
Abstract
With the goal of creating two genetically identical daughter cells, cell division culminates in the equal segregation of sister chromatids. This phase of cell division is monitored by a cell cycle checkpoint known as the spindle assembly checkpoint (SAC). The SAC actively prevents chromosome segregation while one or more chromosomes, or more accurately kinetochores, remain unattached to the mitotic spindle. Such unattached kinetochores recruit SAC proteins to assemble a diffusible anaphase inhibitor. Kinetochores stop production of this inhibitor once microtubules (MTs) of the mitotic spindle are bound, but productive attachment of all kinetochores is required to satisfy the SAC, initiate anaphase, and exit from mitosis. Although mechanisms of kinetochore signaling and SAC inhibitor assembly and function have received the bulk of attention in the past two decades, recent work has focused on the principles of SAC silencing. Here, we review the mechanisms that silence SAC signaling at the kinetochore, and in particular, how attachment to spindle MTs and biorientation on the mitotic spindle may turn off inhibitor generation. Future challenges in this area are highlighted towards the goal of building a comprehensive molecular model of this process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitosis represents the last stage in the life of an individual cell. With the duplication of the genome, the cell embarks upon the distribution of these copies to two new cellular progeny. This distribution is not itself without complications. Each genome is packaged into chromosomes and these packages are individually delivered to each daughter cell. Neither the mitotic spindle which segregates the chromosomes, nor the chromosomes themselves have the capacity to coordinate the proper delivery. Instead a biochemical and biophysical system, called the spindle assembly checkpoint (SAC), has evolved to monitor local spindle attachment to chromosomes and link it to the global signaling machinery ensuring that each daughter cell receives one, and only one, copy of each chromosome (Musacchio and Salmon 2007). While many of the players in this process have been identified, an integrative view of the SAC mechanism has remained elusive.
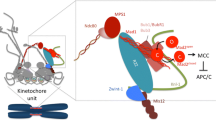
The SAC performs this surveillance function via the generation of a signal that delays the onset of genetic segregation or anaphase. This signal, which is produced at unattached kinetochores, the chromosomal site of spindle attachment, inhibits the activity of the aptly named E3 ubiquitin ligase, the anaphase-promoting complex (APC/C) (Fig. 1) (Pines 2011). This E3 enzyme ubiquitinates and thereby tags for destruction a number of targets that permit the transition to anaphase. The most important targets are: (1) cyclin B, the mitotic cyclin, that when degraded results in the loss of cyclin-dependent kinase 1 (Cdk1) activity and mitotic exit and (2) securin, that when degraded results in the activation of the CD clan protease separase, which cleaves the cohesin complex that links duplicated sister chromatids together. Together, the loss of cyclin B and Cdk1 activity and activation of separase initiates the events of genetic segregation of duplicated chromatids to the daughter cells.

The role of MT attachment in the Spindle Assembly Checkpoint. Unattached kinetochores (gray) catalyze production of a soluble inhibitor (MCC) that prevents the APC/C from targeting Cyclin B and Securin for destruction. Production of MCC requires the kinetochore-bound Mad1/2 heterotetramer and Mps1 kinase activity. Attached kinetochores (green) illustrated by engagement of the Ndc80 complex with MTs, have lost the Mad1/2 scaffold and no longer generate the soluble inhibitor
The SAC prevents anaphase through the production of a stoichiometric inhibitor of the APC/C. While the exact mechanism of the inhibitor generation is still being worked out, some elements have found consensus support. The unattached kinetochore recruits a number of proteins to a catalytic scaffold that has as its core a heterotetramer of the proteins Mad1 and Mad2, two proteins identified in one of the original yeast screens for mitotic regulators (Fig. 1) (Li and Murray 1991). Mad2 exists in two conformational states: open and closed (Luo and Yu 2008; Mapelli and Musacchio 2007). The closed state is a ligand-bound state as in the case of the kinetochore Mad1-bound pool. The open state has the property of being able to dimerize with the closed form which initiates the open to closed transition. Using this conformational transfer property, the kinetochore-bound scaffold recruits open Mad2 and converts it into a closed form (De Antoni et al. 2005; Luo et al. 2002; Mapelli et al. 2006; Simonetta et al. 2009; Xia et al. 2004). This closed form binds to the APC/C activator Cdc20 and along with two other proteins, BubR1/Mad3 and Bub3 forms the major anaphase inhibitor, the mitotic checkpoint complex, or MCC (Fig. 1) (Hardwick et al. 2000; Sudakin et al. 2001; Kulukian et al. 2009; Chao et al. 2012). This complex may be formed on the kinetochore itself or in the cytoplasm and can bind to and inhibit the APC/C. Moreover, the closed form of Mad2, now present in the MCC, can in principle convert more cytoplasmic open Mad2 into a closed form and generate even more MCC complexes (Simonetta et al. 2009). Although as yet unproven, this “cytoplasmic amplification” has the potential of tightly inhibiting anaphase onset, even in the presence of a single unattached kinetochore.
Although the Mad1/2 heterotetramer forms the heart of the checkpoint machinery at unattached kinetochores, several proteins critically contribute to its function. Chief among these is the serine/threonine kinase Mps1, a checkpoint kinase originally identified in budding yeast (Hardwick et al. 1996; Weiss and Winey 1996). Both Mad1 recruitment to unattached kinetochores and Mad2 dimerization depend on the catalytic activity of Mps1 (Fig. 1) (Abrieu et al. 2001; Jelluma et al. 2008; Hewitt et al. 2010; Jelluma et al. 2010; Maciejowski et al. 2010; Santaguida et al. 2010; Sliedrecht et al. 2010; Kwiatkowski et al. 2010) . As a result, Mps1 inhibition or mutation incapacitates the SAC in all model organisms (with the possible exception of Caenorhabditis elegans, the genome of which does not encode an obvious Mps1 homologue). Mad1 localization (Meraldi and Sorger 2005) and spindle checkpoint function (Johnson et al. 2004) also depend on Bub1, another serine/threonine kinase identified in one of the original yeast screens (Hoyt et al. 1991). It is debated, however, whether Bub1 catalytic activity is required for these functions (Klebig et al. 2009; Warren et al. 2002; Yamaguchi et al. 2003; Perera and Taylor 2010).
In metazoan cells, Mad1 also requires the presence of a complex of Rod, Zwilch and Zw10, termed the RZZ complex, for kinetochore localization. While not obviously maintained in all branches of the eukaryotic tree, loss of this complex in flies and vertebrates does prevent spindle checkpoint activity (Chan et al. 2000; Kops et al. 2005; Williams et al. 2003; Basto et al. 2004; Buffin et al. 2005). In addition to ensuring establishment of the Mad1/2 core at unattached kinetochores, RZZ recruits the dynein complex to kinetochores via direct interactions with a subunit of dynactin (Starr et al. 1998), as well as via the recruitment of the kinetochore protein Spindly (Chan et al. 2009). The inner centromere kinase Aurora B has also been shown to be responsible for targeting Mps1, RZZ, Mad1/2, and Bub1 to the unattached kinetochore (Ditchfield et al. 2003; Famulski and Chan 2007; Morrow et al. 2005; Saurin et al. 2011; Santaguida et al. 2011).
The core catalytic scaffold and the molecular elements that recruit it to the kinetochore form the central signaling platform that generates the anaphase inhibitor. A critical event in mitotic progression is the silencing of this generator. Since chromosomes are delivered to daughter cells by the mitotic spindle, coupling the attachment of spindle MTs to the kinetochore is a natural choice for locally shutting down the inhibitor generation. This is supported by overwhelming data demonstrating the loss or diminished recruitment of checkpoint proteins to attached kinetochores. By attached, we mean kinetochores that have engaged interactions with spindle microtubules in end-on or possibly even lateral fashion but that are not necessarily on a bioriented chromosome (and therefore not under tension). Notably, Mad1 and Mad2 are undetectable at attached kinetochores demonstrating a loss of the catalytic scaffold (Chen et al. 1998; Chen et al. 1996; Howell et al. 2000; Waters et al. 1998). One study reported that Cdc20 and BubR1 were diminished at attached kinetochores (Howell et al. 2004), although others have reported little difference (Jablonski et al. 1998; Taylor et al. 2001). Changes in Mps1 and Bub1 have also been reported upon MT attachment (Howell et al. 2004; Skoufias et al. 2001; Taylor et al. 2001) (Table 1).
To make matters more complicated early models of the SAC pointed out that the attachment of kinetochores to the mitotic spindle is insufficient for proper chromosome segregation (McIntosh 1991). Both sister chromatids have their own kinetochores to ensure delivery to a daughter cell. To ensure that each pair is split one to each daughter requires that MTs from one pole of the spindle attach to one sister and the other pole to the other sister. This bipolar, or bioriented, arrangement is thought to generate physical tension between the sister kinetochores due to the MT dynamics that ultimately deliver the sister chromatids to the daughter cells. This critical issue is the origin of the role of tension in SAC signaling. Since the first proposal of tension a multitude of models and molecular components have been proposed to sense tension and signal to the SAC machinery (for a detailed review, see (Maresca and Salmon 2010), (Nezi and Musacchio 2009) and (Khodjakov and Pines 2010)). The term has almost become, and will be used here, interchangeable with biorientation, but dissecting the role of physical forces away from molecular signaling remains a central challenge in the field. Interestingly, while Mad1/2 disappear from kinetochores in an attachment-dependent manner (i.e., not requiring tension), many other kinetochore SAC proteins dissociate only after chromosomes are properly bioriented when sister kinetochores are under physical tension. For example, Mps1, BubR1, Bub1, and the RZZ complex have all been reported to be reduced or completely disappear from kinetochores only after proper biorientation (Famulski and Chan 2007; Howell et al. 2004; Taylor et al. 2001; Williams et al. 1996; Skoufias et al. 2001) (Table 1). Protein dissociation upon biorientation requires a more complex mechanism for protein displacement than simple MT attachment since it implies some communication between the sister kinetochores. This communication has been proposed to be mediated by Aurora B. This kinase resides in the inner centromere, between the two sister kinetochores, as part of the chromosomal passenger complex (CPC) (Ruchaud et al. 2007) and plays an essential role in preventing the persistence of syntelic attachments, where both sister kinetochores are attached to the same spindle pole and lack inter-kinetochore tension (Kelly and Funabiki 2009; Lampson and Cheeseman 2011). These data implicate Aurora B as the tension- (or biorientation-) responsive element of the SAC.
