
Abstract
To evaluate the stability of phlogopite in the presence of carbonate in the Earth’s mantle, we conducted a series of experiments in the KMAS–H2O–CO2 system. A mixture consisting of synthetic phlogopite (phl) and natural magnesite (mag) was prepared (phl90-mag10; wt%) and run at pressures from 4 to 8 GPa at temperatures ranging from 1,150 to 1,550°C. We bracketed the solidus between 1,200 and 1,250°C at pressures of 4, 5 and 6 GPa and between 1,150 and 1,200°C at a pressure of 7 GPa. Below the solidus, phlogopite coexists with magnesite, pyrope and a fluid. At the solidus, magnesite is the first phase to react out, and enstatite and olivine appear. Phlogopite melts over a temperature range of ~150°C. The amount of garnet increases above solidus from ~10 to ~30 modal% to higher pressures and temperatures. A dramatic change in the composition of quench phlogopite is observed with increasing pressure from similar to primary phlogopite at 4 GPa to hypersilicic at pressures ≥5 GPa. Relative to CO2-free systems, the solidus is lowered such, that, if carbonation reactions and phlogopite metasomatism take place above a subducting slab in a very hot (Cascadia-type) subduction environment, phlogopite will melt at a pressure of ~7.5 GPa. In a cold (40 mWm−2) subcontinental lithospheric mantle, phlogopite is stable to a depth of 200 km in the presence of carbonate and can coexist with a fluid that becomes Si-rich with increasing pressure. Ascending kimberlitic melts that are produced at greater depths could react with peridotite at the base of the subcontinental lithospheric mantle, crystallizing phlogopite and carbonate at a depth of 180–200 km.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phlogopite is commonly found in mantle xenoliths in kimberlites (Erlank et al. 1987; van Achterbergh et al. 2001) or basalts (Nixon 1987) and is a main constituent of mantle-derived magmas such as group II kimberlites (orangeites) (Edwards et al. 1992; Mitchell 1995) and carbonatites (e.g. McCormick and Le Bas 1996). Inclusions of phlogopite in diamonds testify to its stability at depths where diamonds grow (Sobolev et al. 1997, 2009; Leost et al. 2003). The breakdown of this water-bearing, alkali-rich mineral will considerably affect mantle processes: Water released will lower the mantle solidus and induce partial melting or stabilize a fluid at subsolidus temperatures. Both, alkali-rich melts and fluids, will infiltrate and metasomatize mantle rocks (cf. review by Thompson 1992). Hence, many experimental studies have been carried out to constrain the stability of phlogopite in the earth’s mantle (cf. review by Frost 2006). Only a few studies focus on melting relations: pure phlogopite systems (Sato et al. 1997; Trønnes 2002), phlogopite + enstatite (Modreski and Boettcher 1972; Sato et al. 1997) and phlogopite + diopside (Luth 1997). Phlogopite in a natural, carbonated spinel lherzolite was studied from 1 to 3 GPa (Wendlandt and Eggler 1980). More information on phlogopite stability comes from multiple saturation experiments on orangeites (Yamashita et al. 1995; Ulmer and Sweeney 2002). The former study reports the occurrence of phlogopite at a temperature of 1,450°C at 4 GPa and a high pressure limit of 6 GPa. Ulmer and Sweeney (2002) found phlogopite at temperatures <1,300°C at ~3 GPa, with pressures of 4–5 GPa as the pressure limit.
Two different melting reactions for phlogopite in carbonate-bearing peridotite have been proposed by (1) Wendlandt and Eggler (1980) and by (2) Ulmer and Sweeney (2002):
As outlined above, no study focuses on melting relations of phlogopite in the presence of carbonates at pressures >3 GPa. The purpose of this study was to constrain phase relations of phlogopite in the presence of magnesite from 4 to 8 GPa by locating the proposed reaction (2), thereby contributing to the understanding of sources for alkali-rich melts and fluids that will be potential metasomatic agents in the mantle.
Experimental and analytical procedures
Starting material
Phlogopite was synthesized from high-purity oxides (MgO, Al2O3 and SiO2 of 99.95, 99.99 and 99.5% purity, respectively) and carbonate (K2CO3 of 99.0% purity) from Alfa Chemicals. A stoichiometric mix was loaded into a 5-mm-outer diameter (OD) Pt capsule along with ~7 wt% distilled H2O. The capsule containing the oxide-carbonate mix was sealed by arc-welding, assembled in a 19 mm talc-Pyrex assembly (Kushiro 1976) and run in an end-loaded piston cylinder for 48 h at 1.5 GPa and a temperature of 1,000°C. Temperature was monitored using a W95Re5–W74Re26 thermocouple without correction for pressure effects on EMF. We did not decarbonate the mix beforehand, so that at run conditions, the CO2 will be dissolved in the fluid, and the resulting H2O–CO2 fluid should contain less solute than a H2O fluid (e.g. Eggler 1987). An aliquot of the synthesized phlogopite was checked by XRD to ensure that only phlogopite is present. Different batches of phlogopite were synthesized to test whether our results were reproducible with slightly changing compositions of the synthetic phlogopite. Chemical analysis by electron microprobe turned out to be problematic because of the extremely small grain size of the synthesized crystals (≤1 μm). This required analysis with a focused beam, which resulted in the loss of potassium during the analysis and, consequently, relatively low K2O contents (Table 1).
Fragments of natural magnesite from Mt. Brussilof, British Columbia, Canada, which were free of visible inclusions, were separated out under a binocular microscope, analyzed by electron microprobe (Table 1) and ground in an agate mortar. A mix of phl90 + mag10 (wt%), consistent with the reaction proposed by Ulmer and Sweeney (2002), was used in the experiments.
