
Abstract
Alterations in the nuclear positioning of chromosomes and specific genes during differentiation and development have suggested strongly the existence of a relationship between non-random organization of the genome and its function. In this study, we have examined the genome organization in interphase nuclei during adipogenesis, using the pig as a model organism. We hypothesized that changes in the gene expression profile and chromatin remodeling which occur during cellular differentiation would elicit repositioning of whole chromosomes, moving specific genes on them to different regions of the nucleus. We established an in vitro adipogenesis differentiation system using mesenchymal stem cells, derived from porcine bone marrow. The nuclear position of seven adipogenesis genes (PPARG, SREBF1, FABP4, CEBPA, CEBPB, CREB, and GATA2), two control genes (SOX9 and MYL1), and six chromosomes carrying these gene loci (SSC4, SSC6, SSC12, SSC13, SSC15, and SSC17) was determined. We found that during adipogenesis, using the in vitro stem cell model system, in contrast to our original hypothesis, the nuclear position of genes involved in adipogenesis was altered radically with the up-regulation of gene expression correlating with these genes becoming more internally located within nuclei. Chromosome territories, containing these genes, were also found to alter their nuclear position during the in vitro adipogenesis model, with the most dramatic repositioning being SSC4 that moved from the nuclear periphery towards the nuclear interior. We found that during in vitro adipogenesis chromosome territories decondensed and the genes were found on loops and projections of chromatin, away from the main body of the chromosomes. From our data, it appears that the temporal repositioning of genes, emanating away from chromosomes, during adipogenesis is correlated with gene activity, supporting models of the involvement of spatial genome repositioning in regulating gene expression and the nuclear interior being an important region of the nucleus for transcription.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genome positioning within the interphase nuclei is non-random, with each chromosome territory occupying a specific radial location (Boyle et al. 2001; Cremer et al. 2006; Meaburn and Misteli 2007). Importantly, for the field of genome biology, the arrangements of chromosomes within the nuclei are being well characterized in a number of different species and cell types (Foster and Bridger 2005). Sub-chromosomal regions and individual genes also display preferential nuclear positions. Recent studies have demonstrated that chromatin domains are organized in nuclei according to local GC content (Hepperger et al. 2008; Saccone et al. 2002), local gene density (Federico et al. 2008; Goetze et al. 2007; Küpper et al. 2007), or replication timing (Grasser et al. 2008) rather than by gene expression level. However, emerging evidence indicates that placing genes in specific nuclear space has a regulatory effect (Hu et al. 2008; Lanctôt et al. 2007; Misteli 2007; Takizawa et al. 2008b). For example, positioning of loci relative to the nuclear periphery or heterochromatin domains has been linked to gene repression (Brown et al. 1999; Finlan et al. 2008; Reddy et al. 2008; Somech et al. 2005), and conversely, repositioning of loci from nuclear periphery to nuclear interior or away from heterochromatin domains is correlated with gene activation (Ballabio et al. 2009; Brown et al. 2001; Kosak et al. 2002; Williams et al. 2006; Zink et al. 2004). Formation of chromatin loops away from chromosome territories has been suggested as a mechanism of genome regulation (Chambeyron and Bickmore 2004; Chambeyron et al. 2005; Volpi et al. 2000; Williams et al. 2002), but genes looping out from chromosome territories may not always be linked to up-regulation of gene expression (Morey et al. 2009). Some genes can migrate to share the same nuclear sub-compartments or transcription factories (Osborne et al. 2004, 2007; Xu and Cook 2008). Recently, it has been shown that distinct alleles of monoallelically expressed genes occupied different nuclear positions and can be taken as direct evidence for correlation between nuclear position and activity status of a gene (Takizawa et al. 2008a).
Several important findings concerning spatial organization of the genome in relation to function have been made during cellular differentiation and development. Among many examples, nuclear organization has been studied during lymphoid differentiation (Brown et al. 1999, 2001; Kosak et al. 2002), erythroid differentiation (Brown et al. 2001, 2008; Ragoczy et al. 2006), epidermal differentiation (Williams et al. 2002), neuronal differentiation (Williams et al. 2006), granulocyte differentiation (Bártová et al. 2002), adipocyte differentiation (Kuroda et al. 2004), embryonic development (Chambeyron and Bickmore 2004; Chambeyron et al. 2005; Morey et al. 2007), and spermatogenesis (Foster et al. 2005; Zalenskaya and Zalensky 2004). Differentiation can be characterized by dynamic changes in cellular morphology, the gene expression profile, and chromatin remodeling, and therefore can be considered as an ideal system for studies correlating nuclear position and gene expression. In vitro differentiation systems using progenitor cells (embryonic or adult stem cells) can be used for understanding epigenetic processes regulating cell lineage commitment.
In this study, we established an in vitro system of adipogenesis differentiation for porcine mesenchymal stem cells. Adipogenesis is a complex process composed of two phases known as determination and terminal differentiation. In the first phase, the mesenchymal precursor cells commit to an adipocyte lineage and traverse to preadipocyte cells. In the second phase, preadipocytes take on characteristics of mature adipocytes (Rosen and MacDougald 2006). Formation of fat cells is regulated by a complex network of transcription factors among which the most important are peroxisome proliferator-activated receptor γ and members of CCAAT/enhancer-binding proteins (C/EBP) family (Farmer 2006). Adipogenesis has been studied extensively in vitro using mainly established mouse cell lines (3T3-L1, 3T3-F442A); however, there are a number of reports on isolating mesenchymal stem cells derived from bone marrow and differentiating them into adipocytes for different species, including pig (Zeng et al. 2006; Zou et al. 2008). Different aspects of adipogenesis have been investigated extensively since excess of adipose mass, i.e., obesity is one of the major health problems, contributing to pathogenesis of obesity-related disorders such as diabetes, hypertension, and coronary heart disease (Formiguera and Cantón 2004). The pig is considered as a model organism for the study of human obesity (Lunney 2007; Schook et al. 2005). Additionally, this species has a central position in agricultural sciences, and its whole genome is now being sequenced and is soon to be completed (Chen et al. 2007). Therefore, detailed knowledge about molecular and cellular events during adipogenesis is important for biomedical research as well as in animal breeding. With respect to the genome behavior and the pig’s usefulness as a model organism for humans, it may be more pertinent to investigate than mouse since it has a range of differently sized chromosomes with differences in centromeric positioning along a chromosome and many expansive syntenic genomic regions.
In the present study, we have examined genome organization in porcine interphase nuclei during adipocyte differentiation. We hypothesized that dynamic changes in gene expression profile, and chromatin remodeling that occur during cellular differentiation would induce changes in the nuclear positions of genes and chromosomes. Therefore, we investigated the nuclear position of nine genes involved in adipogenesis and the chromosomes they are located on, in progenitor mesenchymal stem cells and during in vitro induced adipogenesis. Here, we show that the radial position of chromosome territories was altered during adipocyte differentiation, apart from one chromosome, but most evident were large long-range changes in gene positioning. This repositioning of genes was related to gene activity, with up-regulation correlating with chromatin looping into the nuclear interior and down-regulation with movement towards the nuclear periphery.