Proper mitotic progression requires that SAC activity is silenced at each kinetochore in response to spindle attachment and tension. Observations have highlighted the role of protein dissociation from kinetochores as a key mechanism for deactivating the MCC generation machinery. The mechanisms by which this change in kinetochore composition is accomplished are only now being studied in detail. Here, we review some of the observations that link the physical binding of kinetochores to spindle MTs to events in SAC silencing. Given the strong evolutionary conservation of the SAC and kinetochore-MT attachment interface, we point out where appropriate, evolutionary similarities and distinctions. We also identify understudied areas in MT attachment-mediated SAC silencing that are likely to shed light on novel research directions.
Kinetochore attachment: loss of the catalytic scaffold
What happens to the SAC machinery once the kinetochore latches on to the MT? Immunofluorescence and live cell imaging studies showed that the core checkpoint components Mad1 and Mad2 are specifically depleted from kinetochores upon attachment (Chen et al. 1998; Chen et al. 1996; Howell et al. 2000; Waters et al. 1998) (Table 1). This depletion is essential for checkpoint silencing, as constitutively tethering Mad1 to kinetochores delays anaphase onset in a Mad2-dependent manner after all kinetochores have attached and bioriented (Maldonado and Kapoor 2011). How then is the Mad1-Mad2 heterodimer removed from kinetochores upon attachment? In a set of elegant experiments, the Salmon lab proposed an appealing hypothesis involving the minus-end directed MT motor dynein that is localized to kinetochores in mitosis. Without a road to ride on, dynein remains kinetochore-bound but is ready to go once a MT “highway” engages the kinetochore. When that happens, dynein is thought to transport itself towards the spindle poles (Howell et al. 2001), where both dynein and its cargo can be released into the cytoplasm (Fig. 2). ATP depletion, via treatment with azide and deoxyglucose (Howell et al. 2001) or treatment with nordihydroguaiaretic acid (NDGA) (Arasaki et al. 2007; Famulski et al. 2011) maintained dynein activity but prevented release of dynein and its cargo from spindle poles, allowing the identification of kinetochore proteins transported poleward by dynein. The spindle checkpoint proteins Mad1, Mad2, BubR1, and Mps1 all accumulate on poles under these conditions (Howell et al. 2001; Famulski et al. 2011), suggesting dynein carries these proteins from attached kinetochores to poles (Fig. 2). In support of this, such “stripping” of checkpoint components has been observed by live cell imaging of fluorescent versions of Mad1, Mad2, and Rod (Buffin et al. 2005; Emre et al. 2011; Howell et al. 2000; Wojcik et al. 2001). Dynein has also been suggested to strip checkpoint components not only from kinetochores engaged in end-on MT attachments (MTs end at a kinetochore), but from lateral attachments (kinetochores bound to MT side) as well (Howell et al. 2000; Howell et al. 2001). Dynein is involved in establishing initial, lateral contacts with a MT after which it transports those chromosomes toward the spindle pole (Li et al. 2007; Vorozhko et al. 2008; Yang et al. 2007b). It is presently unclear how dynein coordinates simultaneous protein stripping and chromosome movement on a single kinetochore. In addition, it is unknown if the checkpoint is silenced on those laterally attached kinetochores.

Removal of SAC proteins from an attached kinetochore: dynein-mediated vs. dynein-independent pathways. MT attachment allows dynein (blue) to transport Mad1/2 complexes, RZZ, and Mps1 poleward, thereby removing essential components of the MCC generator from the attached kinetochore (green) (b). Dynein also removes Spindly, a protein that may prevent a dynein-independent Mad1/2 removal pathway (indicated by a question mark) (a). Low opacity in (b) indicates that a fraction of a designated protein/complex is engaged in that pathway
Although dynein-mediated stripping of SAC components is firmly established, its contribution to checkpoint silencing is less so. Conclusive answers using RNA interference or mutations in dynein subunits have been frustrated by the fact that dynein has a variety of cellular functions, including in mitosis. Nevertheless, this can be circumvented to some extent by timed inhibition of dynein. Injection of the disruptive dynactin subunit p50/dynamitin during early prometaphase in PtK1 cells delays mitotic progression in a SAC-dependent manner without disturbing kinetochore–MT interactions and chromosome congression. This observation supports the hypothesis that dynein-dependent stripping contributes directly to checkpoint silencing (Howell et al. 2001). A 25-fold increase of Mad2 at attached kinetochores under dynein perturbation was observed (in comparison to attached kinetochores without dynein perturbation), which represented ∼30 % of Mad2 normally present on unattached kinetochores. This suggests that dynein-dependent and -independent checkpoint removal pathways exist and that inhibition of the former is sufficient to prevent checkpoint silencing.
An alternative approach to inhibiting kinetochore-dynein has been to deplete proteins responsible for dynein kinetochore recruitment. The RZZ complex (Kops et al. 2005; Starr et al. 1998; Yang et al. 2007b) and NudE-like proteins (Vergnolle and Taylor 2007; Li et al. 2005; Niethammer et al. 2000; Sasaki et al. 2000; Stehman et al. 2007) bind dynactin and dynein, respectively, and are essential for the kinetochore localization of dynein. When properly targeted, dynein acts to strip itself and its recruiting proteins from the attached kinetochores as seen by their accumulation on poles upon ATP depletion or NDGA treatment. Depletion or inhibition of any of these dynein recruiters resulted in a mitotic delay in vertebrate cells (Vergnolle and Taylor 2007; Yang et al. 2007b; Barisic et al. 2010; Gassmann et al. 2010; Chan et al. 2000). Unlike direct dynein inhibition by p50 injection, however, depletion of dynein recruiters reduced K-fiber stability potentially eliciting a SAC response (Yang et al. 2007b), making their role in the direct removal of SAC proteins complex to interpret.
The RZZ complex also recruits Spindly, which is indispensable for dynein kinetochore binding (Griffis et al. 2007; Chan et al. 2009; Barisic et al. 2010; Gassmann et al. 2010). The role of Spindly in dynein recruitment is still poorly understood, as it is itself dependent on the RZZ complex that directly binds dynactin. Spindly itself has not been shown to interact with dynein or dynein-associated complexes. In worms, Spindly RNA interference has been reported to disrupt Mad1 and Mad2 recruitment resulting in loss of checkpoint activity (Gassmann et al. 2008; Yamamoto et al. 2008). However, silencing of Spindly in human cells acts much like the loss of other dynein recruiters, causing a SAC-dependent mitotic delay with defective kinetochore MT attachments (Chan et al. 2009; Barisic et al. 2010; Gassmann et al. 2010), making a specific function for Spindly difficult to ascertain. Two recent studies by the Desai and Geley labs elegantly circumvented this issue. Mutations in Spindly in a conserved region known as the Spindly box prevented association of dynein with kinetochores but, as opposed to Spindly depletion, allowed normal MT attachments, chromosome biorientation, and congression (Barisic et al. 2010; Gassmann et al. 2010). Cells expressing this mutant form of Spindly were strongly delayed in metaphase, supporting the notion that dynein is required for SAC silencing at the kinetochores. In fact, the level of Mad2 remaining at bioriented kinetochores in the Spindly mutant was similar to that of p50 injection in PtK1 cells. So unlike RNAi-mediated depletion of Spindly, replacement with the mutant form clearly demonstrates a critical role for dynein-mediated removal of Mad1/Mad2 from attached kinetochores. Interestingly, depletion of Spindly did eliminate detectable kinetochore localization of dynein but permitted Mad1/Mad2 removal from the subset of bioriented chromosomes, indicating the existence of a dynein-independent removal pathway. It should be noted that it is possible that incomplete depletion of Spindly allowed low, undetectable amounts of dynein to remain at kinetochores and eventually strip the SAC proteins. Disruption of dynein function in Spindly-depleted cells will be required to unequivocally prove existence of a dynein-independent removal pathway.
A key unresolved issue is the identity of such a dynein-independent Mad1/Mad2 removal pathway that kicks in when a MT attaches to the kinetochore (Fig. 2). One possibility is that attachment is coupled to activation/localization of a phosphatase that reverts phosphorylations essential for the maintenance of Mad1 at kinetochores. Another possibility is that the MT directly removes a Mad1 regulator or Mad1 receptor, for instance by competition, steric hindrance or electrostatic repulsion, as may be the case with Knl1 (Espeut et al. 2012). Three Mad1 regulators, Mps1, Bub1, and RZZ, bind kinetochore components required for MT attachment, Hec1 for Mps1, and Knl1 for Bub1 and RZZ (Kemmler et al. 2009; Martin-Lluesma et al. 2002; Kiyomitsu et al. 2011), and perhaps this binding is incompatible with Hec1/Knl1–microtubule interactions. Constitutive tethering of Mps1 to bioriented kinetochores, independent of Hec1, maintains Mad1 at attached kinetochores and like the constitutive tethering of Mad1 itself, delays anaphase onset, suggesting that Mps1, like Mad1, needs to be removed from kinetochores for checkpoint silencing to occur (Jelluma et al. 2010; Maldonado and Kapoor 2011). Although the delay by constitutive kinetochore Mps1 may be explained if Mps1 is normally removed by dynein, an intriguing possibility is that it is related to the dynein-independent pathway. Removal of Spindly may be required to delocalize Mps1, or vice versa. Resolving where Mps1 and Mad1 bind to kinetochores, how Spindly might affect this localization, and how MTs influence these events will be critical in resolving, in detail, how attachments to the mitotic spindle are coupled to checkpoint silencing at the level of individual kinetochores.