Experimental setup and analytical methods
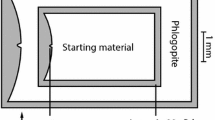
Capsules were fabricated from 4 mm lengths of 1.5-mm-OD Pt tubing. One end was triple-crimped, sealed by arc-welding and flattened. The starting material was loaded so that the capsule contains a thin layer of around 1–2 mm. After cleaning any powder from inside the top end of the capsule, it was triple-crimped, placed in an oven to dry overnight at 120°C and then sealed by arc-welding. A dampened tissue was wrapped around the lower part of the capsule during welding to keep the starting material cool. The sealed capsule was then compressed into cylindrical shape. At this point, the integrity of the capsule was tested by placing it into a water bath for several hours. If the capsule gained weight, it was not used.
All experiments were carried out using our standard 18/11 high-T assemblies (Walter et al. 1995). ZrO2 sleeves with 7 mm outer and 3 mm inner diameter were inserted into semi-sintered, Cr2O3-doped (5%) MgO octahedra. MgO spacers were used to center the capsules, which were placed within a MgO sleeve. W95Re5–W74Re26 thermocouple wires (0.254 mm diameter) were inserted axially into the assembly, encased in a crushable Al2O3 four-bore sleeve. All experiments were heated by stepped graphite furnaces. Each sample assembly was dried at 120°C overnight and then fired without the sample capsule at 1,000°C for 1 h in a N2–H2 (2%) gas that prevents the thermocouple wires from oxidizing.
In experiments at pressures of up to 7 GPa, we did not need to protect the thermocouple wires with copper coils. At higher pressures, however, the thermocouple wires always were torn apart by the deformation of the pyrophyllite gaskets, resulting in failure of the experiment. Consequently, we were using Cu coils in experiments at pressures of 8 GPa, knowing that this may affect the EMF readings so that temperatures could be underestimated (Nishihara et al. 2006). Our EMF readings were not corrected for any pressure effects on the thermocouples.
All experiments were performed using the UHP-2000 uniaxial split-sphere multi-anvil apparatus at the University of Alberta. Samples were pressurized first and heated after at a rate of 60 mV per hour (~60°C min−1) at pressures from 4 to 7 GPa. The heating rate was reduced to 30 mV per hour (~30°C min−1) at 8 GPa, which helped to reduce thermocouple breaks. Experiments were quenched by cutting off the power to the furnaces, dropping the temperature below 300°C in 2–5 s, and then decompressed over 2.5–4 h. Experimental charges were mounted in epoxy (Petropoxy 154), ground open, impregnated under vacuum and polished. Samples were polished using a rotating disk and Al2O3 powder of 5, 1 and 0.05 μm grain size suspended in oil, initially, to preserve potentially water-soluble phases. Precipitation of solute from a fluid phase (see below) made further impregnation with epoxy impossible leading to plucking. The polishing was improved by using the same corundum powder suspended in water instead. Samples were cleaned in an ultrasonic bath, dried overnight at 60°C and then carbon-coated for microprobe analysis.
Analyses were done using a JEOL 8900 microprobe (EPMA) at the University of Alberta with an acceleration voltage of 15 kV and a cup beam current of 15 nA. Standards were sanidine (K), Fo93 olivine (Mg), pyrope (Al, Si), diopside (Ca), albite (Na), willemite (Mn) and hematite (Fe). Counting time was set to 20 and 10 s on peak and background, respectively. A focused beam was used to analyze pyroxene, olivine, magnesite and garnet; beam diameters of 0–3 μm, depending on grain size, were chosen for phlogopite grains. Raw data were corrected using the Phi–Rho–Z program provided by JEOL. NORM was used to normalize and recalculate mineral compositions (P. Ulmer, personal communication, 2007).
Results
General observations
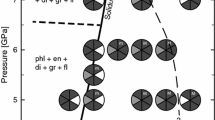
The synthetic phlogopite reacts to form phlogopite plus garnet and a fluid at subsolidus conditions and coexists with magnesite to 1,250, 1,200, 1,200 and 1,150°C at 4, 5, 6 and 7 GPa, respectively (Table 2; Fig. 1). A similar subsolidus reaction was observed by Sato et al. (1997) and Luth (1997). This phlogopite in equilibrium with garnet, magnesite and fluid is referred to as “primary” in this paper.

Phase relations of phlogopite with magnesite. Light gray quadrants indicate primary phlogopite that coexists with melt. Numbers indicate the duration of the experiment in hours. En enstatite, f/m fluid present if quadrant dark gray/dark gray quadrant with m to M for increasing melt portion; Mag magnesite, Ol olivine, Phl phlogopite, Py pyrope, sp spinel
To constrain the amount of fluid and vapor present at subsolidus conditions, we weighed several capsules, froze them at −20°C and cut them open with a razor blade. Fetid-smelling gas escaped, but the amount lost was too little to measurably change the weight of the frozen capsule. Capsules were then heated at 120°C for 20 min and lost ~0.03 mg of weight (~2–3 wt%), which reflects the amount of water present.
A change in texture is obvious above 1,200, 1,200, 1,200 and 1,150°C at 4, 5, 6 and 7 GPa, respectively, and at 1,150°C at 8 GPa (Fig. 2): Over a temperature range of ~150°C the amount of primary phlogopite decreases and elongated, dendritic crystals become common, which were identified to be phlogopite by X-ray diffraction in combination with the EPMA data. This dendritic phlogopite is interpreted to have grown upon quench of the experiment from a hydrous melt (e.g. Yoder and Kushiro 1969). In this ~150°C temperature interval, primary-textured phlogopite is also present and coexists with melt. Magnesite has reacted out at these conditions, and olivine and enstatite have formed and coexist with garnet.