Materials and methods
Stem cell culture and induction of differentiation
Mesenchymal stem cells were isolated from porcine bone marrow samples. The bone marrow was submerged for 1 h in a 1:5 dilution of antibiotics (10 U/ml penicillin, 50 μg/ml streptomycin) and 1:20 dilution of amphotericin B (250 μg/ml) in MesenCult® basal medium for human mesenchymal stem cells, with mesenchymal stem cells stimulatory supplements (StemCell Technologies, The Cell Experts™, USA). The bone marrow was then washed in fresh medium before being finely cut with a sterile scalpel in a 90-mm sterile culturing dish. The mesenchymal stem cells (MSCs) were cultured in complete MesenCult® basal medium with mesenchymal stem cells stimulatory supplements and antibiotics (10 U/ml penicillin, 50 μg/ml streptomycin) at 37°C with 5% CO2. Cells were expanded by passage using standard procedures until cell differentiation was to be induced. To confirm that cells isolated from porcine bone marrow were MSCs, indirect immunofluorescence was performed using antibodies directed against CD34 (monoclonal mouse anti-human CD34 QBEnd 10 antibody diluted 1:25; Dako, USA), CD45 (monoclonal mouse anti-human CD45 2B11+ PD7/26 antibody diluted 1:50; Dako, USA), and Thy 1.2 (monoclonal rat anti-mouse Thy 1.2 FF-10 antibody diluted 1:5, Serotec) diluted with 1% (v/v) NCS in phosphate-buffered saline (PBS). Cells were incubated in the primary antibody at 4°C for 24 h. The cells were washed with PBS before application of the appropriate fluorochrome-conjugated secondary antibody for 30 min at 37°C (donkey anti-mouse fluorescein isothiocyanate (FITC) diluted 1:60, Jackson Laboratory; rabbit anti-rat FITC diluted 1:40, Dako). To stimulate adipogenesis, MSCs were grown to confluency and cultured with MesenCult® basal medium with adipogenic stimulatory supplements (StemCell Technologies, The Cell Experts™, USA). The cells were cultured for 14 days in adipogenic complete medium. The adipocyte differentiation was confirmed by phase contrast microscopy and indirect immunofluorescence with anti-SREBP-1 antibodies, polyclonal anti-SREBP-1 H-160 (a kind gift from Dr. Sue Shackleton, Leicester and Santa Cruz diluted 1:100), and SREBP-1 2A4 (monoclonal anti-mouse SREBP-1 2A4 antibody, diluted 1:10, BD Pharmingen™). The primary antibodies were incubated for 24 h at 4°C, washed with PBS, and then incubated with the appropriate fluorochrome-conjugated secondary antibody (donkey anti-mouse Cy3 diluted 1:100, Jackson Laboratory or swine anti-rabbit TRITC diluted 1:30, Sigma-Aldrich) for 30 min at 37°C. MSCs undergoing adipogenesis were also monitored for lipid accumulation before differentiation and during differentiation at days 3, 7, 11, and 14 using oil red O staining. Briefly, cells were fixed with 3.7% formaldehyde for 10 min at room temperature. The cells were then rinsed with double distilled water before incubating with 0.3% oil red O (Sigma-Aldrich) diluted in water (3:2 v/v) at room temperature for 1 h. The oil red O was decanted, rinsed with water, and left in fresh double distilled water for examination using a phase contrast microscope.
Indirect immunofluorescence
Cells were fixed in 4% paraformaldehyde in PBS (w/v) for 10 min at room temperature, followed by washing in PBS. The cells were then treated with 1% Triton-X 100 in PBS (v/v) for 15 min. Primary mouse monoclonal antibody for fibrillarin (a kind gift from Dr. John Aris dilution 1:1,000) was applied to the cells and incubated for 1 h at room temperature. The cells were washed three times in PBS and then incubated for 1 h at room temperature with secondary antibodies–donkey anti-mouse FITC, dilution 1:70 (Jackson Immunoresearch Laboratories). After further washing in PBS, slides were mounted with Vectashield (Vector Laboratories) containing 4′,6-diamidino-2-phenylindole (DAPI).
DNA probes
Whole porcine chromosome painting probes (SSC4, SSC6, SSC13, SSC15, SSC17) isolated by flow sorting were kindly provided by Prof. M.A. Ferguson-Smith (Cambridge Resource Centre for Comparative Genomics, Cambridge University, UK). The chromosome templates underwent primary and secondary amplification by performing degenerate-oligonucleotide-primed polymerase chain reaction (DOP-PCR) and were subsequently labeled with biotin-16-dUTP (Roche). Labeled probe for SSC12 was kindly provided by Prof. F. Yang (Wellcome Trust Sanger Institute, Cambridge, UK).
Bacterial artificial chromosomes (BACs) clones were obtained from available porcine libraries: the RPCI-44 Male Porcine BAC Library, the CHORI-242 Porcine BAC Library (http://bacpac.chori.org/libraries.php), and the PigE BAC library (Anderson et al. 2000; http://www.ark-genomics.org/clonesVectors/ Table 1). BAC probes for adipogenic genes were selected based on previous information about the gene localization (Szczerbal and Chmurzynska 2008; Szczerbal et al. 2007a, b). BAC probes for control genes (MYL1, SOX9) were used for the first time in this study and were verified by PCR using specific primers (primer sequences available on request) and by fluorescence in situ hybridization (FISH) on metaphase chromosomes. BAC DNA was labeled using random-priming method with biotin-16-dUTP (Roche) or digoxigenin-11-dUTP (Roche).
Labeled BAC probes and paint probes (150–300 ng), with excess of porcine genomic competitor DNA (3 µg) and salmon sperm DNA (10 µg) were ethanol-precipitated and dissolved in 10 µl hybridization mixture (10% dextran sulfate, 50% formamide/2× SSC (0.3 M NaCl, 0.03 M sodium citrate), and 1% Tween20).
2D FISH
Cells were subjected to hypotonic buffer and fixed in 3:1 (v/v) methanol–acetic acid (Szczerbal et al. 2007b) and prepared for FISH by incubation in 100 µg/ml RNase/2× SSC at 37°C for 1 h, followed by washing in 2× SSC and further treated with a pepsin solution (0.005% pepsin/0.01 N HCl {w/v}) at 37°C for 10 min, followed by a wash in PBS/1 M MgCl2. Nuclei were subjected to standard 2D FISH as described in Meaburn et al. (2007). Probes were denaturated at 70°C for 10 min and then were preannealed at 37°C for 20 min. For hybridization, slides were incubated in a humid environment at 37°C overnight. After hybridization, slides were washed three times in 50% formamide/2× SSC at 43°C for 5 min and three times in 2× SSC at 43°C. The slides were placed in 0.05% Tween20/4× SSC at room temperature (RT) for 10 min and blocked in 3% BSA/4× SSC at RT for 30 min. Biotin-labeled probes were detected using streptavidin Cy3 (Amersham) diluted 1:200 and digoxigenin-labeled probes were detected with antidigoxigenin-fluorescein Fab fragments (Roche) diluted 1:200. Cells were mounted in Vectashield (Vector Laboratories) with DAPI counterstain.