Bioriented attachment: tension and complete removal of SAC components
If attachment is sufficient to remove Mad1/2 from the kinetochore, how is chromosome biorientation integrated into SAC silencing? Live cell imaging studies have shown that while kinetochore levels of many SAC components are diminished upon attachment, they are further diminished upon biorientation, to virtually undetectable in some instances (Howell et al. 2004) (Table 1). A key difference between the attached and bioriented states is the change in phosphorylation of kinetochore-bound Aurora B substrates (Lampson and Cheeseman 2011). The prevailing hypothesis posits that tension on bioriented chromosomes separates the kinetochores of the sister chromatids from inner-centromere-localized Aurora B (Liu et al. 2009). The contribution of Aurora B to SAC signaling has been hotly debated but recent studies have provided important new insights. Previous work, first in budding yeast and later in vertebrate cells, established the role of Aurora B in MT destabilization, also called “error correction”, at kinetochore attachments without tension (Tanaka et al. 2002; Ditchfield et al. 2003; Hauf et al. 2003; Lampson et al. 2004). The hypothesis that destabilization of MT attachments by Aurora B reactivates the SAC signaling machinery present at the newly unattached kinetochore is supported by much evidence, but many observations imply that the mechanism may be more complex.
A variety of studies have demonstrated a role for Aurora B in the localization of SAC components to kinetochores, in addition to modulating the kinetochore–MT interface (Ditchfield et al. 2003; Vigneron et al. 2004; Santaguida et al. 2011; Saurin et al. 2011). While Aurora B inhibition accelerates exit from a mitotic delay due to nocodazole (in the absence of polymerized MTs) in mammalian cells (from ∼20 to ∼8–10 h) (Ditchfield et al. 2003; Hauf et al. 2003), this acceleration is modest when compared to the rapid exit seen upon Aurora B inhibition in taxol- or monastrol-induced arrests (i.e., with some MT attachments). The inability to abrogate the checkpoint in the absence of Aurora B activity in conditions where MTs are depolymerized is seen in many cells, from budding yeast to mammalian cells, with the notable exception of fission yeast (Petersen and Hagan 2003). Together, this paints a picture of Aurora B as an enhancer of SAC activity rather than an essential element.
Using live cell analysis of cyclin B levels rather than time of mitotic delays as a measure for checkpoint activity, one study has shown that Aurora B promotes checkpoint establishment in early mitosis by recruiting Mps1, thereby ensuring strong checkpoint signaling upon nuclear envelope breakdown (Saurin et al. 2011). Maximal Aurora B inhibition by combining RNAi with small molecule inhibitors delayed but could not prevent establishment of the SAC, nor could it prevent its maintenance in prometaphase, supporting an enhancement role for Aurora B in checkpoint signaling. Furthermore, weakening checkpoint signaling using doses of Mps1 inhibitors well below those used for SAC abrogation, allowed Aurora B inhibitors to delay checkpoint establishment sufficiently long enough for cells to exit mitosis prematurely (Santaguida et al. 2011; Saurin et al. 2011).
How does Aurora B promote checkpoint signaling? Various studies have reported that Mps1 localization to kinetochores is controlled by Aurora B (Jelluma et al. 2010; Santaguida et al. 2011; Saurin et al. 2011). Indeed, tethering Aurora B near the kinetochore (via a CENP-B-INCENP fusion) dramatically increases Mps1 recruitment (Saurin et al. 2011). Moreover, re-establishing Mps1 kinetochore localization in the absence of Aurora B activity by tethering Mps1 to kinetochores rescued the establishment delay caused by Aurora B inhibition during mitotic entry. If Mps1 recruitment depends on Aurora B activity throughout mitosis, we might expect that tension on sister kinetochores due to biorientation would diminish the levels of kinetochore-bound Mps1. In support of this, Mps1 levels decrease upon attachment and are further reduced upon biorientation (Howell et al. 2004). Interestingly, inhibition of either Aurora B or Mps1 activity was sufficient to abrogate the metaphase delay in cells expressing kinetochore-tethered Mad1 (Maldonado and Kapoor 2011). This may be explained if residual Mps1 activity, which supports the tethered Mad1-mediated arrest, is present at these attached kinetochores in an Aurora B-dependent manner. This is in agreement with the observations that residual Mps1 is present on metaphase chromosomes (Howell et al. 2004; Stucke et al. 2004).
As a kinase, any downstream substrates of Aurora B would also be excellent candidates for the enhancement effect on the SAC. In budding yeast, the MCC protein Mad3 (functionally related to BubR1 in metazoa) is phosphorylated by Ipl1, the Aurora B homologue, and is important in the response to a lack of tension (King et al. 2007). This site and its associated signaling function may not be conserved since homologous sites on BubR1 have yet to be identified (Hengeveld et al. 2012; Hegemann et al. 2011). Interestingly, the potent SAC activity present in the absence of microtubules can be abrogated by Aurora B inhibition via the knockdown of the Ndc80 complex member, Hec1 (Saurin et al. 2011), or the knockdown of centromeric protein network proteins, CENP-I, -H, or -N (Matson et al. 2012). In both cases, there is a reduction of SAC proteins at kinetochores due to the knockdown, but this is insufficient to abrogate the checkpoint activity without further Aurora B inhibition. Identification of pathways that sensitize the SAC to Aurora B inhibition will provide an understanding of how Aurora B regulates SAC establishment in early mitosis and those pathways by which biorientation further extinguishes inhibitor generation subsequent to spindle attachment.
Removing the phosphates: the contribution of phosphatases to SAC silencing
While removal of proteins from attached kinetochores is required to silence the SAC, so is dephosphorylation of checkpoint kinase substrates. In both the budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe, a PP1-like phosphatase is required to exit from mitosis after prior engagement of the checkpoint. Arrested S. pombe cells that are forced to exit mitosis by inhibition of the Aurora B orthologue Ark1 require PP1Dis2 to do so (Vanoosthuyse and Hardwick 2009). And S. cerevisiae cells recovering from mitotic arrest induced by Mps1-overexpression can exit mitosis only in the presence of the PP1-like Glc7 phosphatase (Pinsky et al. 2009). PP1 acts to counter Aurora B activity (Francisco et al. 1994; Hsu et al. 2000; Murnion et al. 2001; Pinsky et al. 2006a; Emanuele et al. 2008), and may simply revert Aurora B-dependent phosphorylation of SAC proteins such as Mad3 (Rancati et al. 2005; King et al. 2007). Moreover, like PP1Dis2 in S. pombe (Meadows et al. 2011), Glc7 in S. cerevisiae requires binding to the KMN network subunit Spc105/Knl1 to allow checkpoint silencing (Rosenberg et al. 2011). Indeed, lethality of budding yeast cells expressing an Spc105 mutant unable to interact with Glc7 was partially rescued by weakening the activity of Aurora kinase orthologue Ipl1 (Rosenberg et al. 2011). It is possible that PP1 is recruited to kinetochores when attachment or biorientation is obtained and proceeds to dephosphorylate the Aurora B-dependent phospho-residues that normally promote checkpoint activity, in addition to PP1's proposed role in stabilizing MT attachments (Liu et al. 2010). It may, however, not be as straightforward as that. Glc7 overexpression alone can bypass a nocodazole-induced arrest, while mutations in Ipl1 do not (Biggins and Murray 2001; Pinsky et al. 2006b; Pinsky et al. 2009), and the Glc7-Spc105/Knl1 interaction required for SAC silencing is likely not regulated by Ipl1 (Rosenberg et al. 2011) as it has been demonstrated in human cells (Liu et al. 2010). Moreover, in addition to the PP1Dis2-Spc7/Knl1 interaction, binding of PP1 to the kinesin-8 family members Klp5-Klp6 (Kif18 in humans) contributes to checkpoint silencing after Ark1 inhibition in fission yeast (Meadows et al. 2011). Interestingly, the requirement of Klp5/6 motors for checkpoint silencing was independent of their motor activity, suggesting that PP1 act without active transport along MTs to attached kinetochores.
The finding that PP1 contributes to checkpoint silencing in S. pombe and S. cerevisiae is an important one, as it may couple attachment to rapid dissociation of the checkpoint signal generator. It will be of significant interest to examine if one of the PP1 family phosphatases is required for SAC silencing in mammalian cells and acts by opposing Aurora B activity and/or the activity of other checkpoint kinases. Identification of the critical PP1 substrates will be needed to comprehend its role in SAC silencing. One candidate substrate is Knl1. Phosphorylation of its MELT motifs by Mps1 in budding and fission yeast potentiates SAC activity by promoting Bub1/Bub3 kinetochore binding (London et al. 2012; Shepperd et al. 2012) which is counteracted by Glc7 (London et al. 2012). Additional candidate PP1 substrates may come from uncovering other Mps1 and Aurora B substrates, such as those identified by recent large scale proteomics efforts (Hengeveld et al. 2012; Alexander et al. 2011; Hegemann et al. 2011; Kettenbach et al. 2011; Koch et al. 2011).
Notably, in vertebrate cells, there exists a reduced dependence on Aurora B for the SAC itself. Loss of Aurora B (either by RNA interference, small molecule inhibition, or a combination of both) does not abrogate a mitotic arrest in which there are no attached kinetochores (e.g., nocodazole) but can abrogate an arrest where some kinetochores are attached (e.g., taxol or monastrol). From this observation, one proposal may be that unlike fission yeast, attached kinetochores in vertebrate cells require the local activity of the phosphatase in addition to the loss of Aurora B kinase activity to achieve SAC silencing. One can also imagine that Mps1-dependent phosphorylations need to be reversed, given the fact that inhibition of Mps1 activity causes rapid SAC silencing and that prolonged Mps1 presence on kinetochores prevents checkpoint silencing. A thorough analysis of checkpoint-specific phosphorylation sites and metaphase-specific dephosphorylation events may provide answers to such questions. Interestingly, PP1 may dephosphorylate dynein intermediate chain (DIC), an event that was recently proposed to allow dynein-mediated poleward transport of checkpoint proteins (Whyte et al. 2008). Finally, vertebrate plus-end directed motors such as the Klp5-Klp6 homologs Kif18A/18B may be important in SAC silencing. It would be of significant interest to examine whether knockdown/inhibition of these motor proteins can prevent forced exit from a checkpoint-dependent mitotic arrest, for instance by addition of Mps1 inhibitors to nocodazole-treated cells. Interestingly, Kif18A (but not Kif18B) has a PP1-binding consensus sequence and can associate with PP1α in vitro (Colland Genome Res 2004), but a role in checkpoint silencing has yet to be reported for these kinesin motors.