Textural changes from a subsolidus, through b solidus to c and d super-solidus. Scale bar in upper left of each image is 150 μm. Enstatite is absent in charges shown. a Subsolidus assemblage: primary phlogopite (p-phl: light gray) in equilibrium with pyrope (py: gray, round grains) and magnesite (mag: dark gray). Magnesite is homogeneously distributed throughout the capsule. Grain size in general is small (1–50 μm) below solidus. b Magnesite has reacted out, except for some relicts enclosed by garnet. Most phlogopite is still primary. Black box highlights the first small melt pocket along the capsule wall, identified by small quench phlogopite needles (q-phl: light gray) and quench magnesite patches (q-mag: black). Note the larger grain size compared to subsolidus conditions. c Increasing the temperature further increases the melt fraction; quench phlogopite is present together with garnet and olivine. d Q-phl (melt) accumulated in the upper half of the capsule as well as interstitially between garnet grains. Olivine grains are located at the bottom of the capsule
Upon opening capsules that were run in the P/T zone of residual primary plus quench phlogopite, a hydrous solution and vapor, probably CO2, bubble out. The solution precipitates a K-rich phase around the notch and within the capsule (Fig. 3).

K-rich precipitates on the capsule surface viewed through a reflected light microscope. See text for details
In experiments run above 1,300 and 1,350°C at 6 and 4 GPa, respectively, the only phlogopite is quench phlogopite, associated with olivine ± enstatite and garnet. At low pressure and high temperature, spinel occurs in some of the run products. In contrast to the above observations, the hydrous solution that escapes these capsules contains little or no solute.
The K-rich precipitates were identified qualitatively to be K-rich by EDS on the microprobe and are highly water soluble. Their small amount prohibited quantitative analysis, but we infer they are probably potassium carbonate.
Garnet
Pyrope grains are euhedral and generally up to 25 μm in diameter in subsolidus experiments, but up to 80 μm in diameter at and above the solidus (Fig. 2). Below the solidus, the modal abundance of garnet increases slightly to higher temperatures and pressures from ~10 to ~15%, whereas above the solidus, the modal abundance increases to ~30%.
As expected from earlier studies (e.g. Ringwood 1967; Kanzaki 1987; Luth 1997), the SiO2 content of the garnet increases with increasing pressure, reflecting an increasing majoritic component. The number of Si cations is greater than 3 per 12 oxygen for some garnets at 4 GPa and for most garnets at pressures >5 GPa (Table 3). The same trend of Si enrichment (by up to 0.13 cations per 12 oxygen) is found with increasing temperatures as well, which may reflect an increase in Si activity of the melt at higher temperatures.
Magnesite
Magnesite grains are prismatic, subhedral to euhedral and up to 50 μm in length. Often, rounded grains are found as inclusions in garnet. Magnesite itself does not contain mineral inclusions. It is non-stoichiometric with rather low MgO ranging from 39 to 44 wt%. Non-stoichiometry in carbonates was also reported in previous high pressure studies; the data available from early research are limited, though, making it impossible to give a general explanation (Katsura and Ito 1990; Luth 2006; Keshav and Gudfinnsson 2010). Perhaps, the EPMA standard chosen for Mg (Forsterite93) is responsible for the offset of the measurements from stoichiometric. At 4 and 5 GPa and 1,250°C, at the solidus, magnesite is intergrown with quench phlogopite (Fig. 2b). We interpret this magnesite to be a quench product from melt. No significant amount of potassium was detected, except for magnesite at 8 GPa (Table 4). This one is difficult to tell apart from primary magnesite by texture, but, as olivine and enstatite are present, it only can be a secondary magnesite that quenched from melt. With increasing pressures, the total MgO content measured increases from 39.4 to 44.9 wt% at 4 and 8 GPa, respectively.
Phlogopite
Primary phlogopite is elongated to equant and measures up to 100 μm in length. Quench phlogopite is of elongated, dendritic habit and is small (<1 μm) and interstitial or up to 500 μm in length, typically with a large length to width ratio. Primary phlogopite sometimes contains inclusions of small garnet grains, whereas quench phlogopite usually is free of mineral inclusions (Fig. 2).
At 1,150 and 1,250°C, primary phlogopite is slightly enriched in MgO and SiO2 and depleted in Al2O3 to higher pressures (Table 5; Fig. 4). A similar trend was observed by Sato et al. (1997). Other studies report a slight enrichment in SiO2 to higher pressures (Luth 1997; Konzett and Fei 2000; Trønnes 2002). Primary phlogopite melts over a temperature interval of ~150°C. One experiment above solidus (ae-58, 1,350°C, 7 GPa) contains, besides residual primary phlogopite, few grains of a nearly anhydrous phase with a water content of ~1 wt% (46.55, 12.68, 28.73 and 11.05 wt% for SiO2, Al2O3, MgO and K2O, respectively, and a total of 99.01 wt%). For quench phlogopite, see “Melt” section below.

Variation of SiO2 and Al2O3 in phlogopite as a function of pressure. Squares (p-phl primary phlogopite) and diamonds (q-phl quench phlogopite) give the average composition at the given pressure over the whole temperature range phlogopite is present. Horizontal lines mark stoichiometric SiO2 and Al2O3 content. Error bars give the standard deviation. a With increasing pressure SiO2 increases and increases dramatically in p-phl and q-phl, respectively. Q-phl at 4 GPa is similar in composition to p-phl. The large standard deviation for q-phl at 5 GPa is due to the presence of residual p-phl coexisting with melt at 1,400°C. With increasing temperature, p-phl disappeared and SiO2 in q-phl decreases. b Al2O3 decreases and decreases dramatically in p-phl and q-phl, respectively. Compositions at 5.5 GPa (ae-22) break the trend, which could be due to variations in the starting material. See text for further details
Pyroxene
Enstatite occurs only above the solidus, is limited to lower temperatures and is prismatic, euhedral, up to 50 μm in length and free of mineral inclusions. Enstatite is rare or absent above 1,400°C or at pressures >6 GPa, and olivine is more common or the only phase coexisting with garnet and melt.
