J, Lim D, Joughin BA, Hegemann B, Hutchins JR, Ehrenberger T, Ivins F, Sessa F, Hudecz O, Nigg EA, Fry AM, Musacchio A, Stukenberg PT, Mechtler K, Peters JM, Smerdon SJ, Yaffe MB (2011) Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci Signal 4:ra42
Alushin GM, Ramey VH, Pasqualato S, Ball DA, Grigorieff N, Musacchio A, Nogales E (2010) The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 467:805–810
Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR (2004) Aurora B regulates MCAK at the mitotic centromere. Dev Cell 6:253–268
Bentley AM, Normand G, Hoyt J, King RW (2007) Distinct sequence elements of cyclin B1 promote localization to chromatin, centrosomes, and kinetochores during mitosis. Mol Biol Cell 18:4847–4858
Bernard P, Maure JF, Javerzat JP (2001) Fission yeast Bub1 is essential in setting up the meiotic pattern of chromosome segregation. Nat Cell Biol 3:522–526
Biggins S, Murray AW (2001) The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev 15:3118–3129
Booher R, Beach D (1989) Involvement of a type 1 protein phosphatase encoded by bws1 + in fission yeast mitotic control. Cell 57:1009–1016
Boyarchuk Y, Salic A, Dasso M, Arnaoutov A (2007) Bub1 is essential for assembly of the functional inner centromere. J Cell Biol 176:919–928
Burton JL, Solomon MJ (2007) Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes Dev 21:655–667
Buttrick GJ, Lancaster TC, Meadows JC, Millar JB (2012) Plo1 phosphorylates Dam1 to promote chromosome bi-orientation in fission yeast. J Cell Sci 125:1645–1651
Cai S, O'Connell CB, Khodjakov A, Walczak CE (2009) Chromosome congression in the absence of kinetochore fibres. Nat Cell Biol 11:832–838
Carmena M, Pinson X, Platani M, Salloum Z, Xu Z, Clark A, Macisaac F, Ogawa H, Eggert U, Glover DM, Archambault V, Earnshaw WC (2012a) The chromosomal passenger complex activates Polo kinase at centromeres. PLoS Biol 10:e1001250
Carmena M, Wheelock M, Funabiki H, Earnshaw WC (2012b) The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 13(12):789–803
Ceulemans H, Vulsteke V, De Maeyer M, Tatchell K, Stalmans W, Bollen M (2002) Binding of the concave surface of the Sds22 superhelix to the alpha 4/alpha 5/alpha 6-triangle of protein phosphatase-1. J Biol Chem 277:47331–47337
Chan GK, Schaar BT, Yen TJ (1998) Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J Cell Biol 143:49–63
Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ (1999) Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol 146:941–954
Chan YW, Jeyaprakash AA, Nigg EA, Santamaria A (2012) Aurora B controls kinetochore–microtubule attachments by inhibiting Ska complex–KMN network interaction. J Cell Biol 196:563–571
Chao WC, Kulkarni K, Zhang Z, Kong EH, Barford D (2012) Structure of the mitotic checkpoint complex. Nature 484:208–213
Cheeseman IM, Desai A (2008) Molecular architecture of the kinetochore–microtubule interface. Nat Rev Mol Cell Biol 9:33–46
Cheeseman IM, Anderson S, Jwa M, Green EM, Js K, Yates JR, Chan CSM, Drubin DG, Barnes G (2002) Phospho-regulation of kinetochore–microtubule attachments by the Aurora kinase Ipl1p. Cell 111:163–172
Cheeseman IM, Niessen S, Anderson S, Hyndman F, Yates JR 3rd, Oegema K, Desai A (2004) A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev 18:2255–2268
Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A (2006) The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127:983–997
Chen RH (2002) BubR1 is essential for kinetochore localization of other spindle checkpoint proteins and its phosphorylation requires Mad1. J Cell Biol 158:487–496
Chen RH (2004) Phosphorylation and activation of Bub1 on unattached chromosomes facilitate the spindle checkpoint. EMBO J 23:3113–3121
Chen R-H, Waters JC, Salmon ED, Murray AW (1996) Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 274:242–246
Chen R-H, Shevchenko A, Mann M, Murray AW (1998) Metaphase arrest induced by an excess of the spindle checkpoint protein Xmad2 is independent of Xmad1. J Cell Biol 143:283–295
Cheng L, Zhang J, Ahmad S, Rozier L, Yu H, Deng H, Mao Y (2011) Aurora B regulates formin mDia3 in achieving metaphase chromosome alignment. Dev Cell 20:342–352
Choi E, Choe H, Min J, Choi JY, Kim J, Lee H (2009) BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J 28:2077–2089
Chung E, Chen RH (2003) Phosphorylation of Cdc20 is required for its inhibition by the spindle checkpoint. Nat Cell Biol 5:748–753
Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, De Luca JG, De Wulf P, Salek M, Rappsilber J, Moores CA, Salmon ED, Musacchio A (2008) Implications for kinetochore–microtubule attachment from the structure of an engineered Ndc80 complex. Cell 133:427–439
Cimini D, Wan X, Hirel CB, Salmon ED (2006) Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol 16:1711–1718
Clute P, Pines J (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol 1:82–87
Coffman VC, Wu P, Parthun MR, Wu JQ (2011) CENP-A exceeds microtubule attachment sites in centromere clusters of both budding and fission yeast. J Cell Biol 195:563–572
D'Angiolella V, Mari C, Nocera D, Rametti L, Grieco D (2003) The spindle checkpoint requires cyclin-dependent kinase activity. Genes Dev 17:2520–2525
Dawlaty MM, Malureanu L, Jeganathan KB, Kao E, Sustmann C, Tahk S, Shuai K, Grosschedl R, van Deursen JM (2008) Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 133:103–115