Getting to anaphase: removing all the inhibitor
Once all chromosomes are bioriented on the spindle, the cell is ready to initiate anaphase. However, the anaphase-promoting complex remains inactive due to the inhibitors generated from the previously unattached kinetochores. Seminal studies support the idea that even a single unattached kinetochore can delay the onset of anaphase (Zirkle 1970; Rieder et al. 1995; Rieder et al. 1994), indicating a strong inhibitory activity emanating from kinetochores. Alongside this observation is the almost immediate activation of APC/C activity once the last chromosome pair has aligned on the metaphase plate (Clute and Pines 1999). How does the SAC achieve both tight inhibition of the APC/C and the rapid release of this inhibition? The dissolution of the cytoplasmic inhibitor remains poorly understood, but here, we briefly discuss some of the current hypotheses regarding the final steps to release inhibition of the APC/C and promote anaphase onset (for more detailed reviews, see (Hardwick and Shah 2010) and (Kim and Yu 2011)). Dissociation of the MCC from the APC/C through auto-ubiquitination aided by the E2 enzyme UbcH10, has been shown to play a critical role in the timing of anaphase onset (Reddy et al. 2007; Stegmeier et al. 2007). In addition, the protein p31comet has been shown to delay anaphase onset downstream of Mad2, and given its affinity for the closed form of Mad2 (Vink et al. 2006; Yang et al. 2007a; Xia et al. 2004), has been proposed to be an anaphase activator (Hagan et al. 2011; Teichner et al. 2011; Westhorpe et al. 2011; Jia et al. 2011; Varetti et al. 2011). One that could itself be inhibited by the unattached kinetochore (Hagan et al. 2011; Fava et al. 2011; Miniowitz-Shemtov et al. 2012). If so, p31 might be “re-activated” upon attachment of the last kinetochore, leading to liberation of APC/C from the MCC via a synergistic p31/UbcH10 pathway. Notably, the ability to generate cytoplasmic extracts without kinetochores, but with extensive APC/C inhibitor activity (Gaglio et al. 1995; Braunstein et al. 2007; Reddy et al. 2007), implies the existence of as yet unrecognized cytoplasmic activities that regulate APC/C reactivation. In the long run, the richness of quantitative measurements for this transition alongside the complex biochemical activities involved all point to computational approaches becoming critical for understanding how the inhibitory milieu, present just after the last kinetochore attaches, is rapidly quenched to promote the onset of anaphase (Ciliberto and Shah 2009).
An integrated model of SAC inactivation at kinetochores
The wealth of data regarding the dynamic localization of SAC components to the kinetochore provides an opportunity to decipher the logic of SAC silencing at the attached kinetochore. Unattached kinetochores start with a repertoire of molecules that act in concert to inhibit the onset of anaphase. While the core signaling platform, i.e., Mad1/2, is completely dissociated from the attached kinetochore, many other SAC proteins remain and are lost in a graded fashion as biorientation is achieved (Table 1) (Fig. 3). With the loss of these proteins, and the gain of others via MT attachment itself, the loss of inhibitor generation results in the cytoplasmic silencing of checkpoint inhibitors and the onset of anaphase (Fig. 3).

A model for SAC silencing at attached and bioriented kinetochores. a While the unattached (gray) sister kinetochore of a chromosome attached in a monotelic fashion maximally recruits the SAC machinery and actively produces MCC, the attached sister (green) experiences increased intrakinetochore stretch and complete or partial (decreased opacity) removal of SAC components. Although the attached sister does not contribute to MCC production, the lack of biorientation prevents APC/C activation. b Amphitelic attachments increase interkinetochore distance, allowing further depletion of the SAC machinery from both sister kinetochores (low opacity) which results in APC/C activity directed against Cyclin B and Securin. See text and Table 1 for more details
How might this graded loss of protein be mediated? Given the role of interkinetochore tension in regulating protein composition at bioriented kinetochores, the partial loss in the monotelic (one kinetochore attached) and syntelic configuration could be mediated by intrakinetochore tension. Recent studies from the Salmon and Hirota labs have demonstrated substantial changes in protein arrangements that occur under monotelic attachments (Uchida et al. 2009; Maresca and Salmon 2009; Wan et al. 2009). Even a small movement of Aurora B substrates away from the inner centromere, as seen in intrakinetochore tension measurements, could reduce the recruitment of proteins. This hypothesis is supported by the dependence of RZZ, Mps1, BubR1, and Mad2 localization on Aurora B kinase activity but disagrees with the observation that loss of Aurora B does not abrogate the SAC under nocodazole treatment. Alternatively, as proposed above, the loss of Aurora B may need to be combined with the activity of phosphatases that themselves arrive via attached MTs, or other attachment-induced changes to kinetochore composition, to sufficiently extinguish kinetochore SAC activity.
A variety of alternatives to the graded response model also exist. Evidence for the direct roles of kinetochore-resident kinases, such as Mps1, in cytoplasmic stability of the MCC (Maciejowski et al. 2010) or Bub1 in the inhibition of Cdc20 (Tang et al. 2004) open the possibility for parallel molecular pathways that can also promote anaphase inhibition and are suppressed by biorientation. These alternative activities are likely to be revealed as we eliminate those known pathways and assess what SAC activities still remain.
Open questions
The current models begin to clarify some of the long-standing observations described in the SAC literature and begin to build a framework for how the increasingly complex set of MT attachment processes can be linked to local silencing of the SAC. Even so, a number of key questions remain and have enjoyed little to no attention in recent studies.
Where do checkpoint proteins bind on the kinetochore?
Given that the regulation of checkpoint protein recruitment is central to the SAC silencing and has been well-studied, it is puzzling that the actual binding sites for many of the checkpoint proteins is not known. For example, deleting a variety of kinetochore proteins can cause the loss of Mad1 and Mad2 at unattached kinetochores. This observation can be interpreted as a lock of the kinetochore in a “MT bound” state, with reduced recruitment of SAC components, or the inability to build the kinetochore in the first place, both of which preclude identification of the binding partner at the kinetochore. Tandem affinity approaches (TAP), like those successful for mapping the kinetochore and centromere components, have been applied to the core SAC proteins with limited success. Mad2 TAP tags have been reported to bring down the MCC-APC/C complex and some components of the kinetochore such as Mad1, Zwint, Rae1, and Bub1 (Kops et al. 2010), and purifications of BubR1 yielded the soluble components of the inhibitory complex as well as proteins essential for its localization to kinetochores, such as Bub1 and Knl1 (Kops et al. 2010). Similar analyses for Mad1, Mps1 (GJPLK, unpublished), and RZZ (Kops et al. 2005), however, have not yielded potential kinetochore receptors. Given the transient nature of kinetochore binding of the MCC components, it may be the case that more stable components of the checkpoint machinery, such as Mad1, will provide some understanding of how and where the catalytic scaffold is localized at the kinetochore.
How is the site of SAC activity regulated?
In addition to the exact site of binding, the regulation of these sites is equally critical. While stripping of the proteins from kinetochores in a dynein/RZZ-mediated pathway is present in metazoans, it only explains part of the loss of the Mad1/2 complex from attached kinetochores (Barisic et al. 2010; Chan et al. 2009; Gassmann et al. 2010). Moreover, in single-celled organisms, specifically fungi, where no apparent RZZ components have been found, this displacement seems to occur in the absence of any MT-based streaming. Apart from Mad1/2 recruitment, BubR1 and Cdc20 are also reduced by about 50 % after attachment (Howell et al. 2004). While a number of binding sites for BubR1 have been proposed (e.g., Knl1/Blinkin, Bub3, CENP-E), it is unclear if these interactions modulate checkpoint activity (Logarinho et al. 2008; Taylor et al. 1998; Krenn et al. 2012; Bolanos-Garcia et al. 2011; Kiyomitsu et al. 2007; Lara-Gonzalez et al. 2011), and a specific kinetochore binding site for Cdc20 is completely unknown. Ultimately, any model of kinetochore attachment-mediated silencing must have a direct mechanism by which the Mad1/2 complex and BubR1 and Cdc20 are no longer actively recruited. Interestingly, it has also been noted in a variety of reports that loss of MT attachment after initial loss of Mad1 and Mad2 can produce a “twinkling” phenomenon indicative of highly dynamic and reversible Mad1/Mad2 recruitment (Kapoor et al. 2000; Bomont et al. 2005). Thus, another key feature of any displacement mechanism is the reappearance of the binding site once MT attachment is lost.
Reactivation of kinetochore signaling upon loss of MT attachment
Central to many models of mitotic progression is an inherent reversibility of the SAC, so that at any time if a chromosome detaches from the spindle, its kinetochore can be re-activated. Clute and Pines demonstrated over a decade ago the stabilization of a GFP fusion of Cyclin B after the addition of taxol to cells that had already begun to degrade Cyclin B (Clute and Pines 1999). Proteins such as Mad2 and Bub1 are re-recruited to kinetochores after attachment or tension is lost demonstrating the re-initiation of SAC signaling (Waters et al. 1998; Skoufias et al. 2001). In addition to drug perturbations, such a loss of attachment can occur under defective MT attachments as seen after depletion of CENP-F (Bomont et al. 2005) or after Aurora B-mediated destabilization of a syntelic attachment (Kapoor et al. 2000). Given the dramatic shedding of all the SAC components that occurs as a kinetochore becomes bioriented, it becomes important to understand what molecular entities might still reside at the kinetochore that could re-initiate the construction of the SAC signaling platform. The molecular requirements for re-activating the SAC could themselves be similar to the initial assembly of the kinetochore that occurs during prophase or perhaps the remaining molecular species at the attached chromosomes (e.g., Mps1 at the kinetochore or Aurora B in the inner centromere), diminished as they are, remain in case of the unlikely but devastating event of a chromosome becoming detached.
Does the kinetochore only generate one inhibitory signal that needs to be silenced?