All orthopyroxenes incorporate Al. At the solidus, at 5 GPa and 1,250°C, the pyroxenes contain ~0.9 wt% of Al2O3, whereas at 1,400°C and the same pressure, they contain 1.5 wt% Al2O3. We interpret this change to result from the dissolution of phlogopite into the melt, increasing the activity of Al2O3 in the melt, thereby increasing the Al content of the orthopyroxene, as observed at lower pressures by Modreski and Boettcher (1972).
Olivine
Olivine only occurs above the solidus and exhibits a prismatic to equant habit, is subhedral to rarely euhedral and grows to up to 200 μm in size (Fig. 2). Olivine contains no mineral inclusions and often is present at the bottom of the capsules. Olivine is stoichiometric and does not significantly substitute Al or K, which is consistent with Luth (1997) and Trønnes (2002).
Melt
Sato et al. (1997) approximate their melt composition by analyzing quench phlogopite. In our experiments, however, it is obvious that the hydrous quench solution carries solute, especially over the temperature range where primary phlogopite melts out (Fig. 3). But this approach becomes more realistic as a valid approximation at higher temperatures, where less or no solute is precipitating from that solution and where less solution seems to be occurring in the presence of more abundant quench phlogopite (Fig. 2d).
Intergrowth of only minor Si-rich quench phlogopite with magnesite along the capsule walls at 4 and 5 GPa at 1,250°C, together with the observation of K-rich precipitates, is taken as evidence that the first melt is K–CO2–H2O–Mg-rich and relatively Si-poor. More Si-rich quench phlogopite present at 5 GPa could reflect an increase in Si activity in the melt at higher pressures.
Within the temperature range where primary phlogopite remains in the capsule, the associated quench phlogopite is rich in SiO2 (~45–48 wt%) and low in Al2O3 (~5–9 wt%). At higher temperatures and 4 GPa, without primary phlogopite present, the average composition of quench phlogopite is close to that of primary phlogopite, but showing a wider standard deviation with respect to Si, Mg and Al content. This variation may be either caused by poor measurements on the small dendritic crystals or by heterogeneity of the melt itself. Potassium remains close to stoichiometric.
There is a clear trend in the quench phlogopite compositions at conditions without residual primary phlogopite and increasing pressure: from 4 GPa with SiO2 ~42 wt%, Al2O3 ~15 wt%, which is similar to primary phlogopite, to 5 GPa with SiO2 ~46 wt%, Al2O3 ~11 wt%, to 6 GPa with SiO2 ~50 wt%, Al2O3 ~6 wt%, to 7 GPa with SiO2 ~51 wt% and Al2O3 ~5 wt% (Table 5; Fig. 4). In our experiments, this trend becomes apparent at temperatures of 1,350, 1,450–1,550°C.
Discussion
Phase relations
The observed phase relations are controlled by two main reactions: reaction (3) and (4):
A similar subsolidus breakdown of phlogopite has been observed at a pressure of ~4.5 and ~6 GPa by Sato et al. (1997) and Luth (1997), respectively. The former formulated the following reaction to explain the occurrence of pyrope and the enrichment of SiO2 and MgO in primary phlogopite to higher pressures (modified by Trønnes 2002):
The hypothetical phlogopite in reaction (3a) requires Mg to occupy tetrahedral sites. Alternatively, magnesian montdiorite (KMg2.5Si4O10(OH)2) component with 2SiIV + \( \square \) VI = 2AlIV + MgVI (Seifert and Schreyer 1971) could explain the observed compositional trend in the phlogopite with Al > 2 cations per 22 oxygen. Primary and quench phlogopite below 6 GPa have Al < 2 cations per 22 O, which points to an eastonite component SiIV + MgVI = AlIV + AlVI (Arai 1984).
In our experiments, the occurrence of pyrope can be explained by formulating a simplified reaction such as (3b) with stoichiometric synthetic phlogopite and with stoichiometric K, (OH), vacancies (\( \square \)) in the octahedral site, all Al in tetrahedral coordination and (Al, Si) IVΣ=4 on the product side:
The fluid phase contains K2O, MgO and SiO2. A higher Si activity of the fluid at higher pressures probably is reflected by the increasing SiO2 in primary phlogopite with increasing pressure. The Mg and CO2 content of the fluid may be buffered by the magnesite, which has higher MgO contents with increasing pressure (see “Magnesite” section above). Pyrope abundance at subsolidus conditions is not increasing much from 4 to 8 GPa, from ~10 to ~15 modal%, respectively. The water of the fluid is mainly produced by the breakdown reaction of phlogopite (3b). A smaller amount of water probably comes from dehydroxylation (e.g. Vedder and Wilkins 1969) of the primary phlogopite itself, which is reflected by a tendency in the primary phlogopite data to have higher totals (>95 wt%) at higher pressures. Reaction (3b), however, does not explain why the synthetic phlogopite is unstable.