3D FISH
Cells grown directly onto glass slides were fixed in 4% PFA/PBS (w/v) for 10 min, washed in PBS, and then permeabilized for 20 min in 0.5% saponin/0.5% TritonX-100/PBS, followed by further washing in PBS (Bridger et al. 1998). The 3D preserved cells were then incubated in 20% glycerol/PBS (v/v) for 1 h and subjected to six repeat freeze–thaw cycles in liquid N2. Before hybridization, cells were treated with 0.1 N HCl for 5 min at room temperature and a pepsin solution (0.002%/0.01 N HCl) for 5 min at 37°C, followed by washing in PBS with 1 M MgCl2 and equilibration in 50%formamide/2× SSC apart from when indirect immunofluorescene was performed. Probes were denaturated at 70°C for 10 min and then were preannealed at 37°C for 20 min. Nuclei were denaturated at 76°C for 3 min in 70% formamide/2× SSC followed by 50% formamide/2× SSC for 1 min. Hybridization was performed over two nights at 37°C. Post-hybridization washes and signal detection were performed as for 2D FISH.
Image analysis
Flattened nuclei were examined with an Olympus epifluorescence microscope and observed under ×100 oil immersion objective. Images of randomly selected nuclei were acquired with a CCD camera and were pseudocolored using Smart Capture VP v1.4. The nuclear position of whole chromosome paint probes and gene-specific probes were analyzed using a script developed by Dr. Paul Perry in IPLab software (Croft et al. 1999) and was a kind gift from Prof. Wendy Bickmore (MRC HGU, Edinburgh). This analysis was performed by ascertaining the distribution of mean proportion (percent) of hybridization signals, normalized by percent of DAPI stain, over five concentric shells (shell 1—periphery, shell 5—center of the nucleus). Approximately, 60 nuclei for positioning of chromosome territories and 100 for gene position were analyzed.
For 3D preserved cells, subjected to 3D FISH, stacks of images through the z-axis of cells were captured with a Nikon Eclipse confocal microscope TE 2000-S with a ×100 oil immersion lens and C1 (EZ-C1) software version 3.00. Stacks of optical sections with an axial distance of 0.2 μm were collected. Stacks of 8-bit gray-scale 2D images were obtained with a pixel dwell of 4.56 and 8 averages were taken for each optical image. The measurements and 3D reconstructions were performed using Imaris 5.7.0 software (Bitplane). The relative distance of hybridization signal from the center of the nucleus to the center of hybridization signal was calculated as a ratio of the nuclear radius (r = total distance from the center of the nucleus to the nuclear periphery through the FISH signal). At least 60 FISH signals were scored per experiment. In Fig. 8, 0% represents the geometric center of the nucleus, and 100% was considered as the nuclear border.
Statistical analysis
Before statistical analysis, the mean proportion of hybridization signals already normalized by percent DAPI stain was added together and the mean for the signals was calculated from five concentric shells for each nucleus. After this adjustment, the signals from five concentric shells within a nucleus add up to 1. Within each concentric shell, the median of adjusted hybridization signals in three cell types/time points (MSC, AD7, AD14) were compared using the Kruskal–Wallis rank sum test. This test was used because the gene signal distribution strongly deviated from a normal distribution, and this test allowed us to compare the three different data sets (i.e., MSC, day 7 and day 14) with each other in each of the five shells. In total, 75 tests were performed.
Student’s t test was used to compare the number of signals co-localized with nucleoli in mesenchymal stem cells and in cells from day 14 of differentiation. P value of <0.05 was considered significant.
Reverse transcription polymerase chain reaction
Total RNA from undifferentiated porcine MSCs and from differentiated cells at days 7 and 14 was isolated using GenElute™ Mammalian Total RNA Miniprep Kit (Sigma Aldrich) according to manufacturer’s specification. RNA samples were treated with DNaseI (Sigma Aldrich). The quality of RNA samples was verified by agarose electrophoresis and measured at A260/280 nm absorbance. The complementary DNA (cDNA) was synthesized using SuperScript™ III reverse transcriptase (Invitrogen). The synthesized cDNA was amplified by PCR in a 50-μl reaction containing 5 U Platinum® Taq polymerase (Invitrogen) and 10 µM of each primer (Table S 1). The β-actin gene was used as an internal control. The following cycling conditions were used: 94°C for 5 min, then 28–30 cycles of 94°C for 30 s, 54–60°C for 30 s, and 72°C for 1 min, finishing with 72°C for 7 min. The annealing temperature and number of PCR cycles were optimized in each case to ensure that the intensity of the product corresponded with the linear phase of amplification. The PCR product was separated by electrophoresis in 1.5% agarose gel, stained, and photographed under UV illumination. Each cDNA was analyzed in duplicate.
Results
Establishment of an in vitro model system for differentiation of porcine mesenchymal stem cells into adipocytes
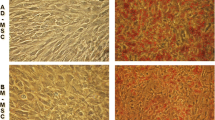
To assess spatial genome organization during adipogenesis, we first established an in vitro system of differentiation of mesenchymal stem cells into adipocytes. Porcine mesenchymal stem cells were successfully isolated from bone marrow (Fig. 1a). The presence of porcine MSCs was verified by indirect immunofluorescence by the lack of anti-CD34 and anti-CD45 staining and the presence of anti-Thy-1 staining (data not shown). Adipogenesis was induced with complete adipogenic medium and cells were cultured for 14 days. Single lipid vesicles were observed from the third day after stimulation and then the size and number of the lipid droplets increased up to day 14 (Fig. 1b). Oil red O staining revealed that ~40% of the cells contained lipid by day 14. Further, anti-SREBP1 staining was evident in the culture after 7 days in adipogenic medium and >80% of cells displayed nuclear foci of SREBP1 by day 14.