De Antoni A, Maffini S, Knapp S, Musacchio A, Santaguida S (2012) A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J Cell Biol 199:269–284
De Wulf P, McAinsh AD, Sorger PK (2003) Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev 17:2902–2921
DeLuca JG, Howell BJ, Canman JC, Hickey JM, Fang G, Salmon ED (2003) Nuf2 and Hec1 are required for retention of the checkpoint proteins Mad1 and Mad2 to kinetochores. Curr Biol 13:2103–2109
DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, Salmon ED (2006) Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127:969–982
DeLuca KF, Lens SM, DeLuca JG (2011) Temporal changes in Hec1 phosphorylation control kinetochore–microtubule attachment stability during mitosis. J Cell Sci 124:622–634
Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS (2003) Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol 161:267–280
Dohadwala M, Da Cruz e Silva EF, Hall FL, Williams RT, Carbonaro-Hall DA, Nairn AC, Greengard P, Berndt N (1994) Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc Natl Acad Sci U S A 91:6408–6412
Doonan JH, Morris NR (1989) The bimG gene of Aspergillus nidulans, required for completion of anaphase, encodes a homolog of mammalian phophoprotein phosphatase 1. Cell 57:987–996
Dumont S, Salmon ED, Mitchison TJ (2012) Deformations within moving kinetochores reveal different sites of active and passive force generation. Science 337:355–358
Dunsch AK, Linnane E, Barr FA, Gruneberg U (2011) The astrin–kinastrin/SKAP complex localizes to microtubule plus ends and facilitates chromosome alignment. J Cell Biol 192:959–968
Elia AE, Cantley LC, Yaffe MB (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299:1228–1231
Elowe S, Hummer S, Uldschmid A, Li X, Nigg EA (2007) Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev 21:2205–2219
Emanuele MJ, Lan W, Jwa M, Miller SA, Chan CSM, Stukenberg PT (2008) Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. J Cell Biol 181:241–254
Espeut J, Gaussen A, Bieling P, Morin V, Prieto S, Fesquet D, Surrey T, Abrieu A (2008) Phosphorylation relieves autoinhibition of the kinetochore motor Cenp-E. Mol Cell 29:637–643
Espeut J, Cheerambathur DK, Krenning L, Oegema K, Desai A (2012) Microtubule binding by KNL-1 contributes to spindle checkpoint silencing at the kinetochore. J Cell Biol 196:469–482
Fernius J, Hardwick KG (2007) Bub1 kinase targets Sgo1 to ensure efficient chromosome biorientation in budding yeast mitosis. PLoS Genet 3:e213
Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O'Donnell M, Gelfand VI, Kapoor TM, Chen JK (2012) Small-molecule inhibitors of the AAA + ATPase motor cytoplasmic dynein. Nature 484:125–129
Fisk HA, Winey M (2001) The mouse Mps1p-like kinase regulates centrosome duplication. Cell 106:95–104
Foley EA, Kapoor TM (2012) Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol 14:25–37
Foley EA, Maldonado M, Kapoor TM (2011) Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol 13:1265–1271
Foster SA, Morgan DO (2012) The APC/C subunit Mnd2/Apc15 promotes Cdc20 autoubiquitination and spindle assembly checkpoint inactivation. Mol Cell 47:921–932
Francisco L, Wang W, Chan CS (1994) Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol 14:4731–4740
Gascoigne KE, Taylor SS (2008) Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14:111–122
Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, Cheeseman IM (2011) Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell 145:410–422
Gassmann R, Holland AJ, Varma D, Wan X, Civril F, Cleveland DW, Oegema K, Salmon ED, Desai A (2010) Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev 24:957–971
Gestaut DR, Graczyk B, Cooper J, Widlund PO, Zelter A, Wordeman L, Asbury CL, Davis TN (2008) Phosphoregulation and depolymerization-driven movement of the Dam1 complex do not require ring formation. Nat Cell Biol 10:407–414
Good MC, Zalatan JG, Lim WA (2011) Scaffold proteins: hubs for controlling the flow of cellular information. Science 332:680–686
Gorbsky GJ, Ricketts WA (1993) Differential expression of a phosphoepitope at the kinetochores of moving chromosomes. J Cell Biol 122:1311–1321
Goto H, Kiyono T, Tomono Y, Kawajiri A, Urano T, Furukawa K, Nigg EA, Inagaki M (2006) Complex formation of Plk1 and INCENP required for metaphase–anaphase transition. Nat Cell Biol 8:180–187
Guimaraes GJ, Dong Y, McEwen BF, DeLuca JG (2008) Kinetochore–microtubule attachment relies on the disordered N-terminal tail domain of Hec1. Curr Biol 18:1778–1784
Guo Y, Kim C, Ahmad S, Zhang J, Mao Y (2012) CENP-E-dependent BubR1 autophosphorylation enhances chromosome alignment and the mitotic checkpoint. J Cell Biol 198:205–217
Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF (2011) In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477:354–358
Hached K, Xie SZ, Buffin E, Cladiere D, Rachez C, Sacras M, Sorger PK, Wassmann K (2011) Mps1 at kinetochores is essential for female mouse meiosis I. Development 138:2261–2271
Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM (2003) The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 161:281–294
Hendrickx A, Beullens M, Ceulemans H, Den Abt T, Van Eynde A, Nicolaescu E, Lesage B, Bollen M (2009) Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem Biol 16:365–371
Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S, Taylor SS (2010) Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1–C-Mad2 core complex. J Cell Biol 190:25–34
Hood EA, Kettenbach AN, Gerber SA, Compton DA (2012) Plk1 regulates the kinesin-13 protein Kif2b to promote faithful chromosome segregation. Mol Biol Cell 23:2264–2274
Hori T, Okada M, Maenaka K, Fukagawa T (2008) CENP-O class proteins form a stable complex and are required for proper kinetochore function. Mol Biol Cell 19:843–854
Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED (2001) Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol 155:1159–1172
Howell BJ, Moree B, Farrar EM, Stewart S, Fang G, Salmon ED (2004) Spindle checkpoint protein dynamics at kinetochores in living cells. Curr Biol 14:953–964
Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, Lin R, Smith MM, Allis CD (2000) Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 102:279–291
Hua S, Wang Z, Jiang K, Huang Y, Ward T, Zhao L, Dou Z, Yao X (2011) CENP-U cooperates with Hec1 to orchestrate kinetochore–microtubule attachment. J Biol Chem 286:1627–1638
Huang H, Hittle J, Zappacosta F, Annan RS, Hershko A, Yen TJ (2008) Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J Cell Biol 183:667–680
Hummer S, Mayer TU (2009) Cdk1 negatively regulates midzone localization of the mitotic kinesin Mklp2 and the chromosomal passenger complex. Curr Biol 19:607–612
Indjeian VB, Murray AW (2007) Budding yeast mitotic chromosomes have an intrinsic bias to biorient on the spindle. Curr Biol 17:1837–1846
Ito D, Saito Y, Matsumoto T (2011) Centromere-tethered Mps1 pombe homolog (Mph1) kinase is a sufficient marker for recruitment of the spindle checkpoint protein Bub1, but not Mad1. Proc Natl Acad Sci U S A 109:209–214
Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, Medema RH, Kops GJ (2008) Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 132:233–246
Jeyaprakash AA, Santamaria A, Jayachandran U, Chan YW, Benda C, Nigg EA, Conti E (2012) Structural and functional organization of the Ska complex, a key component of the kinetochore–microtubule interface. Mol Cell 46:274–286
Jia L, Li B, Warrington RT, Hao X, Wang S, Yu H (2011) Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31(comet). Mol Biol Cell 22:4227–4235
Johnson VL, Scott MI, Holt SV, Hussein D, Taylor SS (2004) Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J Cell Sci 117:1577–1589
Kallio MJ, McCleland ML, Stukenberg PT, Gorbsky GJ (2002) Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr Biol 12:900–905
Kang YH, Park JE, Yu LR, Soung NK, Yun SM, Bang JK, Seong YS, Yu H, Garfield S, Veenstra TD, Lee KS (2006) Self-regulated Plk1 recruitment to kinetochores by the Plk1–PBIP1 interaction is critical for proper chromosome segregation. Mol Cell 24:409–422
Kapoor TM, Lampson MA, Hergert P, Cameron L, Cimini D, Salmon ED, McEwen BF, Khodjakov A (2006) Chromosomes can congress to the metaphase plate before biorientation. Science 311:388–391
Kasuboski JM, Bader JR, Vaughan PS, Tauhata SB, Winding M, Morrissey MA, Joyce MV, Boggess W, Vos L, Chan GK, Hinchcliffe EH, Vaughan KT (2011) Zwint-1 is a novel Aurora B substrate required for the assembly of a dynein-binding platform on kinetochores. Mol Biol Cell 22:3318–3330
Kawashima SA, Yamagishi Y, Honda T, Ishiguro K, Watanabe Y (2010) Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 327:172–177
Kelly AE, Funabiki H (2009) Correcting aberrant kinetochore microtubule attachments: an Aurora B-centric view. Curr Opin Cell Biol 21:51–58
Kelly AE, Sampath SC, Maniar TA, Woo EM, Chait BT, Funabiki H (2007) Chromosomal enrichment and activation of the aurora B pathway are coupled to spatially regulate spindle assembly. Dev Cell 12:31–43
Kelly AE, Ghenoiu C, Xue JZ, Zierhut C, Kimura H, Funabiki H (2010) Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 330:235–239
Kemmler S, Stach M, Knapp M, Ortiz J, Pfannstiel J, Ruppert T, Lechner J (2009) Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. EMBO J 28:1099–1110
Kim Y, Holland AJ, Lan W, Cleveland DW (2010) Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 142:444–455
King EM, Rachidi N, Morrice N, Hardwick KG, Stark MJ (2007a) Ipl1p-dependent phosphorylation of Mad3p is required for the spindle checkpoint response to lack of tension at kinetochores. Genes Dev 21:1163–1168
King EM, van der Sar SJ, Hardwick KG (2007b) Mad3 KEN boxes mediate both Cdc20 and Mad3 turnover, and are critical for the spindle checkpoint. PLoS One 2:e342
Kitajima TS, Sakuno T, Ishiguro K, Iemura S, Natsume T, Kawashima SA, Watanabe Y (2006) Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature 441:46–52
Kitajima TS, Ohsugi M, Ellenberg J (2011) Complete kinetochore tracking reveals error-prone homologous chromosome biorientation in mammalian oocytes. Cell 146:568–581
Kiyomitsu T, Obuse C, Yanagida M (2007) Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev Cell 13:663–676
Kiyomitsu T, Murakami H, Yanagida M (2011) Protein interaction domain mapping of human kinetochore protein Blinkin reveals a consensus motif for binding of spindle assembly checkpoint proteins Bub1 and BubR1. Mol Cell Biol 31:998–1011
Klebig C, Korinth D, Meraldi P (2009) Bub1 regulates chromosome segregation in a kinetochore-independent manner. J Cell Biol 185:841–858