As we learn more about proteins localizing to the kinetochore, more and more of these proteins seem to play a direct role in the SAC. The kinase Mps1 is a key regulator of checkpoint activity and while its loss before the onset of mitosis precludes the localization of Mad1/2 and a rapid exit phenotype, loss of activity during mitosis causes rapid exit, with some Mad1 still present at kinetochores (Hewitt et al. 2010). Like Mad1/2, BubR1, and Cdc20, the site of Mps1 localization at the unattached kinetochore remains unknown. But more importantly, the mechanism by which Mps1 regulates the SAC signal is not completely described. The turnover of Mps1, which requires its own kinase activity, may itself represent another kinetochore-derived signal that informs the cytoplasm of an unattached kinetochore (Jelluma et al. 2010). How might such a signal regulate SAC activity? While the cytoplasmic activity of Mps1 is still being actively identified, some indirect evidence points to its ability to stabilize the MCC (Maciejowski et al. 2010). We also see that p31comet, a molecule involved in MCC dissociation and APC/C re-activation also localizes to and turns over at unattached kinetochores but is absent from attached kinetochores (Hagan et al. 2011). Its turnover has been proposed to lead to a change in its Mad2 affinity thereby reducing its ability to turn off the checkpoint while unattached kinetochores remain. The Mps1 or p31comet signals could act to prolong the lifetime of the inhibitory complex rather than increase inhibitor production. It would be interesting to see if in fact the kinetochore produces more signals than simply the MCC and if some may “inhibit APC/C activation” in addition to “activating the APC/C inhibitor”.
While the last two decades have observed steady progress in the field of kinetochore and SAC biology, each unearthed stone reveals a multitude of new mechanisms. Recent developments in high-resolution imaging, quantitative probes of local kinase activity, and advances in correlative ultrastructural studies promise to dig even deeper into these questions. Moreover, our understanding of these processes in an evolutionary continuum and quantitative modeling provide an integrative view of how these many molecules accomplish such a complex task so robustly. Even as it is one of the oldest cellular processes documented, mitosis seems to remain one with questions at the ready for every answer we provide.
Abbreviations
- SAC:
-
Spindle assembly checkpoint
- MT:
-
Microtubule
- RZZ:
-
Rod/Zw10/Zwilch
- CENP:
-
Centromere protein
- KMN:
-
Knl1/Mis12/Ndc80 complex
- CPC:
-
Chromosome passenger complex
- TAP:
-
Tandem affinity purification
- Cdk1:
-
Cyclin-dependent kinase 1
- APC/C:
-
Anaphase promoting complex/cyclosome
- MCC:
-
Mitotic checkpoint complex
- DIC:
-
Dynein intermediate chain
References
Abrieu A, Magnaghi-Jaulin L, Kahana JA, Peter M, Castro A, Vigneron S, Lorca T, Cleveland DW, Labbé JC (2001) Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell 106(1):83–93
Alexander J, Lim D, Joughin BA, Hegemann B, Hutchins JRA, Ehrenberger T, Ivins F, Sessa F, Hudecz O, Nigg EA, Fry AM, Musacchio A, Stukenberg PT, Mechtler K, Peters J-M, Smerdon SJ, Yaffe MB (2011) Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci Signal 4(179):ra42. doi:10.1126/scisignal.2001796
Amaro AC, Samora CP, Holtackers R, Wang E, Kingston IJ, Alonso M, Lampson M, McAinsh AD, Meraldi P (2010) Molecular control of kinetochore-microtubule dynamics and chromosome oscillations. Nat Cell Biol 12(4):319–329. doi:10.1038/ncb2033
Arasaki K, Tani K, Yoshimori T, Stephens DJ, Tagaya M (2007) Nordihydroguaiaretic acid affects multiple dynein-dynactin functions in interphase and mitotic cells. Mol Pharmacol 71(2):454–460. doi:10.1124/mol.106.029611
Arnaud L, Pines J, Nigg EA (1998) GFP tagging reveals human Polo-like kinase 1 at the kinetochore/centromere region of mitotic chromosomes. Chromosoma 107(6–7):424–429
Barisic M, Sohm B, Mikolcevic P, Wandke C, Rauch V, Ringer T, Hess M, Bonn G, Geley S (2010) Spindly/CCDC99 is required for efficient chromosome congression and mitotic checkpoint regulation. Mol Biol Cell 21(12):1968. doi:10.1091/mbc.E09-04-0356
Basto R, Scaerou F, Mische S, Wojcik E, Lefebvre C, Gomes R, Hays T, Karess R (2004) In vivo dynamics of the rough deal checkpoint protein during Drosophila mitosis. Curr Biol 14(1):56–61
Bentley AM, Normand G, Hoyt J, King RW (2007) Distinct sequence elements of cyclin B1 promote localization to chromatin, centrosomes, and kinetochores during mitosis. Mol Biol Cell 18(12):4847–4858. doi:10.1091/mbc.E06-06-0539
Biggins S, Murray AW (2001) The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev 15(23):3118–3129. doi:10.1101/gad.934801
Bolanos-Garcia VM, Lischetti T, Matak-Vinković D, Cota E, Simpson PJ, Chirgadze DY, Spring DR, Robinson CV, Nilsson J, Blundell TL (2011) Structure of a Blinkin-BUBR1 complex reveals an interaction crucial for kinetochore-mitotic checkpoint regulation via an unanticipated binding Site. Struct (London, England: 1993 19(11):1691–1700. doi:10.1016/j.str.2011.09.017
Bomont P, Maddox P, Shah JV, Desai AB, Cleveland DW (2005) Unstable microtubule capture at kinetochores depleted of the centromere-associated protein CENP-F. EMBO J 24(22):3927–3939
Braunstein I, Miniowitz S, Moshe Y, Hershko A (2007) Inhibitory factors associated with anaphase-promoting complex/cylosome in mitotic checkpoint. Proc Natl Acad Sci U S A 104(12):4870–4875
Buffin E, Lefebvre C, Huang J, Gagou ME, Karess RE (2005) Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr Biol 15(9):856–861. doi:10.1016/j.cub.2005.03.052
Chan GK, Jablonski SA, Starr DA, Goldberg ML, Yen TJ (2000) Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat Cell Biol 2(12):944–947. doi:10.1038/35046598
Chan YW, Fava LL, Uldschmid A, Schmitz MHA, Gerlich DW, Nigg EA, Santamaria A (2009) Mitotic control of kinetochore-associated dynein and spindle orientation by human Spindly. J Cell Biol 185(5):859–874. doi:10.1083/jcb.200812167
Chan YW, Jeyaprakash AA, Nigg EA, Santamaria A (2012) Aurora B controls kinetochore-microtubule attachments by inhibiting Ska complex-KMN network interaction. J Cell Biol 196(5):563–571. doi:10.1083/jcb.201109001
Chao WCH, Kulkarni K, Zhang Z, Kong EH, Barford D (2012) Structure of the mitotic checkpoint complex. Nature. doi:10.1038/nature10896
Cheeseman IM, Niessen S, Anderson S, Hyndman F, Yates JR 3rd, Oegema K, Desai A (2004) A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev 18(18):2255–2268
Chen RH, Waters JC, Salmon ED, Murray AW (1996) Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 274(5285):242–246
Chen RH, Shevchenko A, Mann M, Murray AW (1998) Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J Cell Biol 143(2):283–295
Chen Q, Zhang X, Jiang Q, Clarke PR, Zhang C (2008) Cyclin B1 is localized to unattached kinetochores and contributes to efficient microtubule attachment and proper chromosome alignment during mitosis. Cell Research 18(2):268–280. doi:10.1038/cr.2008.11
Ciliberto A, Shah JV (2009) A quantitative systems view of the spindle assembly checkpoint. EMBO J 28(15):2162–2173
Clute P, Pines J (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol 1(2):82–87
Daum JR, Wren JD, Daniel JJ, Sivakumar S, McAvoy JN, Potapova TA, Gorbsky GJ (2009) Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Cur Biol: CB 19(17):1467–1472. doi:10.1016/j.cub.2009.07.017
De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, Musacchio A (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15(3):214–225
DeLuca KF, Lens SMA, Deluca JG (2011) Temporal changes in Hec1 phosphorylation control kinetochore-microtubule attachment stability during mitosis. J Cell Sci 124(Pt 4):622–634. doi:10.1242/jcs.072629
Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS (2003) Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol 161(2):267–280
Dunsch AK, Linnane E, Barr FA, Gruneberg U (2011) The astrin-kinastrin/SKAP complex localizes to microtubule plus ends and facilitates chromosome alignment. J Cell Biol 192(6):959–968. doi:10.1083/jcb.201008023
Emanuele MJ, Lan W, Jwa M, Miller SA, Chan CSM, Stukenberg PT (2008) Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. J Cell Biol 181(2):241–254. doi:10.1083/jcb.200710019
Emre D, Terracol R, Poncet A, Rahmani Z, Karess RE (2011) A mitotic role for Mad1 beyond the spindle checkpoint. J Cell Sci 124(Pt 10):1664–1671. doi:10.1242/jcs.081216
Espeut J, Cheerambathur DK, Krenning L, Oegema K, Desai A (2012) Microtubule binding by KNL-1 contributes to spindle checkpoint silencing at the kinetochore. J Cell Biol. doi:10.1083/jcb.201111107
Famulski JK, Chan GK (2007) Aurora B kinase-dependent recruitment of hZW10 and hROD to tensionless kinetochores. Curr Biol 17(24):2143–2149. doi:10.1016/j.cub.2007.11.037
Famulski JK, Vos LJ, Rattner JB, Chan GK (2011) Dynein/dynactin-mediated transport of kinetochore components off kinetochores and onto spindle poles induced by nordihydroguaiaretic acid. PLoS One 6(1):e16494. doi:10.1371/journal.pone.0016494
Fava LL, Kaulich M, Nigg EA, Santamaria A (2011) Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J. doi:10.1038/emboj.2011.239
Foley EA, Maldonado M, Kapoor TM (2011) Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol 13(10):1265–1271. doi:10.1038/ncb2327
Francisco L, Wang W, Chan CS (1994) Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol 14(7):4731–4740