Sato et al. (1997) use natural, Fe-bearing phlogopite in their starting material and perform experiments using Pt capsules. In their study, phlogopite breakdown could result from Fe loss to the platinum capsule, since Pt is known to alloy with iron (Isaacs and Tamman 1907; Bowen and Schairer 1932). Phlogopite in their starting material contains 1.83 wt% FeO, whereas in the run products, it only contains 0.01–0.31 wt% FeO. Why this breakdown is observed ≥4.5 GPa is unclear, though, and implies that another mechanism is involved as well, since Fe is also lost to the capsule at 4 GPa without phlogopite being destabilized. To illustrate the Fe loss, a simplified reaction (3c) can be formulated. Phlogopite compositions chosen ((K) XIIΣ=1 (Mg, Fe2+) VIΣ=3 (Al, Si) IVΣ=4 ) are similar to Sato et al. (1997) with respect to Mg and Fe cations:
The breakdown in our experiments is not caused by Fe loss, and pure synthetic phlogopite experiments we performed rule out the presence of carbonate as the trigger as well. Trønnes (2002) did not observe this breakdown of synthetic phlogopite in his experiments. Unfortunately, grains were too small for a chemical analysis of his starting material. More likely, a non-stoichiometric nature of the synthetic phlogopite causes this breakdown. As mentioned above, it was difficult to get a chemical analysis of the ≤1 μm-sized phlogopite crystals. K is low (1.87 cations per 22 O) because we had to probe with a focused beam, and Si is higher (6.19 cations per 22 O), possibly a result of homogenizing the starting material in an agate mortar. Si/Al is close to the ideal 3/1 with Al being 2.05 cations per 22 O. Mg is low with 5.89 cations per 22 oxygen. Luth’s (1997) synthetic phlogopite has 6.04, 2.02, 5.90 and 2.00 for Si, Al, Mg and K cations per 22 O, respectively. Luth (1997) has some vacancy (~0.1) in the octahedral site, whereas the other sites are full and stoichiometric. Sato et al. (1997) have vacancies of 0.117 in their octahedral site. In our case, the vacancy is 0.11, and we might have vacancies of <0.13 per 22 O in the I-site. Perhaps, vacancies in the crystal structure are the trigger for such a breakdown.
More garnet, relative to the amount resulting from (3b), is produced in this reaction; thus, we assume reaction (4) to be univariant in the six-component system and think that the divariant appearance is caused by thermal gradients within the Pt capsule. We show in this study that reaction (4) is sensitive to temperature rather than pressure and found that phlogopite, in the presence of carbonate, is stable to pressures up to 8 GPa and temperatures <1,150°C. This is in contrast to reaction (2) that was proposed by Ulmer and Sweeney (2002), which predicts that phlogopite melts >4 GPa in the presence of carbonate. They, however, use a different experimental approach (multiple saturation), which can be problematic in the presence of K–H2O phases that exhibit incongruent melting (Yoder and Kushiro 1969). In addition, they use a more complex starting mix (synthetic kimberlite), which would reduce the phlogopite stability compared to simple or end-member systems.
An increase in the amount of garnet is observed to higher temperatures and pressures (Sato et al. 1997; Trønnes 2002; this study) and is coupled with the composition of primary and quench phlogopite, which get enriched in Si and depleted in Al. Sato et al. (1997) did not find exactly the same trend in their quench phlogopite compositions of the phlogopite–enstatite system. Al2O3 decreases to high pressures with 12.03, 12.23, 9.24 and 6.25 wt% at 4, 5, 6 and 8 GPa, respectively. In agreement with our study, Al2O3 decreases more rapidly at pressures >5 GPa. SiO2 in their study, on the other hand, is high already at 4 GPa (~50 wt%) and varies from 45 to 48 wt% to higher pressures.
In our study, quench phlogopite, identified by texture at 4 GPa, shows compositions similar to primary phlogopite to temperatures as high as 1,550°C. Probably, residual primary phlogopite was still present at low pressures in Sato et al. (1997) (see “Melt” section above). This similarity in the composition of quench to primary phlogopite could lead to misinterpretations in studies that only use chemical arguments to distinguish between them. Other studies within the same pressure range either do not observe quench phlogopite or do not report its composition.
Enrichment of Si in phlogopite compositions was also observed in the previous studies. Trønnes (2002) did experiments in a pure synthetic phlogopite system and reports compositions containing 42.8–43.4 wt% SiO2 at pressures of 4.5–7.4 GPa, respectively. Luth (1997) ran synthetic phlogopite with diopside and measured phlogopite with 42.2–42.5 wt% SiO2 at pressures of 3–5 GPa, respectively. Fluid was absent at the above conditions, and phlogopite compositions remain constant. The following studies contain a fluid below the solidus: Above 6 GPa, Luth’s (1997) phlogopite breaks down releasing a fluid, and the remaining phlogopite compositions are enriched in SiO2, ranging from 43.6 to 43.7 wt% at a pressure of 7.5 GPa. Sato et al. (1997) performed experiments in the pure natural phlogopite system and report SiO2 contents of 41.04, 40.73 and 41.69 wt% at pressures of 4, 6 and 8 GPa, respectively. Further experiments using natural phlogopite with synthetic enstatite result in SiO2 contents of 41.71, 45.95 and 44.71 wt% at pressures of 4, 6 and 8 GPa, respectively. Experiments using synthetic phlogopite with natural magnesite (this study) result in SiO2 contents of 42.1–44.5 wt% at pressures of 4–8 GPa, respectively. Konzett and Ulmer (1999) did experiments in KNCMASH with 1.22 wt% of H2O resulting in SiO2 contents in phlogopite varying from 42.1 to 45.2 at pressures of 4–8 GPa, respectively. In the modified lherzolite system (BRIAN2), 0.21 wt% H2O is present and SiO2 ranges from 41.7 to 43.5 wt% at pressures of 6–6.5 GPa, respectively (Fig. 5). Konzett and Fei (2000) have 4.76 wt% water in KNCMASH, and their phlogopite contains 43.3–43.9 wt% SiO2 at a pressure of 10 GPa.

Comparison of phlogopite data. Systems with fluid at subsolidus conditions show a wider spread in Al and Si apfu. Modal abundance of garnet increases to higher pressures from ~10 to 15% and ~10–30% at subsolidus and super-solidus conditions, respectively. See text for discussion. BRIAN2 modified natural lherzolite, di diopside, en enstatite, K&U Konzett and Ulmer, KNCMASH K2O–Na2O–CaO–MgO–Al2O3–SiO2–H2O, p primary phlogopite, phl phlogopite, py pyrope, q quench phlogopite
Quench phlogopite compositions (this study) show a dramatic increase in SiO2 above the solidus of 41.2, 44.6, 49.1 and 50.7 wt% at pressures of 4, 5, 6 and 7 GPa, respectively. Although this quenched phlogopite does not represent an exact melt composition, the increase in silicon may reflect a higher Si activity in the melt at pressures ≥5 GPa.