Kline SL, Cheeseman IM, Hori T, Fukagawa T, Desai A (2006) The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J Cell Biol 173:9–17
Knowlton AL, Lan W, Stukenberg PT (2006) Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr Biol 16:1705–1710
Knowlton AL, Vorozhko VV, Lan W, Gorbsky GJ, Stukenberg PT (2009) ICIS and Aurora B coregulate the microtubule depolymerase Kif2a. Curr Biol 19:758–763
Koivomagi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ER, Rubin SM, Morgan DO, Loog M (2011) Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 480:128–131
Kops GJ, Shah JV (2012) Connecting up and clearing out: how kinetochore attachment silences the spindle assembly checkpoint. Chromosoma 121:509–525
Kulukian A, Han JS, Cleveland DW (2009) Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell 16:105–117
Labit H, Fujimitsu K, Bayin NS, Takaki T, Gannon J, Yamano H (2012) Dephosphorylation of Cdc20 is required for its C-box-dependent activation of the APC/C. EMBO J 31:3351–3362
Lampert F, Hornung P, Westermann S (2010) The Dam1 complex confers microtubule plus end-tracking activity to the Ndc80 kinetochore complex. J Cell Biol 189:641–649
Lampson MA, Cheeseman IM (2011) Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol 21(3):133–140
Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM (2004) Correcting improper chromosome-spindle attachments during cell division. Nat Cell Biol 6:232–237
Lan W, Zhang X, Kline-Smith SL, Rosasco SE, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Walczak CE, Stukenberg PT (2004) Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol 14:273–286
Lara-Gonzalez P, Scott MI, Diez M, Sen O, Taylor SS (2011) BubR1 blocks substrate recruitment to the APC/C in a KEN-box-dependent manner. J Cell Sci 124:4332–4345
Lawrimore J, Bloom KS, Salmon ED (2011) Point centromeres contain more than a single centromere-specific Cse4 (CENP-A) nucleosome. J Cell Biol 195:573–582
Lee S, Thebault P, Freschi L, Beaufils S, Blundell TL, Landry CR, Bolanos-Garcia VM, Elowe S (2012) Characterization of spindle checkpoint kinase Mps1 reveals domain with functional and structural similarities to tetratricopeptide repeat motifs of Bub1 and BubR1 checkpoint kinases. J Biol Chem 287:5988–6001
Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM (2007) The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 17:304–315
Lens SM, Rodriguez JA, Vader G, Span SW, Giaccone G, Medema RH (2006) Uncoupling the central spindle-associated function of the chromosomal passenger complex from its role at centromeres. Mol Biol Cell 17:1897–1909
Li Y, Benezra R (1996) Identification of a human mitotic checkpoint gene: hsMAD2. Science 274:246–248
Li H, Liu XS, Yang X, Wang Y, Wang Y, Turner JR, Liu X (2010) Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore–microtubule attachments. EMBO J 29:2953–2965
Lin YT, Chen Y, Wu G, Lee WH (2006) Hec1 sequentially recruits Zwint-1 and ZW10 to kinetochores for faithful chromosome segregation and spindle checkpoint control. Oncogene 25:6901–6914
Liu ST, Chan GK, Hittle JC, Fujii G, Lees E, Yen TJ (2003) Human MPS1 kinase is required for mitotic arrest induced by the loss of CENP-E from kinetochores. Mol Biol Cell 14:1638–1651
Liu D, Vader G, Vromans MJM, Lampson MA, Lens SMA (2009) Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science 323:1350–1353
Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, Lampson MA (2010) Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J Cell Biol 188:809–820
Liu D, Davydenko O, Lampson MA (2012) Polo-like kinase-1 regulates kinetochore–microtubule dynamics and spindle checkpoint silencing. J Cell Biol 198:491–499
Logarinho E, Resende T, Torres C, Bousbaa H (2008) The human spindle assembly checkpoint protein bub3 is required for the establishment of efficient kinetochore–microtubule attachments. Mol Biol Cell 19:1798–1813
London N, Ceto S, Ranish JA, Biggins S (2012) Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr Biol 22:900–906
Luo X, Yu H (2008) Protein metamorphosis: the two-state behavior of Mad2. Structure 16:1616–1625
Maciejowski J, George KA, Terret ME, Zhang C, Shokat KM, Jallepalli PV (2010) Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J Cell Biol 190:89–100
Magidson V, O'Connell CB, Loncarek J, Paul R, Mogilner A, Khodjakov A (2011) The spatial arrangement of chromosomes during prometaphase facilitates spindle assembly. Cell 146:555–567
Maldonado M, Kapoor TM (2011) Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat Cell Biol 13:475–482
Malureanu LA, Jeganathan KB, Hamada M, Wasilewski L, Davenport J, van Deursen JM (2009) BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev Cell 16:118–131
Manning AL, Bakhoum SF, Maffini S, Correia-Melo C, Maiato H, Compton DA (2010) CLASP1, astrin and Kif2b form a molecular switch that regulates kinetochore–microtubule dynamics to promote mitotic progression and fidelity. EMBO J 29:3531–3543
Mansfeld J, Collin P, Collins MO, Choudhary JS, Pines J (2011) APC15 drives the turnover of MCC-CDC20 to make the spindle assembly checkpoint responsive to kinetochore attachment. Nat Cell Biol 13:1234–1243
Mao Y, Abrieu A, Cleveland DW (2003) Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell 114:87–98
Mao Y, Desai A, Cleveland DW (2005) Microtubule capture by CENP-E silences BubR1-dependent mitotic checkpoint signaling. J Cell Biol 170:873–880