Gaglio T, Saredi A, Compton DA (1995) NuMA is required for the organization of microtubules into aster-like mitotic arrays. J Cell Biol 131(3):693–708
Gassmann R, Essex A, Hu J-S, Maddox PS, Motegi F, Sugimoto A, O'Rourke SM, Bowerman B, McLeod I, Yates JR, Oegema K, Cheeseman IM, Desai A (2008) A new mechanism controlling kinetochore-microtubule interactions revealed by comparison of two dynein-targeting components: SPDL-1 and the Rod/Zwilch/Zw10 complex. Genes Dev 22(17):2385–2399. doi:10.1101/gad.1687508
Gassmann R, Holland AJ, Varma D, Wan X, Civril F, Cleveland DW, Oegema K, Salmon ED, Desai A (2010) Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev 24(9):957–971. doi:10.1101/gad.1886810
Giet R, Glover DM (2001) Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol 152(4):669–682
Griffis ER, Stuurman N, Vale RD (2007) Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J Cell Biol 177(6):1005–1015. doi:10.1083/jcb.200702062
Habu T, Kim SH, Weinstein J, Matsumoto T (2002) Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J 21(23):6419–6428
Hagan RS, Manak MS, Buch HK, Meier MG, Meraldi P, Shah JV, Sorger PK (2011) p31(comet) acts to ensure timely spindle checkpoint silencing subsequent to kinetochore attachment. Mol Biol Cell 22(22):4236–4246. doi:10.1091/mbc.E11-03-0216
Hanisch A, Silljé HHW, Nigg EA (2006) Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J 25(23):5504–5515. doi:10.1038/sj.emboj.7601426
Hardwick KG, Shah JV (2010) Spindle checkpoint silencing: ensuring rapid and concerted anaphase onset. F1000 biology reports 2:55. doi:10.3410/B2-55
Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW (1996) Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science 273(5277):953–956
Hardwick KG, Johnston RC, Smith DL, Murray AW (2000) MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol 148(5):871–882
Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM (2003) The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 161(2):281–294
Hegemann B, Hutchins JRA, Hudecz O, Novatchkova M, Rameseder J, Sykora MM, Liu S, Mazanek M, Lénárt P, Hériché J-K, Poser I, Kraut N, Hyman AA, Yaffe MB, Mechtler K, Peters J-M (2011) Systematic phosphorylation analysis of human mitotic protein complexes. Sci Signal 4(198):rs12. doi:10.1126/scisignal.2001993
Hengeveld RCC, Hertz NT, Vromans MJM, Zhang C, Burlingame AL, Shokat KM, Lens SMA (2012) Development of a chemical genetic approach for human Aurora B kinase identifies novel substrates of the chromosomal passenger complex. Molecular & Cellular Proteomics. doi:10.1074/mcp.M111.013912
Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S, Taylor SS (2010) Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol 190(1):25–34. doi:10.1083/jcb.201002133
Holt SV, Vergnolle MAS, Hussein D, Wozniak MJ, Allan VJ, Taylor SS (2005) Silencing Cenp-F weakens centromeric cohesion, prevents chromosome alignment and activates the spindle checkpoint. J Cell Sci 118(Pt 20):4889–4900. doi:10.1242/jcs.02614
Hori T, Haraguchi T, Hiraoka Y, Kimura H, Fukagawa T (2003) Dynamic behavior of Nuf2-Hec1 complex that localizes to the centrosome and centromere and is essential for mitotic progression in vertebrate cells. J Cell Sci 116(Pt 16):3347–3362
Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED (2000) Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol 150(6):1233–1250
Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED (2001) Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol 155(7):1159–1172. doi:10.1083/jcb.200105093
Howell BJ, Moree B, Farrar EM, Stewart S, Fang G, Salmon ED (2004) Spindle checkpoint protein dynamics at kinetochores in living cells. Curr Biol 14(11):953–964
Hoyt MA, Totis L, Roberts BT (1991) S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66(3):507–517
Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, Lin R, Smith MM, Allis CD (2000) Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 102(3):279–291
Jablonski SA, Chan GK, Cooke CA, Earnshaw WC, Yen TJ (1998) The hBUB1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma 107(6–7):386–396
Jelluma N, Brenkman AB, van den Broek NJF, Cruijsen CWA, van Osch MHJ, Lens SMA, Medema RH, Kops GJPL (2008) Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 132(2):233–246. doi:10.1016/j.cell.2007.11.046
Jelluma N, Dansen TB, Sliedrecht T, Kwiatkowski NP, Kops GJPL (2010) Release of Mps1 from kinetochores is crucial for timely anaphase onset. J Cell Biol 191(2):281–290. doi:10.1083/jcb.201003038
Jia L, Li B, Warrington RT, Hao X, Wang S, Yu H (2011) Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31(comet). Mol Biol Cell 22(22):4227–4235. doi:10.1091/mbc.E11-05-0389
Johnson VL, Scott MIF, Holt SV, Hussein D, Taylor SS (2004) Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J Cell Sci 117(Pt 8):1577–1589. doi:10.1242/jcs.01006
Kallio MJ, Beardmore VA, Weinstein J, Gorbsky GJ (2002) Rapid microtubule-independent dynamics of Cdc20 at kinetochores and centrosomes in mammalian cells. J Cell Biol 158(5):841–847
Kang YH, Park J-E, Yu L-R, Soung N-K, Yun S-M, Bang JK, Seong Y-S, Yu H, Garfield S, Veenstra TD, Lee KS (2006) Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol Cell 24(3):409–422. doi:10.1016/j.molcel.2006.10.016
Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ (2000) Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol 150(5):975–988
Kasuboski JM, Bader JR, Vaughan PS, Tauhata SBF, Winding M, Morrissey MA, Joyce MV, Boggess W, Vos L, Chan GK, Hinchcliffe EH, Vaughan KT (2011) Zwint-1 is a novel Aurora B substrate required for the assembly of a dynein-binding platform on kinetochores. Mol Biol Cell 22(18):3318–3330. doi:10.1091/mbc.E11-03-0213
Kelly AE, Funabiki H (2009) Correcting aberrant kinetochore microtubule attachments: an Aurora B-centric view. Curr Opin Cell Biol 21(1):51–58. doi:10.1016/j.ceb.2009.01.004
Kemmler S, Stach M, Knapp M, Ortiz J, Pfannstiel J, Ruppert T, Lechner J (2009) Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. EMBO J 28(8):1099–1110. doi:10.1038/emboj.2009.62
Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA (2011) Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 4(179):rs5. doi:10.1126/scisignal.2001497
Khodjakov A, Pines J (2010) Centromere tension: a divisive issue. Nat Cell Biol 12(10):919–923. doi:10.1038/ncb1010-919
Kim S, Yu H (2011) Mutual regulation between the spindle checkpoint and APC/C. Semin Cell Dev Biol 22(6):551–558. doi:10.1016/j.semcdb.2011.03.008
King JM, Hays TS, Nicklas RB (2000) Dynein is a transient kinetochore component whose binding is regulated by microtubule attachment, not tension. J Cell Biol 151(4):739–748
King EMJ, Rachidi N, Morrice N, Hardwick KG, Stark MJR (2007) Ipl1p-dependent phosphorylation of Mad3p is required for the spindle checkpoint response to lack of tension at kinetochores. Genes Dev 21(10):1163–1168. doi:10.1101/gad.431507
Kiyomitsu T, Obuse C, Yanagida M (2007) Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev Cell 13(5):663–676. doi:10.1016/j.devcel.2007.09.005
Kiyomitsu T, Murakami H, Yanagida M (2011) Protein interaction domain mapping of human kinetochore protein Blinkin reveals a consensus motif for binding of spindle assembly checkpoint proteins Bub1 and BubR1. Mol Cell Biol 31(5):998–1011. doi:10.1128/MCB.00815-10
Klebig C, Korinth D, Meraldi P (2009) Bub1 regulates chromosome segregation in a kinetochore-independent manner. J Cell Biol 185(5):841–858. doi:10.1083/jcb.200902128
Koch A, Krug K, Pengelley S, Macek B, Hauf S (2011) Mitotic substrates of the kinase aurora with roles in chromatin regulation identified through quantitative phosphoproteomics of fission yeast. Sci Signal 4(179):rs6. doi:10.1126/scisignal.2001588
Kops GJPL, Kim Y, Weaver BAA, Mao Y, McLeod I, Yates JR, Tagaya M, Cleveland DW (2005) ZW10 links mitotic checkpoint signaling to the structural kinetochore. J Cell Biol 169(1):49–60. doi:10.1083/jcb.200411118
Kops GJPL, van der Voet M, Manak MS, van Osch MHJ, Naini SM, Brear A, McLeod IX, Hentschel DM, Yates JR, Van Den Heuvel S, Shah JV (2010) APC16 is a conserved subunit of the anaphase-promoting complex/cyclosome. J Cell Sci 123(Pt 10):1623–1633. doi:10.1242/jcs.061549
Krenn V, Wehenkel A, Li X, Santaguida S, Musacchio A (2012) Structural analysis reveals features of the spindle checkpoint kinase Bub1-kinetochore subunit Knl1 interaction. The Journal of Cell Biology. doi:10.1083/jcb.201110013
Kulukian A, Han J, Cleveland D (2009) Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell 16(1):105–117. doi:10.1016/j.devcel.2008.11.005
Kwiatkowski N, Jelluma N, Filippakopoulos P, Soundararajan M, Manak MS, Kwon M, Choi HG, Sim T, Deveraux QL, Rottmann S, Pellman D, Shah JV, Kops GJPL, Knapp S, Gray NS (2010) Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat Chem Biol 6(5):359–368. doi:10.1038/nchembio.345
Lampson MA, Cheeseman IM (2011) Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol 21(3):133–140. doi:10.1016/j.tcb.2010.10.007
Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM (2004) Correcting improper chromosome-spindle attachments during cell division. Nat Cell Biol 6(3):232–237
Lara-Gonzalez P, Scott MIF, Diez M, Sen O, Taylor SS (2011) BubR1 blocks substrate recruitment to the APC/C in a KEN-box-dependent manner. J Cell Sci 124(Pt 24):4332–4345. doi:10.1242/jcs.094763