Generally, a hydrous fluid or melt at subsolidus or super-solidus conditions, respectively, coupled with and may be controlled by garnet-forming reactions such as (3b), enhances a montdioritic substitution in phlogopite with increasing pressure, leading to hypersilicic phlogopite compositions.
Implications for subduction environments
Phlogopite can be produced above a subducting slab by reactions of hydrous K-rich fluid with peridotite (Sato et al. 1997; Konzett and Ulmer 1999) or with metasediments (Massonne 1992; ii in Fig. 7a). K-rich melts and fluids originate from the melting of phengite (Vielzeuf and Schmidt 2001) and successive dehydration of phengite, respectively (Schmidt 1996; Hermann and Spandler 2008). Furthermore, reactions of hydrous silicate melt from the slab with peridotite in the mantle wedge will result in the precipitation of phlogopite and enstatite (Wyllie and Sekine 1982). Assuming common subduction zone geometries with geotherms parallel to a subducting slab (Davies and Stevenson 1992; Schmidt and Poli 1998), with an ambient mantle temperature of 1,250°C at ~160 km depth, resulting in slab surface temperatures of <1,000°C below the arc (Kincaid and Sacks 1997; van Keken et al. 2002), and considering solidi of previous studies (see “Introduction” for references; Fig. 6), phlogopite remains stable above the slab. In this case, phlogopite is dragged to greater depths, until it breaks down at subsolidus conditions to K-richterite over the pressure range of 7–11 GPa (Sudo and Tatsumi 1990), 9–11 GPa (Luth 1997) and 6.5–8.5 GPa (Konzett and Ulmer 1999).

Compilation of phlogopite breakdown curves with continental geotherms (dashed) and Cascadia-type subduction adiabat (shaded region). Addition of CO2 shifts the solidus significantly to lower temperatures. See text for discussion
It is unclear how far above the slab phlogopite would form. In the following discussion, slab surface temperatures, thus, are taken to be the minimum temperature at which phlogopite could occur. The further away from the surface, the hotter will be temperatures in the phlogopite-bearing peridotite. Conder (2005) shows that older subduction models use parameters that will underestimate a slab surface temperature by 200–300°C. Furthermore, slab surface temperatures may vary locally within a slab, depending on subduction style and slab-mantle wedge interaction, by up to 200°C (Kincaid and Griffiths 2003). Syracuse et al. (2010) compare different models, and hottest slab surface temperatures calculated at a depth of 240 km are ~1,150°C. Solidi of previous phlogopite studies are located at too high temperatures (≥1,250°C), and phlogopite remains stable above the slab.
In contrast, LaTourrette et al. (1995) conclude that phlogopite should be involved in the melting process to produce the common geochemical arc magma signature with respect to LILE. K-rich lamprophyric rocks found in arcs of young subduction zones (e.g. Esperança and Holloway 1987; Hochstaedter et al. 1996; Maria and Luhr 2008; Owen 2008; Vigouroux et al. 2008) support the presence and melting of phlogopite within the mantle wedge. Previous CO2-free studies cannot support this idea, because phlogopite is stable to too high temperatures (Fig. 6). CO2, however, should be considered because carbonates are present in most subduction environments and are subducted into the mantle (Rea and Ruff 1996; Plank and Langmuir 1998). Modeling shows that more than 70% of carbonates in a slab will be stable in the typical volcanic arc region and will be transported to greater depths (Thomsen and Schmidt 2008). Metamorphic devolatilization of carbonates is promoted by hydrous fluids infiltrating carbonate sediments (Kerrick and Connolly 2001), and resulting CO2-bearing fluids will react with peridotite to form carbonates (i in Fig. 7a). The stability of carbonates has been studied extensively to date (cf. review by Luth 1999), and different peridotite carbonation reactions have been proposed: e.g. olivine + diopside + CO2 = dolomite + enstatite (Wyllie et al. 1983); magnesite + diopside = dolomite + enstatite (Brey et al. 1983), with magnesite being the stable carbonate at higher pressures, in the diamond stability field.

Sketch of a Cascadia-type subduction zone (numbers from Syracuse et al. 2010). Slab temperatures and mantle isotherms in °C. Blue and brown denote the presence of carbonate and phlogopite, respectively. Phlogopite with carbonate melts at a depth of ~240 km (star). Resulting melts rise and enrich the mantle wedge in K and CO2. Melt could react with cooler mantle underneath the continent (iii), possibly crystallizing phlogopite and carbonate, which could be transported back into the mantle wedge. See text for i–ii. b Melt from below infiltrates the subcontinental lithospheric mantle and reacts with enstatite, olivine and pyrope to phlogopite, magnesite and a fluid. en enstatite, fl fluid (dark blue), G/D graphite/diamond transition, mag magnesite (light blue), melt (gray), ol olivine, phl phlogopite (brown), py pyrope
If carbonation reactions and phlogopite metasomatism take place within the mantle wedge, our study shows that the presence of carbonate will lower the temperature stability of phlogopite considerably. If not broken down to K-richterite yet, phlogopite will melt at pressures of ~7.5 GPa (this study) in the presence of carbonate in a Cascadia-type subduction zone (shaded region in Fig. 6; star in Fig. 7a). Alternatively, assuming phlogopite forms away from the slab surface, or, considering a tendency to underestimate slab surface temperatures and local variations in temperatures on the slab surface, the solidus may be reached in the hotter region above a slab in a very hot, young and slow subduction environment at lower pressures already. In this case, phlogopite melting above the slab may happen locally, on a smaller scale, whereas phlogopite in cooler regions would be subducted to greater depths.