In vitro culture of adipogenesis using porcine mesenchymal cells. Representative images of porcine mesenchymal stem cells stimulated to undergo adipogenesis. a MSCs prior to differentiation; b cells induced to undergo adipogenesis, showing the presence of lipid droplets. Cell cultures were imaged using phase contrast optics, with ×40 lens, on a Zeiss Cell Observer. Scale bar, 10 μm
Chromosome territory behavior during in vitro porcine adipogenesis from mesenchymal stem cells
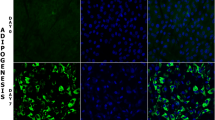
In order to determine if adipogenesis genes and the chromosomes they are housed upon undergo nuclear repositioning during differentiation, we investigated the nuclear positioning of a set of adipogenic genes and controls, along with their associated chromosomes. We selected nine genes—seven of which are involved in adipogenesis (PPARG, SREBF1, CEBPA, CEBPB, CREB, FABP4, GATA2) and two that were used as control genes (SOX9, MYL1; Table 1). The genes were mapped by FISH to different porcine chromosomes, such as SSC4, SSC6, SSC12, SSC13, SSC15, and SSC17 (Szczerbal and Chmurzynska 2008; Szczerbal et al. 2007a, b). We selected the genes so that adipogenic and control genes or pro- and anti-adipogenic genes were located on the same chromosome (Fig. S 1). Radial positioning of the nine genes and six chromosomes was determined in nuclei derived from MSC and from cells induced to differentiate for 7 days (AD7) and for 14 days (AD14; Table 2). This was performed by preparing cells for 2D FISH whereby standard hypotonic treatment and fixation with methanol/acetic acid were used. Digital images were captured of random cells containing the chromosome territories (Fig. 2) and gene signals (Fig. 4). These images were subjected to erosion analysis using a bespoke script in IPLab, such that the nucleus is delineated and divided into five shells of equal area, and the fluorescent intensity of the FISH signal (gene or chromosome territory) is calculated. This value is normalized by the amount of DAPI signal found in each radial shell (see Boyle et al. 2001; Croft et al. 1999). These radial distribution data are plotted as histograms for chromosomes (Fig. 3) and genes (Fig. 5). Figure 3 displays the distributions of whole chromosome territories during induced adipogenesis. Shell 1 represents the periphery of nuclei and shell 5 the nuclear interior. To explain the location of chromosomes (or genes) in the nucleus, we have selected four terms: peripheral (P) to refer to a situation where signal was located preferentially at the nuclear periphery, interior (I) where the signal was preferentially observed towards the nuclear interior, intermediate (IM) to define a situation where signal was positioned between the nuclear periphery and the nuclear interior, and equally distributed (EQ) to describe a random distribution of signal. Throughout adipogenesis, chromosome territories demonstrated non-random positioning within the nuclei, i.e., in MSC SSC4, 13, and 15 are located at the nuclear periphery, while SSC6 is located in an intermediate location with chromosomes 12 and 17 being found in the nuclear interior. In MSC, SSC4 territories were located at the nuclear periphery and relocated to the nuclear interior at day 7 of adipogenesis. An equal distribution of chromosome signal for SSC4 was observed at day 14. This distribution indicates a more random positioning of this chromosome. SSC13 also appears to be relocated during adipogenesis since day 0, and day 14 SSC13 displays a more peripheral distribution, but at day 7, the shape of the graph indicates a more intermediate nuclear location (Fig. 3, Table 2). All the other chromosomes demonstrate lesser changes in overall nuclear location. Table S 2 reveals the results of a Kruskal–Wallis test, a non-parametric analysis of variance, where all data sets for the normalized chromosome position have been analyzed for the normalized signal within each individual shell for all three time points, i.e., days 0, 7, and 14. The data for SSC4 display significant differences between shells 1, 2, 4, and 5 for chromosome position at all three time points. SSC13 displays significant differences in shells 1, 2, and 5, whereas SSC17 only displays a significant difference for shells 1 and 2, SSC12 and 15 only demonstrate significant difference in shell 1, and SSC6 displays no significant differences at all.

Nuclear distribution of whole chromosomes in interphase nuclei of porcine mesenchymal stem cells induced to differentiate into preadipocytes and adipocytes. Representative images of nuclear positioning of whole chromosome territories in mesenchymal stem cells (MSC) and in cells induced to become adipocytes from day 7 (AD7) and day 14 (AD14). Cells were fixed with methanol/acetic acid and subjected to 2D-fluorescence in situ hybridization. Chromosome territories were delineated using flow-sorted porcine whole chromosome painting probes for chromosomes SSC4, SSC6, SSC12, SSC13, SSC15, and SSC17. Digital images were collected on an Olympus epifluorescence microscope via Smart Capture VP v1.4 software. Chromosome territories are green; DNA in the nuclei is counterstained with DAPI (blue). Scale bar, 10 μm

Nuclear positioning of chromosome territories in interphase nuclei of porcine mesenchymal stem cells induced to differentiate into preadipocytes and adipocytes. The distribution of whole chromosome territories within the interphase nuclei was analyzed by erosion analysis of at least 50 digital images of cells subjected to 2D FISH. The script divides the nuclei into five shells of equal area and measures the intensity of the DNA and FISH signal in each shell. Each measurement for the FISH signal is normalized by the DAPI measurement for that shell. The histograms represent the mean proportion of the normalized chromosome signal (y-axis), across the five concentric shells of the nuclei (x-axis), from the nuclear periphery (shell 1) to the nuclear interior (shell 5). Statistical difference (P < 0.05) was assessed by the non-parametric Kruskal–Wallis rank sum test and indicated by asterisks (for details, see Table S 2)
Major nuclear repositioning of adipogenic specific genes during differentiation
When we analyzed gene loci position in nuclei during adipogenesis, we found much more dramatic differences with major relocations of adipogenesis genes than for the chromosomes (Fig. 4), revealing genes moving from the nuclear periphery to the nuclear interior (PPARG, FABP4, GATA2), from an intermediate position to the nuclear interior (SREBF1, CEBPB) and from a peripheral to an intermediate location (CREB). Thus, six out of the seven adipogenesis genes have significantly changed their nuclear location during adipogenesis. Of the two control genes, SOX9 changes location from an interior location to a more random distribution at day 7 and then returns to a more interior location by day 14. The other control gene MYL1 does not change its nuclear distribution in 14 days. Again, we have performed a Kruskal–Wallis test for all three data sets for each time point for the individual shells (Table S 3) and reveal significance difference in gene loci positioning for four out of the five shells in four genes (PPARG1, SREBF1, FABP4, and GATA2); two genes (CEBPA and CREB) display significant difference in two shells for the three time points, and one gene (CEBPB) displays a significant difference in shell 4 (Fig. 5).

Nuclear distribution of gene loci in interphase nuclei of porcine mesenchymal stem cells induced to differentiate into preadipocytes and adipocytes. Representative images of the fluorescent gene signals of the seven adipogenesis genes and two controls in mesenchymal stem cells (MSC) and preadipocytes and adipocytes from day 7 (AD7) and day 14 (AD14) after induction of adipogenesis. Cells were fixed with methanol/acetic acid and subjected to 2D FISH. Gene loci were delineated using BAC probes. Digital images were collected on an Olympus epifluorescence microscope via Smart Capture VP v1.4 software. Gene signals are green; DNA in the nuclei is counterstained with DAPI (blue). Scale bar, 10 μm
From the skew of the histograms displayed in Fig. 5, it can be seen that a number of the adipogenesis genes do indeed alter their nuclear location, quite dramatically and are located in disparate nuclear compartments over the time period of the experiments.