Li R, Murray AW (1991) Feedback control of mitosis in budding yeast. Cell 66(3):519–531
Li J, Lee W-L, Cooper JA (2005) NudEL targets dynein to microtubule ends through LIS1. Nat Cell Biol 7(7):686–690. doi:10.1038/ncb1273
Li Y, Yu W, Liang Y, Zhu X (2007) Kinetochore dynein generates a poleward pulling force to facilitate congression and full chromosome alignment. Cell Res 17(8):701–712. doi:10.1038/cr.2007.65
Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ (1995) CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol 130(3):507–518
Liu D, Vader G, Vromans MJM, Lampson MA, Lens SMA (2009) Sensing chromosome biorientation by spatial separation of aurora B kinase from kinetochore substrates. Sci (New York, NY) 323(5919):1350–1353. doi:10.1126/science.1167000
Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, Lampson MA (2010) Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J Cell Biol 188(6):809–820. doi:10.1083/jcb.201001006
Logarinho E, Resende T, Torres C, Bousbaa H (2008) The human spindle assembly checkpoint protein Bub3 is required for the establishment of efficient kinetochore-microtubule attachments. Mol Biol Cell 19(4):1798–1813. doi:10.1091/mbc.E07-07-0633
London N, Ceto S, Ranish JA, Biggins S (2012) Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr Biol: CB 22(10):900–906. doi:10.1016/j.cub.2012.03.052
Luo X, Yu H (2008) Protein metamorphosis: the two-state behavior of Mad2. Struct/Fold Des 16(11):1616–1625. doi:10.1016/j.str.2008.10.002
Luo X, Tang Z, Rizo J, Yu H (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell 9(1):59–71
Maciejowski J, George KA, Terret M-E, Zhang C, Shokat KM, Jallepalli PV (2010) Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J Cell Biol 190(1):89–100. doi:10.1083/jcb.201001050
Mack GJ, Compton DA (2001) Analysis of mitotic microtubule-associated proteins using mass spectrometry identifies astrin, a spindle-associated protein. Proc Natl Acad Sci U S A 98(25):14434–14439. doi:10.1073/pnas.261371298
Maiato H, Fairley EAL, Rieder CL, Swedlow JR, Sunkel CE, Earnshaw WC (2003) Human CLASP1 is an outer kinetochore component that regulates spindle microtubule dynamics. Cell 113(7):891–904
Maldonado M, Kapoor TM (2011) Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat Cell Biol. doi:doi:10.1038/ncb2223
Manning AL, Bakhoum SF, Maffini S, Correia-Melo C, Maiato H, Compton DA (2010) CLASP1, astrin and Kif2b form a molecular switch that regulates kinetochore-microtubule dynamics to promote mitotic progression and fidelity. EMBO J 29(20):3531–3543. doi:10.1038/emboj.2010.230
Mapelli M, Musacchio A (2007) MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr Opin Struct Biol 17(6):716–725. doi:10.1016/j.sbi.2007.08.011
Mapelli M, Filipp FV, Rancati G, Massimiliano L, Nezi L, Stier G, Hagan RS, Confalonieri S, Piatti S, Sattler M, Musacchio A (2006) Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J 25(6):1273–1284
Maresca TJ, Salmon ED (2009) Intrakinetochore stretch is associated with changes in kinetochore phosphorylation and spindle assembly checkpoint activity. J Cell Biol 184(3):373–381. doi:10.1083/jcb.200808130
Maresca TJ, Salmon ED (2010) Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. J Cell Sci 123(Pt 6):825–835. doi:10.1242/jcs.064790
Martin-Lluesma S, Stucke VM, Nigg EA (2002) Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science 297(5590):2267–2270
Matson DR, Demirel PB, Stukenberg PT, Burke DJ (2012) A conserved role for COMA/CENP-H/I/N kinetochore proteins in the spindle checkpoint. Genes Dev 26(6):542–547. doi:10.1101/gad.184184.111
McCleland ML, Gardner RD, Kallio MJ, Daum JR, Gorbsky GJ, Burke DJ, Stukenberg PT (2003) The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev 17(1):101–114. doi:10.1101/gad.1040903
McIntosh JR (1991) Structural and mechanical control of mitotic progression. Cold Spring Harb Symp Quant Biol 56:613–619
Meadows JC, Shepperd LA, Vanoosthuyse V, Lancaster TC, Sochaj AM, Buttrick GJ, Hardwick KG, Millar JBA (2011) Spindle checkpoint silencing requires association of PP1 to both Spc7 and kinesin-8 motors. Dev Cell 20(6):739–750. doi:10.1016/j.devcel.2011.05.008
Meraldi P, Sorger PK (2005) A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J 24(8):1621–1633
Miniowitz-Shemtov S, Eytan E, Ganoth D, Sitry-Shevah D, Dumin E, Hershko A (2012) Role of phosphorylation of Cdc20 in p31comet-stimulated disassembly of the mitotic checkpoint complex. Proc Natl Acad Sci U S A. doi:10.1073/pnas.1204081109
Morrow CJ, Tighe A, Johnson VL, Scott MI, Ditchfield C, Taylor SS (2005) Bub1 and Aurora B cooperate to maintain BubR1-mediated inhibition of APC/CCdc20. J Cell Sci 118(Pt 16):3639–3652
Murata-Hori M, Tatsuka M, Wang Y-L (2002) Probing the dynamics and functions of aurora B kinase in living cells during mitosis and cytokinesis. Mol Biol Cell 13(4):1099–1108. doi:10.1091/mbc.01-09-0467
Murnion ME, Adams RR, Callister DM, Allis CD, Earnshaw WC, Swedlow JR (2001) Chromatin-associated protein phosphatase 1 regulates aurora-B and histone H3 phosphorylation. J Biol Chem 276(28):26656–26665. doi:10.1074/jbc.M102288200
Musacchio A, Salmon ED (2007) The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8(5):379–393
Nezi L, Musacchio A (2009) Sister chromatid tension and the spindle assembly checkpoint. Curr Opin Cell Biol 21(6):785–795. doi:10.1016/j.ceb.2009.09.007
Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH (2000) NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28(3):697–711
Nishihashi A, Haraguchi T, Hiraoka Y, Ikemura T, Regnier V, Dodson H, Earnshaw WC, Fukagawa T (2002) CENP-I is essential for centromere function in vertebrate cells. Dev Cell 2(4):463–476
Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M (2004) A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biolo 6(11):1135–1141. doi:10.1038/ncb1187
Perera D, Taylor SS (2010) Sgo1 establishes the centromeric cohesion protection mechanism in G2 before subsequent Bub1-dependent recruitment in mitosis. J Cell Sci 123(5):653–659. doi:10.1242/jcs.059501
Petersen J, Hagan IM (2003) S. pombe aurora kinase/survivin is required for chromosome condensation and the spindle checkpoint attachment response. Curr Biol 13(7):590–597
Pines J (2011) Cubism and the cell cycle: the many faces of the APC/C. Nature Reviews Molecular Cell Biology. doi:10.1038/nrm3132
Pinsky BA, Kotwaliwale CV, Tatsutani SY, Breed CA, Biggins S (2006a) Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol Cell Biol 26(7):2648–2660. doi:10.1128/MCB.26.7.2648-2660.2006
Pinsky BA, Kung C, Shokat KM, Biggins S (2006b) The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol 8(1):78–83. doi:10.1038/ncb1341
Pinsky BA, Nelson CR, Biggins S (2009) Protein phosphatase 1 regulates exit from the spindle checkpoint in budding yeast. Curr Biol 19(14):1182–1187. doi:10.1016/j.cub.2009.06.043
Posch M, Khoudoli GA, Swift S, King EM, Deluca JG, Swedlow JR (2010) Sds22 regulates aurora B activity and microtubule–kinetochore interactions at mitosis. J Cell Biol 191(1):61–74. doi:10.1083/jcb.200912046
Raaijmakers JA, Tanenbaum ME, Maia AF, Medema RH (2009) RAMA1 is a novel kinetochore protein involved in kinetochore-microtubule attachment. J Cell Sci 122(Pt 14):2436–2445. doi:10.1242/jcs.051912
Rancati G, Crispo V, Lucchini G, Piatti S (2005) Mad3/BubR1 phosphorylation during spindle checkpoint activation depends on both Polo and Aurora kinases in budding yeast. Cell cycle (Georgetown, Tex) 4(7):972–980
Reddy SK, Rape M, Margansky WA, Kirschner MW (2007) Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature 446(7138):921–925. doi:10.1038/nature05734
Rieder CL, Schultz A, Cole R, Sluder G (1994) Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol 127(5):1301–1310
Rieder CL, Cole RW, Khodjakov A, Sluder G (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol 130(4):941–948
Rosenberg JS, Cross FR, Funabiki H (2011) KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr Biol: CB 21(11):942–947. doi:10.1016/j.cub.2011.04.011
Ruchaud S, Carmena M, Earnshaw WC (2007) Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol 8(10):798–812. doi:10.1038/nrm2257
Salimian KJ, Ballister ER, Smoak EM, Wood S, Panchenko T, Lampson MA, Black BE (2011) Feedback control in sensing chromosome biorientation by the Aurora B kinase. Current biology: CB 21(13):1158–1165. doi:10.1016/j.cub.2011.06.015
Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A (2010) Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 190(1):73–87. doi:10.1083/jcb.201001036
Santaguida S, Vernieri C, Villa F, Ciliberto A, Musacchio A (2011) Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J 30(8):1508–1519. doi:10.1038/emboj.2011.70
Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S (2000) A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 28(3):681–696. doi:10.1016/S0896-6273(00)00146-X
Saurin AT, van der Waal MS, Medema RH, Lens SMA, Kops GJPL (2011) Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat Commun 2:316. doi:10.1038/ncomms1319
Scaërou F, Starr DA, Piano F, Papoulas O, Karess RE, Goldberg ML (2001) The ZW10 and Rough Deal checkpoint proteins function together in a large, evolutionarily conserved complex targeted to the kinetochore. J Cell Sci 114(Pt 17):3103–3114
Schmidt JC, Kiyomitsu T, Hori T, Backer CB, Fukagawa T, Cheeseman IM (2010) Aurora B kinase controls the targeting of the Astrin-SKAP complex to bioriented kinetochores. J Cell Biol 191(2):269–280. doi:10.1083/jcb.201006129