Maresca TJ, Salmon ED (2009) Intrakinetochore stretch is associated with changes in kinetochore phosphorylation and spindle assembly checkpoint activity. J Cell Biol 184:373–381
Maresca TJ, Salmon ED (2010) Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. J Cell Sci 123:825–835
Martinez-Exposito MJ, Kaplan KB, Copeland J, Sorger PK (1999) Retention of the BUB3 checkpoint protein on lagging chromosomes. Proc Natl Acad Sci U S A 96:8493–8498
Martin-Lluesma S, Stucke VM, Nigg EA (2002) Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science 297:2267–2270
Matson DR, Demirel PB, Stukenberg PT, Burke DJ (2012) A conserved role for COMA/CENP-H/I/N kinetochore proteins in the spindle checkpoint. Genes Dev 26:542–547
Matsumura S, Toyoshima F, Nishida E (2007) Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J Biol Chem 282:15217–15227
Maure JF, Kitamura E, Tanaka TU (2007) Mps1 kinase promotes sister-kinetochore bi-orientation by a tension-dependent mechanism. Curr Biol 17:2175–2182
Mayer C, Filopei J, Batac J, Alford L, Paluh JL (2006) An extended anaphase signaling pathway for Mad2p includes microtubule organizing center proteins and multiple motor-dependent transitions. Cell Cycle 5:1456–1463
Meadows JC, Shepperd LA, Vanoosthuyse V, Lancaster TC, Sochaj AM, Buttrick GJ, Hardwick KG, Millar JB (2011) Spindle checkpoint silencing requires association of PP1 to both Spc7 and kinesin-8 motors. Dev Cell 20:739–750
Meraldi P, Sorger PK (2005) A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J 24:1621–1633
Meraldi P, Draviam VM, Sorger PK (2004) Timing and checkpoints in the regulation of mitotic progression. Dev Cell 7:45–60
Miller SA, Johnson ML, Stukenberg PT (2008) Kinetochore attachments require an interaction between unstructured tails on microtubules and Ndc80(Hec1). Curr Biol 18:1785–1791
Minoshima Y, Hori T, Okada M, Kimura H, Haraguchi T, Hiraoka Y, Bao YC, Kawashima T, Kitamura T, Fukagawa T (2005) The constitutive centromere component CENP-50 is required for recovery from spindle damage. Mol Cell Biol 25:10315–10328
Minshull J, Sun H, Tonks NK, Murray AW (1994) MAP-kinase dependent mitotic feedback arrest in Xenopus egg extracts. Cell 79:475–486
Morrow CJ, Tighe A, Johnson VL, Scott MI, Ditchfield C, Taylor SS (2005) Bub1 and aurora B cooperate to maintain BubR1-mediated inhibition of APC/CCdc20. J Cell Sci 118:3639–3652
Musacchio A, Salmon ED (2007) The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8:379–393
Nagaoka SI, Hassold TJ, Hunt PA (2012) Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 13:493–504
Nicklas RB, Ward SC, Gorbsky GJ (1995) Kinetochore chemistry is sensitive to tension and may link mitotic forces to a cell cycle checkpoint. J Cell Biol 130:929–939
Nishino M, Kurasawa Y, Evans R, Lin SH, Brinkley BR, Yu-Lee LY (2006) NudC is required for Plk1 targeting to the kinetochore and chromosome congression. Curr Biol 16:1414–1421
Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M (2004) A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol 6:1135–1141
Ohi R, Sapra T, Howard J, Mitchison TJ (2004) Differentiation of cytoplasmic and meiotic spindle assembly MCAK functions by Aurora B-dependent phosphorylation. Mol Biol Cell 15:2895–2906
Ohkura H, Yanagida M (1991) S. pombe gene sds22+ essential for a midmitotic transition encodes a leucine-rich repeat protein that positively modulates protein phosphatase-1. Cell 64:149–157
Ohkura H, Noriyuki K, Miyatani S, Toda T, Yanagida M (1989) The fission yeast dis2 + gene required for chromosome disjoining encodes one of two putative type 1 protein phosphatases. Cell 57:997–1007
Park JE, Erikson RL, Lee KS (2011) Feed-forward mechanism of converting biochemical cooperativity to mitotic processes at the kinetochore plate. Proc Natl Acad Sci U S A 108:8200–8205
Parry DH, Hickson GR, O'Farrell PH (2003) Cyclin B destruction triggers changes in kinetochore behavior essential for successful anaphase. Curr Biol 13:647–653
Perera D, Taylor SS (2010) Sgo1 establishes the centromeric cohesion protection mechanism in G2 before subsequent Bub1-dependent recruitment in mitosis. J Cell Sci 123:653–659
Perpelescu M, Fukagawa T (2011) The ABCs of CENPs. Chromosoma 120:425–446
Peters JM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7:644–656
Petersen J, Hagan IM (2003) S. pombe aurora kinase/survivin is required for chromosome condensation and the spindle checkpoint attachment response. Curr Biol 13:590–597
Petrovic A, Pasqualato S, Dube P, Krenn V, Santaguida S, Cittaro D, Monzani S, Massimiliano L, Keller J, Tarricone A, Maiolica A, Stark H, Musacchio A (2010) The MIS12 complex is a protein interaction hub for outer kinetochore assembly. J Cell Biol 190:835–852
Pinsky BA, Kotwaliwale CV, Tatsutani SY, Breed CA, Biggins S (2006a) Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol Cell Biol 26:2648–2660
Pinsky BA, Kung C, Shokat KM, Biggins S (2006b) The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol 8:78–83
Pinsky BA, Nelson CR, Biggins S (2009) Protein phosphatase 1 regulates exit from the spindle checkpoint in budding yeast. Curr Biol 19:1182–1187
Pomerening JR, Sontag ED, Ferrell JE Jr (2003) Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat Cell Biol 5:346–351