Nuclear positioning of gene loci in interphase nuclei of porcine mesenchymal stem cells induced to differentiate into preadipocytes and adipocytes. The distribution of specific gene loci within the interphase nuclei was analyzed by erosion analysis of at least 50 digital images of cells subjected to 2D FISH. The script divides the nuclei into five shells of equal area and measures the intensity of the DNA and FISH signal in each shell. Each measurement for the FISH signal is normalized by the DAPI measurement for that shell. The histograms represent the mean proportion of the normalized chromosome signal (y-axis), across the five concentric shells of the nuclei (x-axis), from the nuclear periphery (shell 1) to the nuclear interior (shell 5). Statistical difference (P < 0.05) was assessed by the non-parametric Kruskal–Wallis rank sum test and indicated by asterisks (for details, see Table S 3). Reverse transcriptase-PCR was performed on total RNA extracted from the cells cultured in vitro using gene-specific primers. β-Actin was used as a control for cDNA synthesis. The primers for MYL1 gene was checked on cDNA from muscle. RNA samples were taken on day 0, day 7, and day 14 of the in vitro cultures. The intensity measurements of the amplified cDNA for each gene at each time point are in Table S 4
Genes associated with adipogenesis loop out from their chromosome territory during differentiation and this is associated with gene expression. Out of the seven adipogenesis genes analyzed, two of them were deemed to reside in different nuclear regions to their parent chromosomes (see Table 2 for comparisons) in MSC at time 0, and these were CREBPA on SSC6 and CEBPB on SSC17. By day 7 (AD7), there are four genes that were deemed to reside in a different nuclear compartment to their parent chromosome, and by day 14, there were still four with SREBF1 being significantly more interiorly located at day 14 than day 7 with no change in its parent chromosome. These data imply that there is a reorganization of the adipogenesis genes studied, to the edge or away from the main bulk of the chromosome territory. In other studies, this behavior of gene loci, i.e., moving away from the chromosome territory, towards the nuclear interior, can be associated with an increase in gene expression. In order to test this postulation, we measured the amount of gene expression by reverse transcriptase (RT)-PCR for each of the nine genes at the three time points, and we have demonstrated that all the genes associated with adipogenesis increase their expression significantly (Fig. 5 and Table S 4) from day 0. The RT-PCR results were in agreement with previous studies with respect to the pattern of expression of the studied genes for human, mouse, and pig (Guo and Liao 2000; Kouadjo et al. 2007; Nakamura et al. 2003; Zou et al. 2008). We found that messenger RNA (mRNA) encoding the adipogenic transcription factors PPARG, CEBPA, and SREBF1 increased significantly during the course of adipogenic differentiation. CEBPB’s expression was up-regulated at day 7 and then down-regulated at day 14. The expression of the CREB gene was present in all the analyzed time points. Expression levels of FABP4, a late marker of adipogenesis, increased during differentiation with the highest level at day 14. The GATA2 gene was highly expressed in cells from day 7 of differentiation and down-regulated at day 14 and is known to be involved in preadipogenesis (Tong et al. 2000). Expression of SOX9, a key regulator of chondrogenic differentiation, was suppressed during adipogenesis. This was expected since it is high in MSC and is switched off during adipogenesis. We did not detect transcripts for MYL1 gene during adipogenesis since this gene is characteristic for skeletal muscle and not expressed in either MSCs or adipocytes.
One of the most interesting gene behaviors belongs to GATA2. This gene is an anti-adipogenic transcription factor and its expression is restricted to preadipocytes. In our study, GATA2’s expression is at its highest at day 7, and by day 7, it is relocated from a peripheral nuclear location to one in the nuclear interior. By day 14, its expression has fallen and the gene is relocalized to the nuclear periphery again. However, for the other gene, CEBPB, that has an up-regulation of expression at day 7, which has diminished by day 14, the loci remain in the nuclear interior. CEBPB is located on porcine chromosome 17q, which is syntenic to human 20q. There are many different genes on 20q in human whose function are associated with adipogenesis, obesity, and diabetes. It is possible that this region remains within the nuclear interior for co-regulation of other important genes for adipogenesis.
From these analyses and data, it seems that the nuclear radial location of whole chromosome territories (see Tables S 2 and 3) is not quite as dramatic, in response to adipogenic stimulants, as the gene loci. This implies that genes may be looping out away from their chromosome territories. This is confirmed in 2D RE-FISH experiments that delineate both the gene loci probes and the parent chromosome territories using whole chromosome painting probes where the gene loci are observed at some distance from the chromosome territories (Fig. 6). Indeed, in MSC (day 0), 14%, 6%, and 10% of chromosome territories have a gene locus at a distance not associated with them for SREBP1:SSC12, PPARG:SSC13, and CREB:SSC15, respectively. Conversely, for AD14 (day 14) cells, these numbers had increased dramatically to 32%, 45%, and 42%, respectively. Further, using 3D FISH analysis, we revealed that during the course of differentiation the architecture of chromosome territories was altered (Fig. 7a–d). In MSCs, more condensed chromosome territories with gene signals buried inside the chromosome territories were observed (Fig. 7a, c), while in differentiated cells, chromosome territories studied were more decondensed and were approximately twice the size (Figs. 7b, d and 9). These decondensed chromosomes displayed loops of chromatin emanating from the chromosome territory with the gene signal on the end of the loop, oriented towards the nuclear interior (Fig. 7b, d). This was confirmed by 3D measurement analyses, plotted as frequency distributions with 0% on the x-axis representing the nuclear interior and 100% the nuclear periphery, for the following gene and chromosome combinations: FABP4:SSC4 (Fig. 8a, b), PPARG:SSC13 (Fig. 8c, d), and CEBPB:SSC17 (Fig. 8e, f). The frequency distribution of the nuclear location of the gene loci and chromosome territories in Fig. 8a, c reveals the co-positioning in MSC (day 0) of the FABP4 and PPARG gene signals with their chromosome territories, SSC4 and SSC13, respectively. CEBPB is not so well co-positioned with the territories of SSC17 and is slightly more peripheral than the chromosome. After 14 days of differentiation, the movement of all three genes to the nuclear interior is confirmed (Fig. 8b, d, f). The chromosome positioning is also confirmed with SSC4 moving towards the nuclear interior during course of differentiation (Fig. 8b) and SSC13 and SSC17 not changing their position very much at all.

a, b Nuclear distribution of gene signals and chromosome territories in cells from day 14 of differentiation. AD14 nuclei that have been subjected to 2D FISH for whole SSC13 delineation were subjected to RE-FISH with probes to delineate PPARG gene loci. Gene loci signals (red) were found frequently located outside of the chromosome territory (green). DNA was counterstained with DAPI (blue). Similar spatial relationships were found for SREBP1 and SSC12 and CREB and SSC15 (not shown). Scale bar, 10 μm

Nuclear distribution of PPARG gene and SSC13 chromosome territories in cells from day 14 of differentiation subjected to 3D FISH. MSC, AD7, and AD14 cells were fixed and permeabilized to preserve the 3D structure of the cells. Dual color FISH experiments were performed with whole chromosome painting probes and BAC probes for specific gene loci. Cells were optically sectioned at 0.2-μm intervals using a Nikon 3-color laser scanning microscope. 3D reconstructions and measurements of the optical sections were performed in Imaris software. a 3D reconstruction of a nucleus from a MSC. b 3D reconstruction of a nucleus from an AD14 cell for the PPARG (green) and SSC13 chromosome territory (red). c, d 3D reconstructions of representative chromosome territories with gene signals of MSC and AD14 cells, respectively. Scale bar, 10 μm

Frequency distribution plots of the nuclear positioning of chromosomes and genes in MSC and AD14 cells. a, c, d Frequency distributions of the radial distance measurement profiles of FABP4 and SSC4 (a), PPARG gene and SSC13 (c), CEBPB and SSC17 in MSCs (e). b, d, f The same gene/chromosome combinations are represented after 14 days in differentiation medium (AD14). Gene positioning is displayed with green lines and chromosome positioning with red lines; 0% of the x-axis represents the nuclear interior, while 100% represents the nuclear periphery
For most of the genes, we observed a strong correlation between gene repositioning and a change in expression levels. The significance of these movements has been tested by Kruskal–Wallis rank sum test (Table S 2), and if the movement is significant, it is signified by asterisks on the graphs in Fig. 5. For down-regulated genes, we observed movement to a more peripheral nuclear position. The GATA2 gene was up-regulated at day 7 of adipogenesis and moved to the nuclear interior and down-regulated at day 14 and became positioned peripherally. The SOX9 genes were down-regulated during the course of differentiation and their position became more peripheral when compared to day 0. The control gene MYL1 did not show expression in our system of differentiation and the position of loci was constant, as was the chromosome containing MYL1. However, for the other three genes, the correlation between gene activity and gene position was not so clear. The CEBPA gene was up-regulated during differentiation but the location changed only slightly, more towards the interior. CEBPB was up-regulated at day 7 and moved to a more interior position; however, at day 14, it was down-regulated, but its position remained unchanged. Although the CREB gene showed the same level of transcriptional activity throughout differentiation, the nuclear position of the gene loci changed from peripheral to intermediate during induction of adipogenesis.