Shah JV, Botvinick E, Bonday Z, Furnari F, Berns M, Cleveland DW (2004) Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol 14(11):942–952
Shepperd LA, Meadows JC, Sochaj AM, Lancaster TC, Zou J, Buttrick GJ, Rappsilber J, Hardwick KG, Millar JBA (2012) Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr Biol: CB 22(10):891–899. doi:10.1016/j.cub.2012.03.051
Simonetta M, Manzoni R, Mosca R, Mapelli M, Massimiliano L, Vink M, Novak B, Musacchio A, Ciliberto A (2009) The influence of catalysis on Mad2 activation dynamics. Plos Biol 7(1):e10. doi:10.1371/journal.pbio.1000010
Skoufias DA, Andreassen PR, Lacroix FB, Wilson L, Margolis RL (2001) Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc Natl Acad Sci U S A 98(8):4492–4497
Sliedrecht T, Zhang C, Shokat KM, Kops GJPL (2010) Chemical genetic inhibition of Mps1 in stable human cell lines reveals novel aspects of Mps1 function in mitosis. PLoS One 5(4):e10251. doi:10.1371/journal.pone.0010251
Starr DA, Williams BC, Hays TS, Goldberg ML (1998) ZW10 helps recruit dynactin and dynein to the kinetochore. J Cell Biol 142(3):763–774
Stegmeier F, Rape M, Draviam VM, Nalepa G, Sowa ME, Ang XL, McDonald ER, Li MZ, Hannon GJ, Sorger PK, Kirschner MW, Harper JW, Elledge SJ (2007) Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature 446(7138):876–881. doi:10.1038/nature05694
Stehman SA, Chen Y, McKenney RJ, Vallee RB (2007) NudE and NudEL are required for mitotic progression and are involved in dynein recruitment to kinetochores. J Cell Biol 178(4):583–594. doi:10.1083/jcb.200610112
Stucke VM, Baumann C, Nigg EA (2004) Kinetochore localization and microtubule interaction of the human spindle checkpoint kinase Mps1. Chromosoma 113(1):1–15
Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154(5):925–936
Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJR, Nasmyth K (2002) Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome biorientation by altering kinetochore-spindle pole connections. Cell 108(3):317–329
Tanenbaum ME, Macůrek L, Galjart N, Medema RH (2008) Dynein, Lis1 and CLIP-170 counteract Eg5-dependent centrosome separation during bipolar spindle assembly. EMBO J 27(24):3235–3245. doi:10.1038/emboj.2008.242
Tang Z, Shu H, Oncel D, Chen S, Yu H (2004) Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell 16(3):387–397
Taylor SS, Ha E, McKeon F (1998) The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J Cell Biol 142(1):1–11
Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ (2001) Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci 114(Pt 24):4385–4395
Teichner A, Eytan E, Sitry-Shevah D, Miniowitz-Shemtov S, Dumin E, Gromis J, Hershko A (2011) p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proceedings of the National Academy of Sciences:1-6. doi:10.1073/pnas.1100023108
Theis M, Slabicki M, Junqueira M, Paszkowski-Rogacz M, Sontheimer J, Kittler R, Heninger A-K, Glatter T, Kruusmaa K, Poser I, Hyman AA, Pisabarro MT, Gstaiger M, Aebersold R, Shevchenko A, Buchholz F (2009) Comparative profiling identifies C13orf3 as a component of the Ska complex required for mammalian cell division. EMBO J 28(10):1453–1465. doi:10.1038/emboj.2009.114
Tirnauer JS, Canman JC, Salmon ED, Mitchison TJ (2002) EB1 targets to kinetochores with attached, polymerizing microtubules. Mol Biol Cell 13(12):4308–4316. doi:10.1091/mbc.E02-04-0236
Trinkle-Mulcahy L, Andrews PD, Wickramasinghe S, Sleeman J, Prescott A, Lam YW, Lyon C, Swedlow JR, Lamond AI (2003) Time-lapse imaging reveals dynamic relocalization of PP1gamma throughout the mammalian cell cycle. Mol Biol Cell 14(1):107–117. doi:10.1091/mbc.E02-07-0376
Uchida KSK, Takagaki K, Kumada K, Hirayama Y, Noda T, Hirota T (2009) Kinetochore stretching inactivates the spindle assembly checkpoint. J Cell Biol 184(3):383–390. doi:10.1083/jcb.200811028
Vanoosthuyse V, Hardwick KG (2009) A novel protein phosphatase 1-dependent spindle checkpoint silencing mechanism. Curr Biol 19(14):1176–1181. doi:10.1016/j.cub.2009.05.060
Varetti G, Guida C, Santaguida S, Chiroli E, Musacchio A (2011) Homeostatic control of mitotic arrest. Mol Cell 44(5):710–720. doi:10.1016/j.molcel.2011.11.014
Vergnolle MAS, Taylor SS (2007) Cenp-F links kinetochores to Ndel1/Nde1/Lis1/dynein microtubule motor complexes. Curr Biol 17(13):1173–1179. doi:10.1016/j.cub.2007.05.077
Vigneron S, Prieto S, Bernis C, Labbe JC, Castro A, Lorca T (2004) Kinetochore localization of spindle checkpoint proteins: who controls whom? Mol Biol Cell 15(10):4584–4596
Vink M, Simonetta M, Transidico P, Ferrari K, Mapelli M, De Antoni A, Massimiliano L, Ciliberto A, Faretta M, Salmon ED, Musacchio A (2006) In vitro FRAP identifies the minimal requirements for Mad2 kinetochore dynamics. Curr Biol 16(8):755–766
Vorozhko VV, Emanuele MJ, Kallio MJ, Stukenberg PT, Gorbsky GJ (2008) Multiple mechanisms of chromosome movement in vertebrate cells mediated through the Ndc80 complex and dynein/dynactin. Chromosoma 117(2):169–179. doi:10.1007/s00412-007-0135-3
Wan X, O'Quinn RP, Pierce HL, Joglekar AP, Gall WE, Deluca JG, Carroll CW, Liu ST, Yen TJ, McEwen BF, Stukenberg PT, Desai A, Salmon ED (2009) Protein architecture of the human kinetochore microtubule attachment site. Cell 137(4):672–684. doi:10.1016/j.cell.2009.03.035
Warren CD, Brady DM, Johnston RC, Hanna JS, Hardwick KG, Spencer FA (2002) Distinct chromosome segregation roles for spindle checkpoint proteins. Mol Biol Cell 13(9):3029–3041. doi:10.1091/mbc.E02-04-0203
Waters JC, Chen RH, Murray AW, Salmon ED (1998) Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol 141(5):1181–1191
Weiss E, Winey M (1996) The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol 132(1–2):111–123
Welburn JPI, Vleugel M, Liu D, Yates JR, Lampson MA, Fukagawa T, Cheeseman IM (2010) Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell 38(3):383–392. doi:10.1016/j.molcel.2010.02.034
Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS (2011) p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. Journal of Cell Science. doi:10.1242/jcs.093286
Whyte J, Bader JR, Tauhata SBF, Raycroft M, Hornick J, Pfister KK, Lane WS, Chan GK, Hinchcliffe EH, Vaughan PS, Vaughan KT (2008) Phosphorylation regulates targeting of cytoplasmic dynein to kinetochores during mitosis. J Cell Biol 183(5):819–834. doi:10.1083/jcb.200804114
Williams BC, Gatti M, Goldberg ML (1996) Bipolar spindle attachments affect redistributions of ZW10, a Drosophila centromere/kinetochore component required for accurate chromosome segregation. J Cell Biol 134(5):1127–1140
Williams BC, Li Z, Liu S, Williams EV, Leung G, Yen TJ, Goldberg ML (2003) Zwilch, a new component of the ZW10/ROD complex required for kinetochore functions. Mol Biol Cell 14(4):1379–1391. doi:10.1091/mbc.E02-09-0624
Wojcik E, Basto R, Serr M, Scaërou F, Karess R, Hays T (2001) Kinetochore dynein: its dynamics and role in the transport of the rough deal checkpoint protein. Nat Cell Biol 3(11):1001–1007. doi:10.1038/ncb1101-1001
Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H (2004) Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J 23(15):3133–3143
Yamaguchi S, Decottignies A, Nurse P (2003) Function of Cdc2p-dependent Bub1p phosphorylation and Bub1p kinase activity in the mitotic and meiotic spindle checkpoint. EMBO J 22(5):1075–1087. doi:10.1093/emboj/cdg100
Yamamoto TG, Watanabe S, Essex A, Kitagawa R (2008) SPDL-1 functions as a kinetochore receptor for MDF-1 in Caenorhabditis elegans. J Cell Biol 183(2):187–194. doi:10.1083/jcb.200805185
Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X (2007a) p31(comet) blocks Mad2 activation through structural mimicry. Cell 131(4):744–755. doi:10.1016/j.cell.2007.08.048
Yang Z, Tulu US, Wadsworth P, Rieder CL (2007b) Kinetochore dynein is required for chromosome motion and congression independent of the spindle checkpoint. Curr Biol 17(11):973–980. doi:10.1016/j.cub.2007.04.056
Yasui Y, Urano T, Kawajiri A, K-i N, Tatsuka M, Saya H, Furukawa K, Takahashi T, Izawa I, Inagaki M (2004) Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J Biol Chem 279(13):12997–13003. doi:10.1074/jbc.M311128200
Yen TJ, Compton DA, Wise D, Zinkowski RP, Brinkley BR, Earnshaw WC, Cleveland DW (1991) CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J 10(5):1245–1254
Zirkle RE (1970) Ultraviolet-microbeam irradiation of newt-cell cytoplasm: spindle destruction, false anaphase, and delay of true anaphase. Radiat Res 41(3):516–537
Acknowledgements
We apologize to the authors whose work was not discussed or cited owing to space limitations. We thank Suzanne Lens, Patrick Meraldi, Michael Lampson and the members of the Kops and Shah laboratories for their insightful comments. Work in the Kops laboratory is supported by the Dutch Cancer Society, by the Netherlands Organization for Scientific Research (NWO), and by the European Research Council (ERC). Work in the Shah laboratory is supported by the US National Institutes of Health (NIH) and the Foundation of the Beckman Laser Institute (BLI).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Erich Nigg
Rights and permissions
About this article
Cite this article
Kops, G.J.P.L., Shah, J.V. Connecting up and clearing out: how kinetochore attachment silences the spindle assembly checkpoint. Chromosoma 121, 509–525 (2012). https://doi.org/10.1007/s00412-012-0378-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-012-0378-5