Implication for the subcontinental lithospheric mantle
The addition of CO2 to the system shifts the solidus significantly to lower temperatures (Fig. 6), whereas the pressure stability is not dramatically reduced in this simple system. A cool 40-mWm−2 geotherm intersects the carbonated solidus above 6 GPa, implying that phlogopite will be stable to a depth of ~200 km in the presence of carbonate. Unlike the suggestion of Ulmer and Sweeney (2002), the presence of a C–O–H fluid does not destabilize the phase assembly present at subsolidus conditions at pressures >4 GPa. In a colder lithospheric mantle (<40 mWm−2), the stability would extend to even higher pressures. The hotter geotherm (44 mWm−2) intersects the solidus at a depth of ~180 km. To this depth, a fluid, that, with increasing pressure becomes enriched in Si (see “Phase relations” section above), is coexisting with phlogopite, magnesite and pyrope. This is in agreement with the data of Mibe et al. (2002) who found an increasing amount on Mg and Si being dissolved in a fluid in the MgO–SiO2–H2O system with increasing pressure. Beyond 180 km depth, at the base of a subcontinental lithospheric mantle, a hydrous, Si-, K- and CO2-bearing silicate melt will be present and could be a potential metasomatic agent. Consequently, potassium- and CO2-rich magmas, such as kimberlites, originate at pressures beyond 5 GPa, which is in agreement with early studies (Wyllie 1980; Canil and Scarfe 1990; Dalton and Presnall 1998). Ascending melts, on the other hand, that were produced deeper could freeze within the subcontinental lithospheric mantle by reacting with garnet, olivine and pyroxene to form phlogopite plus carbonate and thus could be trapped (Fig. 7b). This freezing of the melt will happen at ~200 km or at ~180 km depth in a cold (40 mWm−2) or in a warmer lithospheric mantle (44 mWm−2), respectively. Resulting fluids will further percolate upwards in that lithospheric mantle. Reactivation of the lithosphere (Foley 2008) then might melt this low temperature assemblage and possibly will produce K-rich magmas. Given that phlogopite and carbonate need to occur in the same potential source region for group II kimberlites, and that carbonate and phlogopite can coexist to a considerable depth (i.e. to 180–200 km depth) in a continental lithospheric mantle, might be responsible for the scarcity of group II kimberlites reaching the surface.
Conclusions
Phlogopite, garnet and a C–O–H-fluid are in equilibrium with magnesite at 4, 5, 6 and 7 GPa to temperatures of 1,250, 1,200, 1,200, and 1,150°C, respectively. At higher temperatures, phlogopite and magnesite react to form enstatite, forsterite, pyrope and a hydrous Si-, K- and CO2-bearing melt. The modal amount of garnet above solidus further increases to higher pressures and temperatures from ~10 to ~30%. Melt quenches to dendritic phlogopite crystals and a hydrous solution. There is a dramatic change in quench phlogopite compositions along isotherms with increasing pressure from close to primary at 4 GPa, to hypersilicic at pressures ≥5 GPa.
In a subduction zone, the simple infiltration of a K-rich fluid and C–O–H fluid will not necessarily induce melting by destabilizing carbonate and phlogopite, respectively, as temperature rather than pressure is the limiting factor of such a phase assembly. A Cascadia-type subduction adiabat intersects the solidus found in the present study at a pressure of ~7.5 GPa, where phlogopite in the presence of carbonate will melt.
In a cold (40 mWm−2) subcontinental lithospheric mantle, phlogopite will be stable to 200 km depth in the presence of carbonate, and to a depth of 180 km assuming a 44-mWm−2 geotherm. Kimberlitic melts could be trapped at the base of a subcontinental lithospheric mantle by reactions with peridotite at ~180–200 km depth to form phlogopite and carbonate. Fluids in phlogopite-bearing peridotite become richer in Si at pressures ≥5 GPa. The same holds for a carbonated phlogopite-bearing lherzolite as long the CO2 is buffered by the stable carbonate phase.
References
Arai S (1984) Pressure-temperature dependent compositional variation of phlogopitic micas in upper mantle peridotites. Contrib Mineral Petrol 87:260–264
Bowen NL, Schairer JF (1932) The system FeO–SiO2. Am J Sci 224(24):177–213
Brey G, Brice WR, Ellis DR, Green DH, Harris KL, Ryabchikov ID (1983) Pyroxene-carbonate reactions in the upper mantle. Earth Planet Sci Lett 62:63–74
Canil D, Scarfe CM (1990) Phase relations in peridotite + CO2 systems to 12 GPa: implications for the origin of kimberlite and carbonate stability in the earth’s upper mantle. J Geophys Res 95:15805–15816
Conder JA (2005) A case for hot slab surface temperatures in numerical viscous flow models of subduction zones with an improved fault zone parameterization. Phys Earth Planet Inter 149:155–164
Dalton JA, Presnall DC (1998) The continuum of primary carbonatitic-kimberlitic melt compositions in equilibrium with lherzolite: data from the system CaO–MgO–Al2O3–SiO2–CO2 at 6 GPa. J Petrol 39:1953–1964
Davies JH, Stevenson DJ (1992) Physical model for the source region of subduction zone volcanics. J Geophys Res 97:2037–2070
Edwards D, Rock NMS, Taylor WR, Griffin BJ, Ramsay RR (1992) Mineralogy and petrology of the Aries diamondiferous kimberlite pipe, Central Kimberley Block, Western Australia. J Petrol 33:1157–1191
Eggler DH (1987) Solubility of major and trace elements in the mantle metasomatic fluids: experimental constraints. In: Menzies MA, Hawkesworth CJ (eds) Mantle metasomatism. Geology series. Academic Press, London, pp 21–41