Discussion
In order to investigate the role of genome organization in differentiation, we have established an in vitro differentiation mesenchymal stem cell model system using porcine stem cells. The pig is an important model organism used to understand adipogenesis, obesity, and diabetes, and its genome is organized in a similar way to humans, with different sized chromosomes with a range of chromosome positions, where the centromere is located. Thus, we investigated the behavior of a panel of important adipogenesis genes and the chromosomes they are housed on in porcine mesenchymal stem cells, preadipocytes, and adipocytes.
There are a number of studies that support the importance of genome/chromosome/gene positioning within interphase nuclei in regulating gene expression (Foster and Bridger 2005; Lanctôt et al. 2007; Takizawa et al. 2008b). There have been examples where chromosomes alter their nuclear location as cells differentiate (Schöfer and Weipoltshammer 2008), for example, in human adipogenesis (Kuroda et al. 2004). Further, other studies have revealed genes being relocated as they are switched on or off. Therefore, it does appear that where a sequence is located within the nucleus, it could affect its expression. It is not clear for a number of genes that are relocated whether it is the gene by itself or whether it is the whole chromosome territory including the gene that moves. Indeed, it appears that both situations may be true. There have been examples of genes being relocated without the chromosome territory moving. These genes come away from the main body of the chromosome territory on chromatin loops (Chambeyron and Bickmore 2004; Chambeyron et al. 2005; Volpi et al. 2000). Recent studies on gene repositioning during erythropoiesis indicate that local chromatin environment plays a crucial role in spatial associations of genes located on different chromosomes (Brown et al. 2008).
Although the higher-order chromatin organization in the nucleus is conserved in vertebrates (Habermann et al. 2001; Tanabe et al 2002a, b), there is also an increasing number of examples demonstrating differential chromosome behavior in different species. Studies on human and murine α-globin and β-globin genes revealed a different nuclear position in these two species, in relation to their transcriptional activity (Brown et al. 2006). A similar situation was observed in the case of CFTR locus and adjacent loci (Sadoni et al. 2008; Zink et al. 2004). The nuclear positioning of these highly conserved loci in human and mouse is therefore species-specific, the murine loci were localized, irrespective of their transcriptional status within the nucleus interior, while the human orthologs became repositioned, depending on their transcription activity. These findings indicate the importance of investigating the nuclear and genome organization and their role in gene regulation in different species. We demonstrate such an example here with our study on porcine adipogenesis. We found that whole chromosomes relocated to different extents over the three time points assessed. One chromosome in particular, SSC4, relocated from the nuclear periphery more towards the nuclear interior, as yet we do not know the reason for this. We do know, however, that SSC4 demonstrates synteny with HSA8q, where a number of genes associated with adipogenesis such as SLUG (SNAI2; Pérez-Mancera et al. 2007), DDEF1 (Ehlers et al. 2005), as well as FABP4 are found. In another study, and to date, the only one on chromosome positioning during adipocyte differentiation in human cells (Kuroda et al. 2004), the authors found changes in chromosome positions. They analyzed two chromosomes HSA12 and HSA16 which are involved in chromosome translocation in liposacroma tumorigenesis and detected close association between chromosome territories in adipocytes when compared with preadipocytes. Additionally, they detected that during human adipogenesis HSA16 territory size increased by 50%. In our study, we did not look at relative positioning of the studied chromosomes. However, we observed very condensed chromosome territories in MSC and more decondensed chromosome territories with extended loops at day 14 of differentiation. It has been suggested that changes in the architecture of chromosome territories are produced by transcription (Croft et al. 1999; Chubb and Bickmore 2003). It is also possible that a more decondensed chromosome territory could slightly affect the number of shells a territory was found within in the 2D analyses.
When we analyzed the nuclear positions of the genes themselves, we found that substantial repositioning had occurred, with a number of genes moving from a peripheral location within nuclei to a more central position. These were genes that we knew were up-regulated in adipogenesis from studies in other organisms, i.e., mouse and human. However, when gene expression was determined using RT-PCR, we found that the movement of genes to a more central nuclear location did indeed correlate with transcriptional up-regulation. The most interesting gene was GATA2, whose expression is up-regulated in preadipocytes and then down-regulated again in adipocytes. The GATA2 gene loci were located in a peripheral position in MSC nuclei and then became interior at day 7, but by day 14 of sampling, they were relocated at the nuclear periphery. This correlates with the gene expression data. Other genes that were located more towards the nuclear interior became significantly more interior when adipogenesis was induced and their expression up-regulated. Thus, this is an important model system in which to analyze the role of gene translocation within the interphase nuclei and provides a number of examples whereby genes appear to change their location with expression. Our data confirm our hypothesis that correlation between gene positioning and expression activity can be observed during differentiation when gene status has changed from silenced form to active form (Takizawa et al. 2008b). However, there are also reports where a lack of correlation between radial position of genes and their transcription activity has been described. For example, in a large study where radial positioning of 11 genes during early tumorigenesis of breast cancer was investigated, no correlation with gene expression was found (Meaburn and Misteli 2008).
There are a number of recent studies proposing that groups of related genes or genes within a certain pathway favor specific transcription factories or interchromatin granules and are found together even though their chromosome territories may be at a distance (Osborne et al. 2004, 2007; Hu et al. 2008). However, we found that four of the adipogenesis genes we positioned by FISH within the same sample were not co-localised but the numbers of genes associated with nucleoli were increased (Fig. S 2). Furthermore, large RNA-FISH and 4C studies would be required to determine if the porcine adipogenesis genes do indeed move to a common nuclear area and/or transcription factories. Interesting data were generated in the study of gene association during erythropoiesis (Brown et al. 2008). The authors found that a tendency of genes to associate depends on the local chromatin environment and not sharing transcription factories. They demonstrated that active genes cluster around common nuclear speckles.

Changes in chromosome territory condensation during porcine adipogenesis. Areas of chromosome territories were measured in methanol/acetic acid-fixed nuclei. Chromosome territories from 30 nuclei were analyzed in Adobe photoshop and their area is expressed as a percentage of the total nuclear area and are displayed as histograms with purple bars representing MSCs, the burgundy bars the AD7 cells, and the lemon bars the AD14 cells. Error bars are SEM
How genes become relocated is not yet clear, but nuclear motors containing nuclear myosin and actins may be involved (Hofmann et al. 2006; Mehta et al. 2008). Given the importance of these genes in adipogenesis, knowing how they behave in situ will help to control their expression by developing interventionist mechanisms to prevent expression. Thus, these types of analysis will help in not only understanding adipogenesis and diseases like obesity but also working towards the control of co-regulated expression of this network of genes, which will be of benefit to the health economy and agriculture.