Posch M, Khoudoli GA, Swift S, King EM, DeLuca JG, Swedlow JR (2010) Sds22 regulates aurora B activity and microtubule–kinetochore interactions at mitosis. J Cell Biol 191:61–74
Przewloka MR, Glover DM (2009) The kinetochore and the centromere: a working long distance relationship. Annu Rev Genet 43:439–465
Qi W, Tang Z, Yu H (2006) Phosphorylation- and polo-box-dependent binding of Plk1 to Bub1 is required for the kinetochore localization of Plk1. Mol Biol Cell 17:3705–3716
Reddy SK, Rape M, Margansky WA, Kirschner MW (2007) Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature 446:921–925
Ricke RM, Jeganathan KB, Malureanu L, Harrison AM, van Deursen JM (2012) Bub1 kinase activity drives error correction and mitotic checkpoint control but not tumor suppression. J Cell Biol 199:931–949
Riedel CG, Katis VL, Katou Y, Mori S, Itoh T, Helmhart W, Galova M, Petronczki M, Gregan J, Cetin B, Mudrak I, Ogris E, Mechtler K, Pelletier L, Buchholz F, Shirahige K, Nasmyth K (2006) Protein phosphatase 2A protects centromeric sister chromatid cohesion during meiosis I. Nature 441:53–61
Rieder CL, Alexander SP (1990) Kinetochores are transported poleward along a single astral microtubule during chromosome attachment to the spindle in newt lung cells. J Cell Biol 110(1):81–95
Rieder CL, Schultz A, Cole R, Sluder G (1994) Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol 127:1301–1310
Rieder CL, Cole RW, Khodjakov A, Sluder G (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol 130:941–948
Rischitor PE, May KM, Hardwick KG (2007) Bub1 is a fission yeast kinetochore scaffold protein, and is sufficient to recruit other spindle checkpoint proteins to ectopic sites on chromosomes. PLoS One 2:e1342
Rosasco-Nitcher SE, Lan W, Khorasanizadeh S, Stukenberg PT (2008) Centromeric Aurora-B activation requires TD-60, microtubules, and substrate priming phosphorylation. Science 319:469–472
Rosenberg JS, Cross FR, Funabiki H (2011) KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr Biol 21:942–947
Ryu H, Furuta M, Kirkpatrick D, Gygi SP, Azuma Y (2010) PIASy-dependent SUMOylation regulates DNA topoisomerase IIalpha activity. J Cell Biol 191:783–794
Sakuno T, Tada K, Watanabe Y (2009) Kinetochore geometry defined by cohesion within the centromere. Nature 458:852–858
Salimian KJ, Ballister ER, Smoak EM, Wood S, Panchenko T, Lampson MA, Black BE (2011) Feedback control in sensing chromosome biorientation by the Aurora B kinase. Curr Biol 21:1158–1165
Santaguida S, Vernieri C, Villa F, Ciliberto A, Musacchio A (2011) Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J 30:1508–1519
Saurin AT, van der Waal MS, Medema RH, Lens SM, Kops GJ (2011) Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat Commun 2:316
Schaar BT, Chan GK, Maddox P, Salmon ED, Yen TJ (1997) CENP-E function at kinetochores is essential for chromosome alignment. J Cell Biol 139:1373–1382
Schmidt JC, Kiyomitsu T, Hori T, Backer CB, Fukagawa T, Cheeseman IM (2010) Aurora B kinase controls the targeting of the Astrin–SKAP complex to bioriented kinetochores. J Cell Biol 191:269–280
Schmidt JC, Arthanari H, Boeszoermenyi A, Dashkevich NM, Wilson-Kubalek EM, Monnier N, Markus M, Oberer M, Milligan RA, Bathe M, Wagner G, Grishchuk EL, Cheeseman IM (2012) The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev Cell 23(5):968–980
Sczaniecka M, Feoktistova A, May KM, Chen JS, Blyth J, Gould KL, Hardwick KG (2008) The spindle checkpoint functions of Mad3 and Mad2 depend on a Mad3 KEN box-mediated interaction with Cdc20-anaphase-promoting complex (APC/C). J Biol Chem 283:23039–23047
Shah JV, Botvinick E, Bonday Z, Furnari F, Berns M, Cleveland DW (2004) Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol 14:942–952
Shapiro PS, Vaisberg E, Hunt AJ, Tolwinski NS, Whalen AM, McIntosh JR, Ahn NG (1998) Activation of the MKK/ERK pathway during somatic cell mitosis: direct interactions of active ERK with kinetochores and regulation of the mitotic 3F3/2 phosphoantigen. J Cell Biol 142:1533–1545
Sharp-Baker H, Chen RH (2001) Spindle checkpoint protein Bub1 is required for kinetochore localization of Mad1, Mad2, Bub3, and CENP-E, independently of its kinase activity. J Cell Biol 153:1239–1250
Shepperd LA, Meadows JC, Sochaj AM, Lancaster TC, Zou J, Buttrick GJ, Rappsilber J, Hardwick KG, Millar JB (2012) Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr Biol 22:891–899
Sliedrecht T, Zhang C, Shokat KM, Kops GJ (2010) Chemical genetic inhibition of Mps1 in stable human cell lines reveals novel aspects of Mps1 function in mitosis. PLoS One 5:e10251
Stegmeier F, Rape M, Draviam VM, Nalepa G, Sowa ME, Ang XL, McDonald ER 3rd, Li MZ, Hannon GJ, Sorger PK, Kirschner MW, Harper JW, Elledge SJ (2007) Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature 446:876–881
Stone EM, Yamano H, Kinoshita N, Yanagida M (1993) Mitotic regulation of protein phosphatases by the fission yeast sds22 protein. Curr Biol 3:13–26
Stucke VM, Sillje HH, Arnaud L, Nigg EA (2002) Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J 21:1723–1732
Stucke VM, Baumann C, Nigg EA (2004) Kinetochore localization and microtubule interaction of the human spindle checkpoint kinase Mps1. Chromosoma 113:1–15
Stumpff J, von Dassow G, Wagenbach M, Asbury C, Wordeman L (2008) The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev Cell 14:252–262
Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154:925–936