References
Anderson SI, Lopez-Corrales NL, Gorick B, Archibald AL (2000) A large-fragment porcine genomie library resource in a BAC vector. Mamm Genome 9:811–814
Ballabio E, Cantarella CD, Federico C, Di Mare P, Hall G, Harbott J, Hughes J, Saccone S, Tosi S (2009) Ectopic expression of the HLXB9 gene is associated with an altered nuclear position in t(7;12) leukaemias. Leukemia 23(6):1179–1182
Bártová E, Kozubek S, Jirsová P, Kozubek M, Gajová H, Lukásová E, Skalníková M, Ganová A, Koutná I, Hausmann M (2002) Nuclear structure and gene activity in human differentiated cells. J Struct Biol 139:76–89
Boyle S, Gilchrist S, Bridger JM, Mahy NL, Ellis JA, Bickmore WA (2001) The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum Mol Genet 10:211–219
Brown KE, Baxter J, Graf D, Merkenschlager M, Fisher AG (1999) Dynamic repositioning of genes in the nucleus of lymphocytes preparing for cell division. Mol Cell 3:207–217
Brown KE, Amoils S, Horn JM, Buckle VJ, Higgs DR, Merkenschlager M, Fisher AG (2001) Expression of alpha- and beta-globin genes occurs within different nuclear domains in haemopoietic cells. Nat Cell Biol 3:602–606
Brown JM, Leach J, Reittie JE, Atzberger A, Lee-Prudhoe J, Wood WG, Higgs DR, Iborra FJ, Buckle VJ (2006) Coregulated human globin genes are frequently in spatial proximity when active. J Cell Biol 172:177–187
Brown JM, Green J, das Neves RP, Wallace HA, Smith AJ, Hughes J, Gray N, Taylor S, Wood WG, Higgs DR, Iborra FJ, Buckle VJ (2008) Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J Cell Biol 182:1083–1097
Bridger JM, Herrmann H, Muenkel C, Lichter P (1998) Identification of an interchromosomal compartment by polymerisation of nuclear-targeted vimentin. J Cell Sci 111:1241–1253
Chambeyron S, Bickmore WA (2004) Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev 18:1119–1130
Chambeyron S, Da Silva NR, Lawson KA, Bickmore WA (2005) Nuclear re-organisation of the Hoxb complex during mouse embryonic development. Development 132:2215–2223
Chen K, Baxter T, Muir WM, Groenen MA, Schook LB (2007) Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int J Biol Sci 3:153–165
Chubb JR, Bickmore WA (2003) Considering nuclear compartmentalization in the light of nuclear dynamics. Cell 112:403–406
Cremer T, Cremer M, Dietzel S, Müller S, Solovei I, Fakan S (2006) Chromosome territories—a functional nuclear landscape. Curr Opin Cell Biol 18:307–316
Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA (1999) Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 145:1119–1131
Ehlers JP, Worley L, Onken MD, Harbour JW (2005) DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res 11:3609–3613
Farmer SR (2006) Transcriptional control of adipocyte formation. Cell Metab 4:263–273
Federico C, Cantarella CD, Di Mare P, Tosi S, Saccone S (2008) The radial arrangement of the human chromosome 7 in the lymphocyte cell nucleus is associated with chromosomal band gene density. Chromosoma 117:399–410
Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA (2008) Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4:e1000039
Formiguera X, Cantón A (2004) Obesity: epidemiology and clinical aspects. Best Pract Res Clin Gastroenterol 18:1125–1146
Foster HA, Bridger JM (2005) The genome and the nucleus: a marriage made by evolution. Genome organisation and nuclear architecture. Chromosoma 114:212–229
Foster HA, Abeydeera LR, Griffin DK, Bridger JM (2005) Non-random chromosome positioning in mammalian sperm nuclei, with migration of the sex chromosomes during late spermatogenesis. J Cell Sci 118:1811–1820
Goetze S, Mateos-Langerak J, Gierman HJ, de Leeuw W, Giromus O, Indemans MH, Koster J, Ondrej V, Versteeg R, van Driel R (2007) The three-dimensional structure of human interphase chromosomes is related to the transcriptome map. Mol Cell Biol 27:4475–4487
Grasser F, Neusser M, Fiegler H, Thormeyer T, Cremer M, Carter NP, Cremer T, Müller S (2008) Replication-timing-correlated spatial chromatin arrangements in cancer and in primate interphase nuclei. J Cell Sci 121:1876–1886
Guo X, Liao K (2000) Analysis of gene expression profile during 3T3-L1 preadipocyte differentiation. Gene 251:45–53
Habermann FA, Cremer M, Walter J, Kreth G, von Hase J, Bauer K, Wienberg J, Cremer C, Cremer T, Solovei I (2001) Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res 9:569–584
Hepperger C, Mannes A, Merz J, Peters J, Dietzel S (2008) Three-dimensional positioning of genes in mouse cell nuclei. Chromosoma . doi:10.1007/s00412-008-0168-2
Hofmann WA, Johnson T, Klapczynski M, Fan JL, de Lanerolle P (2006) From transcription to transport: emerging roles for nuclear myosin I. Biochem Cell Biol 84:418–426
Hu Q, Kwon YS, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, Fu XD (2008) Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci U S A 105:19199–19204
Kosak ST, Skok JA, Medina KL, Riblet R, Le Beau MM, Fisher AG, Singh H (2002) Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 296:158–162
Kouadjo KE, Nishida Y, Cadrin-Girard JF, Yoshioka M, St-Amand J (2007) Housekeeping and tissue-specific genes in mouse tissues. BMC Genomics 8:127
Küpper K, Kölbl A, Biener D, Dittrich S, von Hase J, Thormeyer T, Fiegler H, Carter NP, Speicher MR, Cremer T, Cremer M (2007) Radial chromatin positioning is shaped by local gene density, not by gene expression. Chromosoma 116:285–306
Kuroda M, Tanabe H, Yoshida K, Oikawa K, Saito A, Kiyuna T, Mizusawa H, Mukai K (2004) Alteration of chromosome positioning during adipocyte differentiation. J Cell Sci 117:5897–5903
Lanctôt C, Cheutin T, Cremer M, Cavalli G, Cremer T (2007) Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet 8:104–115
Lunney JK (2007) Advances in swine biomedical model genomics. Int J Biol Sci 3:179–184
Meaburn KJ, Misteli T (2007) Cell biology: chromosome territories. Nature 445:379–781
Meaburn KJ, Misteli T (2008) Locus-specific and activity-independent gene repositioning during early tumorigenesis. J Cell Biol 180:39–50
Meaburn KJ, Cabuy E, Bonne G, Levy N, Morris GE, Novelli G, Kill IR, Bridger JM (2007) Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell 6:139–153
Mehta IS, Elcock LS, Amira M, Kill IR, Bridger JM (2008) Nuclear motors and nuclear structures containing A-type lamins and emerin: is there a functional link? Biochem Soc Trans 36:1384–1388
Misteli T (2007) Beyond the sequence: cellular organization of genome function. Cell 128:787–800
Morey C, Da Silva NR, Perry P, Bickmore WA (2007) Nuclear reorganisation and chromatin decondensation are conserved, but distinct, mechanisms linked to Hox gene activation. Development 134:909–919
Morey C, Kress C, Bickmore WA (2009) Lack of bystander activation shows that localization exterior to chromosome territories is not sufficient to upregulate gene expression. Genome Res (in press)