Suijkerbuijk SJ, van Dam TJ, Karagoz GE, von Castelmur E, Hubner NC, Duarte AM, Vleugel M, Perrakis A, Rudiger SG, Snel B, Kops GJ (2012a) The vertebrate mitotic checkpoint protein BUBR1 is an unusual pseudokinase. Dev Cell 22:1321–1329
Suijkerbuijk SJ, Vleugel M, Teixeira A, Kops GJ (2012b) Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore–microtubule attachments. Dev Cell 23:745–755
Suzuki A, Hori T, Nishino T, Usukura J, Miyagi A, Morikawa K, Fukagawa T (2011) Spindle microtubules generate tension-dependent changes in the distribution of inner kinetochore proteins. J Cell Biol 193:125–140
Takeda DY, Wohlschlegel JA, Dutta A (2001) A bipartite substrate recognition motif for cyclin-dependent kinases. J Biol Chem 276:1993–1997
Takenaka K, Gotoh Y, Nishida E (1997) MAP kinase is required for the spindle assembly checkpoint but is dispensable for the normal M phase entry and exit in Xenopus egg cell cycle extracts. J Cell Biol 136:1091–1097
Takeuchi K, Fukagawa T (2012) Molecular architecture of vertebrate kinetochores. Exp Cell Res 318:1367–1374
Tanaka TU (2010) Kinetochore-microtubule interactions: steps towards bi-orientation. EMBO J 29:4070–4082
Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJ, Nasmyth K (2002) Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore–spindle pole connections. Cell 108:317–329
Tanaka K, Mukae N, Dewar H, van Breugel M, James EK, Prescott AR, Antony C, Tanaka TU (2005) Molecular mechanisms of kinetochore capture by spindle microtubules. Nature 434:987–994
Tang Z, Bharadwaj R, Li B, Yu H (2001) Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell 1:227–237
Tang Z, Shu H, Oncel D, Chen S, Yu H (2004) Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell 16:387–397
Tang Z, Shu H, Qi W, Mahmood NA, Mumby MC, Yu H (2006) PP2A is required for centromeric localization of Sgo1 and proper chromosome segregation. Dev Cell 10:575–585
Tanno Y, Kitajima TS, Honda T, Ando Y, Ishiguro K, Watanabe Y (2010) Phosphorylation of mammalian Sgo2 by Aurora B recruits PP2A and MCAK to centromeres. Genes Dev 24:2169–2179
Taylor SS, McKeon F (1997) Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell 89:727–735
Taylor SS, Ha E, McKeon F (1998) The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J Cell Biol 142:1–11
Teichner A, Eytan E, Sitry-Shevah D, Miniowitz-Shemtov S, Dumin E, Gromis J, Hershko A (2011) p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc Natl Acad Sci U S A 108:3187–3192
Tien JF, Umbreit NT, Gestaut DR, Franck AD, Cooper J, Wordeman L, Gonen T, Asbury CL, Davis TN (2010) Cooperation of the Dam1 and Ndc80 kinetochore complexes enhances microtubule coupling and is regulated by aurora B. J Cell Biol 189:713–723
Tighe A, Staples O, Taylor S (2008) Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol 181:893–901
Tooley JG, Miller SA, Stukenberg PT (2011) The Ndc80 complex uses a tripartite attachment point to couple microtubule depolymerization to chromosome movement. Mol Biol Cell 22:1217–1226
Trinkle-Mulcahy L, Andrews PD, Wickramasinghe S, Sleeman J, Prescott A, Lam YW, Lyon C, Swedlow JR, Lamond AI (2003) Time-lapse imaging reveals dynamic relocalization of PP1gamma throughout the mammalian cell cycle. Mol Biol Cell 14:107–117
Trinkle-Mulcahy L, Andersen J, Lam YW, Moorhead G, Mann M, Lamond AI (2006) Repo-Man recruits PP1 gamma to chromatin and is essential for cell viability. J Cell Biol 172:679–692
Tseng BS, Tan L, Kapoor TM, Funabiki H (2010) Dual detection of chromosomes and microtubules by the chromosomal passenger complex drives spindle assembly. Dev Cell 18:903–912
Tsukahara T, Tanno Y, Watanabe Y (2010) Phosphorylation of the CPC by Cdk1 promotes chromosome bi-orientation. Nature 467:719–723
Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO (2003) Targets of the cyclin-dependent kinase Cdk1. Nature 425:859–864
Uchida KS, Takagaki K, Kumada K, Hirayama Y, Noda T, Hirota T (2009) Kinetochore stretching inactivates the spindle assembly checkpoint. J Cell Biol 184:383–390
Uzunova K, Dye BT, Schutz H, Ladurner R, Petzold G, Toyoda Y, Jarvis MA, Brown NG, Poser I, Novatchkova M, Mechtler K, Hyman AA, Stark H, Schulman BA, Peters JM (2012) APC15 mediates CDC20 autoubiquitylation by APC/C(MCC) and disassembly of the mitotic checkpoint complex. Nat Struct Mol Biol 19:1116–1123
van der Waal MS, Hengeveld RC, van der Horst A, Lens SM (2012) Cell division control by the chromosomal passenger complex. Exp Cell Res 318:1407–1420
Vanoosthuyse V, Hardwick KG (2009) A novel protein phosphatase 1-dependent spindle checkpoint silencing mechanism. Curr Biol 19:1176–1181
Varma D, Monzo P, Stehman SA, Vallee RB (2008) Direct role of dynein motor in stable kinetochore–microtubule attachment, orientation, and alignment. J Cell Biol 182:1045–1054
Vigneron S, Prieto S, Bernis C, Labbe JC, Castro A, Lorca T (2004) Kinetochore localization of spindle checkpoint proteins: who controls whom? Mol Biol Cell 15:4584–4596
Vorozhko VV, Emanuele MJ, Kallio MJ, Stukenberg PT, Gorbsky GJ (2008) Multiple mechanisms of chromosome movement in vertebrate cells mediated through the Ndc80 complex and dynein/dynactin. Chromosoma 117:169–179
Wan X, O'Quinn RP, Pierce HL, Joglekar AP, Gall WE, DeLuca JG, Carroll CW, Liu ST, Yen TJ, McEwen BF, Stukenberg PT, Desai A, Salmon ED (2009) Protein architecture of the human kinetochore microtubule attachment site. Cell 137:672–684