Nakamura T, Shiojima S, Hirai Y, Iwama T, Tsuruzoe N, Hirasawa A, Katsuma S, Tsujimoto G (2003) Temporal gene expression changes during adipogenesis in human mesenchymal stem cells. Biochem Biophys Res Commun 303:306–312
Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, Fraser P (2004) Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 36:1065–1071
Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P (2007) Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol 5:e192
Pérez-Mancera PA, Bermejo-Rodríguez C, González-Herrero I, Herranz M, Flores T, Jiménez R, Sánchez-García I (2007) Adipose tissue mass is modulated by SLUG (SNAI2). Hum Mol Genet 16:2972–2986
Ragoczy T, Bender MA, Telling A, Byron R, Groudine M (2006) The locus control region is required for association of the murine beta-globin locus with engaged transcription factories during erythroid maturation. Genes Dev 20:1447–1457
Reddy KL, Zullo JM, Bertolino E, Singh H (2008) Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452:243–247
Rosen ED, MacDougald OA (2006) Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 7:885–896
Saccone S, Federico C, Bernardi G (2002) Localization of the gene-richest and the gene-poorest isochores in the interphase nuclei of mammals and birds. Gene 300:169–178
Sadoni N, Targosz BS, Englmann A, Fesser S, Koch J, Schindelhauer D, Zink D (2008) Transcription-dependent spatial arrangements of CFTR and conserved adjacent loci are not conserved in human and murine nuclei. Chromosoma 117:381–397
Schöfer C, Weipoltshammer K (2008) Gene dynamics and nuclear architecture during differentiation. Differentiation 76:41–56
Schook L, Beattie C, Beever J, Donovan S, Jamison R, Zuckermann F, Niemi S, Rothschild M, Rutherford M, Smith D (2005) Swine in biomedical research: creating the building blocks of animal models. Anim Biotechnol 16:183–190
Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, Gal-Yam EN (2005) The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci 118:4017–4025
Szczerbal I, Chmurzynska A (2008) Chromosomal localization of nine porcine genes encoding transcription factors involved in adipogenesis. Cytogenet Genome Res 121:50–54
Szczerbal I, Chmurzynska A, Switonski M (2007a) Cytogenetic mapping of eight genes encoding fatty acid binding proteins (FABPs) in the pig genome. Cytogenet Genome Res 118:63–66
Szczerbal I, Lin L, Stachowiak M, Chmurzynska A, Mackowski M, Winter A, Flisikowski K, Fries R, Switonski M (2007b) Cytogenetic mapping of DGAT1, PPARA, ADIPOR1 and CREB genes in the pig. J Appl Genet 48:73–76
Takizawa T, Gudla PR, Guo L, Lockett S, Misteli T (2008a) Allele-specific nuclear positioning of the monoallelically expressed astrocyte marker GFAP. Genes Dev 22:489–498
Takizawa T, Meaburn KJ, Misteli T (2008b) The meaning of gene positioning. Cell 135:9–13
Tanabe H, Habermann FA, Solovei I, Cremer M, Cremer T (2002a) Non-random radial arrangements of interphase chromosome territories: evolutionary considerations and functional implications. Mutat Res 504:37–45
Tanabe H, Müller S, Neusser M, von Hase J, Calcagno E, Cremer M, Solovei I, Cremer C, Cremer T (2002b) Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc Natl Acad Sci U S A 99:4424–4429
Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS (2000) Function of GATA transcription factors in preadipocyte–adipocyte transition. Science 290:134–138
Volpi EV, Chevret E, Jones T, Vatcheva R, Williamson J, Beck S, Campbell RD, Goldsworthy M, Powis SH, Ragoussis J, Trowsdale J, Sheer D (2000) Large-scale chromatin organization of the major histocompatibility complex and other regions of human chromosome 6 and its response to interferon in interphase nuclei. J Cell Sci 113:1565–1576
Williams RR, Broad S, Sheer D, Ragoussis J (2002) Subchromosomal positioning of the epidermal differentiation complex (EDC) in keratinocyte and lymphoblast interphase nuclei. Exp Cell Res 272:163–175
Williams RR, Azuara V, Perry P, Sauer S, Dvorkina M, Jørgensen H, Roix J, McQueen P, Misteli T, Merkenschlager M, Fisher AG (2006) Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J Cell Sci 119:132–140
Xu M, Cook PR (2008) The role of specialized transcription factories in chromosome pairing. Biochim Biophys Acta 1783:2155–2160
Zalenskaya IA, Zalensky AO (2004) Non-random positioning of chromosomes in human sperm nuclei. Chromosome Res 12:163–173
Zeng L, Rahrmann E, Hu Q, Lund T, Sandquist L, Felten M, O'Brien TD, Zhang J, Verfaillie C (2006) Multipotent adult progenitor cells from swine bone marrow. Stem Cells 24:2355–2366
Zink D, Amaral MD, Englmann A, Lang S, Clarke LA, Rudolph C, Alt F, Luther K, Braz C, Sadoni N, Rosenecker J, Schindelhauer D (2004) Transcription-dependent spatial arrangements of CFTR and adjacent genes in human cell nuclei. J Cell Biol 166:815–825
Zou L, Zou X, Chen L, Li H, Mygind T, Kassem M, Bünger C (2008) Multilineage differentiation of porcine bone marrow stromal cells associated with specific gene expression pattern. J Orthop Res 26:56–64
Acknowledgments
We thank Prof. Wendy Bickmore and Dr. Paul Perry (MRC HGU, Edinburgh) for erosion script, Prof. Malcom A. Ferguson-Smith (Cambridge Resource Centre for Comparative Genomics, Cambridge University, UK) for flow-sorted pig chromosome paints, and Prof. Fengtang Yang (Wellcome Trust Sanger Institute, Cambridge, UK) for paint probe for porcine chromosome 12. We also thank Dr. Maciej Szydlowski (Poznan University of Life Sciences, Poland) for helping with statistical analysis. This study was financed by the Polish Ministry of Science and Higher Education, grant N301 3381 33, fellowship 9/MOB/2007/0 and monies awarded to JMB from Brunel University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by T. Misteli
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Table S1
(DOC 35 kb)
Table S2
(DOC 32 kb)
Table S3
(DOC 29 kb)
Table S4
(DOC 30 kb)
Fig. S1
Ideograms of porcine chromosomes (SSC4, SSC6, SSC12, SSC13, SSC15, SSC17) showing the location of selected genes. (JPG 145 kb)
Fig. S2
Relative positioning of four gene loci—PPARG, SREBF1, CEBPA, and FABP4 with nucleoli. Nucleoli were revealed with anti-fibrillarin (green) and gene probes were delineated in red. a MSC; b AD14. c The graph shows the percentage of total gene signals co-localized with nucleoli (means ± SEM). Scale bar, 10 μm (JPG 234 kb)
Rights and permissions
About this article
Cite this article
Szczerbal, I., Foster, H.A. & Bridger, J.M. The spatial repositioning of adipogenesis genes is correlated with their expression status in a porcine mesenchymal stem cell adipogenesis model system. Chromosoma 118, 647–663 (2009). https://doi.org/10.1007/s00412-009-0225-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-009-0225-5