Wang XM, Zhai Y, Ferrell JE Jr (1997) A role for mitogen-activated protein kinase in the spindle assembly checkpoint in XTC cells. J Cell Biol 137:433–443
Wang HW, Ramey VH, Westermann S, Leschziner AE, Welburn JP, Nakajima Y, Drubin DG, Barnes G, Nogales E (2007) Architecture of the Dam1 kinetochore ring complex and implications for microtubule-driven assembly and force-coupling mechanisms. Nat Struct Mol Biol 14:721–726
Wang F, Dai J, Daum JR, Niedzialkowska E, Banerjee B, Stukenberg PT, Gorbsky GJ, Higgins JM (2010) Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 330:231–235
Wang F, Ulyanova NP, van der Waal MS, Patnaik D, Lens SM, Higgins JM (2011) A positive feedback loop involving Haspin and Aurora B promotes CPC accumulation at centromeres in mitosis. Curr Biol 21:1061–1069
Wang F, Ulyanova NP, Daum JR, Patnaik D, Kateneva AV, Gorbsky GJ, Higgins JM (2012) Haspin inhibitors reveal centromeric functions of Aurora B in chromosome segregation. J Cell Biol 199:251–268
Warren CD, Brady DM, Johnston RC, Hanna JS, Hardwick KG, Spencer FA (2002) Distinct chromosome segregation roles for spindle checkpoint proteins. Mol Biol Cell 13:3029–3041
Waters JC, Chen RH, Murray AW, Salmon ED (1998) Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol 141:1181–1191
Weaver BA, Cleveland DW (2006) Does aneuploidy cause cancer? Curr Opin Cell Biol 18:658–667
Wei RR, Al-Bassam J, Harrison SC (2007) The Ndc80/HEC1 complex is a contact point for kinetochore–microtubule attachment. Nat Struct Mol Biol 14:54–59
Wei R, Ngo B, Wu G, Lee WH (2011) Phosphorylation of the Ndc80 complex protein, HEC1, by Nek2 kinase modulates chromosome alignment and signaling of the spindle assembly checkpoint. Mol Biol Cell 22:3584–3594
Welburn JPI, Vleugel M, Liu D, Iii JRY, Lampson MA, Fukagawa T, Cheeseman IM (2010) Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore–microtubule interface. Mol Cell 38:383–392
Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS (2011) p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci 124:3905–3916
Wong OK, Fang G (2007) Cdk1 phosphorylation of BubR1 controls spindle checkpoint arrest and Plk1-mediated formation of the 3F3/2 epitope. J Cell Biol 179:611–617
Wood KW, Sakowicz R, Goldstein LS, Cleveland DW (1997) CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment. Cell 91:357–366
Wurzenberger C, Held M, Lampson MA, Poser I, Hyman AA, Gerlich DW (2012) Sds22 and Repo-Man stabilize chromosome segregation by counteracting Aurora B on anaphase kinetochores. J Cell Biol 198:173–183
Yamagishi Y, Honda T, Tanno Y, Watanabe Y (2010) Two histone marks establish the inner centromere and chromosome bi-orientation. Science 330:239–243
Yamagishi Y, Yang CH, Tanno Y, Watanabe Y (2012) MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat Cell Biol 14:746–752
Yamano H, Ishii K, Yanagida M (1994) Phosphorylation of dis2 protein phosphatase at the C-terminal cdc2 consensus and its potential role in cell cycle regulation. EMBO J 13:5310–5318
Yang M, Li B, Liu CJ, Tomchick DR, Machius M, Rizo J, Yu H, Luo X (2008) Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol 6:e50
Yasuda S, Oceguera-Yanez F, Kato T, Okamoto M, Yonemura S, Terada Y, Ishizaki T, Narumiya S (2004) Cdc42 and mDia3 regulate microtubule attachment to kinetochores. Nature 428:767–771
Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K (1995) Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharomyces cerevisiae. J Cell Biol 130:687–700
Yue Z, Carvalho A, Xu Z, Yuan X, Cardinale S, Ribeiro S, Lai F, Ogawa H, Gudmundsdottir E, Gassmann R, Morrison CG, Ruchaud S, Earnshaw WC (2008) Deconstructing Survivin: comprehensive genetic analysis of Survivin function by conditional knockout in a vertebrate cell line. J Cell Biol 183:279–296
Zecevic M, Catling AD, Eblen ST, Renzi L, Hittle JC, Yen TJ, Gorbsky GJ, Weber MJ (1998) Active MAP kinase in mitosis: localization at kinetochores and association with the motor protein CENP-E. J Cell Biol 142:1547–1558
Zhang J, Ahmad S, Mao Y (2007) BubR1 and APC/EB1 cooperate to maintain metaphase chromosome alignment. J Cell Biol 178:773–784
Zhao Y, Chen RH (2006) Mps1 phosphorylation by MAP kinase is required for kinetochore localization of spindle-checkpoint proteins. Curr Biol 16:1764–1769
Zhou T, Aumais JP, Liu X, Yu-Lee LY, Erikson RL (2003) A role for Plk1 phosphorylation of NudC in cytokinesis. Dev Cell 5:127–138
Zich J, Sochaj AM, Syred HM, Milne L, Cook AG, Ohkura H, Rappsilber J, Hardwick KG (2012) Kinase activity of fission yeast Mph1 is required for Mad2 and Mad3 to stably bind the anaphase promoting complex. Curr Biol 22:296–301
Acknowledgments
HF is supported by a National Institutes of Health (NIH) grant (R01GM075249), and DW is supported by a NIH Ruth L. Kirschstein National Research Service Award Postdoctoral Fellowship (1F32GM103147). We thank Sahand Jamal Rahi for critical comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 457 kb)
Rights and permissions
About this article
Cite this article
Funabiki, H., Wynne, D.J. Making an effective switch at the kinetochore by phosphorylation and dephosphorylation. Chromosoma 122, 135–158 (2013). https://doi.org/10.1007/s00412-013-0401-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-013-0401-5