Erlank AJ, Waters FG, Hawkesworth CJ, Haggerty SE, Allsopp HL, Rckard RS, Menzies M (1987) Evidence for mantle metasomatism in peridotite nodules from the Kimberley pipes, South Africa. In: Menzies MA, Hawkesworth CJ (eds) Mantle metasomatism. Geology series. Academic Press, London, pp 221–311
Esperança S, Holloway JR (1987) On the origin of some mica-lamprophyres: experimental evidence from a mafic minette. Contrib Mineral Petrol 95:207–216
Foley SF (2008) Rejuvenation and erosion of the cratonic lithosphere. Nat Geosci 1:503–510
Frost DJ (2006) The stability of hydrous mantle phases. In: Keppler H, Smyth JR (eds) Water in nominally anhydrous minerals. Mineral Soc Am 62:243–271
Hermann J, Spandler CJ (2008) Sediment melts at sub-arc depths: an experimental study. J Petrol 49(4):717–740
Hochstaedter AG, Ryan JG, Luhr JF, Hasenaka T (1996) On B/Be ratios in the Mexican volcanic belt. Geochim Cosmochim Acta 60(4):613–628
Isaacs E, Tamman G (1907) Über die Legierungen des Eisens mit Platin. Z Anorg Chem 55:63–71
Kanzaki M (1987) Ultrahigh-pressure phase relations in the system Mg4Si4O12–Mg3Al2Si3O12. Phys Earth Planet Inter 49:168–175
Katsura T, Ito E (1990) Melting and subsolidus phase relations in the MgSiO3–MgCO3 system at high pressures: implications to evolution of the earth’s atmosphere. Earth Planet Sci Lett 99:110–117
Kerrick DM, Connolly JAD (2001) Metamorphic devolatilization of subducted marine sediments and the transport of volatiles into the earth’s mantle. Nature 411:293–296
Keshav S, Gudfinnsson G (2010) Experimentally dictated stability of carbonated oceanic crust to moderately great depths in the earth: results from the solidus determination in the system CaO–MgO–Al2O3–SiO2–CO2. J Geophys Res 115:20
Kincaid C, Griffiths RW (2003) Laboratory models of the thermal evolution of the mantle during rollback subduction. Nature 425:58–62
Kincaid C, Sacks IS (1997) Thermal and dynamical evolution of the upper mantle in subduction zones. J Geophys Res 102:12295–12315
Konzett J, Fei Y (2000) Transport and storage of potassium in the earth’s upper mantle and transition zone: an experimental study to 23 GPa in simplified and natural bulk compositions. J Petrol 41(4):583–603
Konzett J, Ulmer P (1999) The stability of hydrous potassic phases in lherzolitic mantle-an experimental study to 9.5 GPa in simplified and natural bulk compositions. J Petrol 40(4):629–652
Kushiro I (1976) Changes in viscosity and structure of melt of NaAlSi2O6 composition at high pressures. J Geophys Res 81:6347–6350
LaTourrette T, Hervig RL, Holloway JR (1995) Trace element partitioning between amphibole, phlogopite, and basanite melt. Earth Planet Sci Lett 135:13–30
Leost I, Stachel T, Brey GP, Harris JW, Ryabchikov ID (2003) Diamond formation and source carbonation: mineral associations in diamonds from Namibia. Contrib Mineral Petrol 145:15–24
Luth RW (1997) Experimental study of the system phlogopite-diopside from 3.5 to 17 GPa. Am Mineral 82:1198–1209
Luth RW (1999) Carbon and carbonates in the mantle. In: Fey Y, Bertka CM, Mysen BO (eds) Mantle petrology: field observations and high pressure experimentation: a tribute to Francis R. (Joe) Boyd. The Geochemical Society, Special Publication 6, pp 297–316
Luth RW (2006) Experimental study of the CaMgSi2O6–CO2 system at 3–8 GPa. Contrib Mineral Petrol 151:141–157
Maria AH, Luhr JF (2008) Lamprophyres, basanites, and basalts of the western Mexican volcanic belt: volatile contents and a vein–wallrock melting relationship. J Petrol 49:2123–2156
Massonne HJ (1992) Evidence for low-temperature ultrapotassic siliceous fluids in subduction zone environments from experiments in the system K2O–MgO–Al2O3–SiO2–H2O (KMASH). Lithos 28:421–434
McCormick GR, Le Bas MJ (1996) Phlogopite crystallization in carbonatitic magmas from Uganda. Can Mineral 34:469–478
Mibe K, Fujii T, Yasuda A (2002) Composition of aqueous fluid coexisting with mantle minerals at high pressure and its bearing on the differentiation of the earth’s mantle. Geochim Cosmochim Acta 66:2273–2285
Mitchell RH (ed) (1995) Kimberlites, orangeites, and related rocks. Plenum Press, New York, p 410
Modreski PJ, Boettcher AL (1972) The stability of phlogopite + enstatite at high pressures: a model for micas in the interior of the earth. Am J Sci 272:852–869
Nishihara Y, Matsukage KN, Karato SI (2006) Effects of metal protection coils on thermocouple EMF in multi-anvil high-pressure experiments. Am Mineral 91:111–114
Nixon PH (ed) (1987) Mantle xenoliths. Wiley, New York
Owen JP (2008) Geochemistry of lamprophyres from the Western Alps, Italy: implications for the origin of an enriched isotopic component in the Italian mantle. Contrib Mineral Petrol 155:341–362
Plank T, Langmuir CH (1998) The chemical composition of subducting sediment and its consequences for the crust and mantle. Chem Geol 145:325–394
Rea DK, Ruff LJ (1996) Composition and mass flux of sediment entering the world’s subduction zones: implications for global sediment budgets, great earthquakes, and volcanism. Earth Planet Sci Lett 140:1–12
Ringwood AE (1967) Pyroxene-garnet transformations in the earth’s mantle. Earth Planet Sci Lett 2:255–263
Sato K, Katsura T, Ito E (1997) Phase relations of natural phlogopite with and without enstatite up to 8 GPa: implication for mantle metasomatism. Earth Planet Sci Lett 146:511–526
Schmidt MW (1996) Experimental constraints on recycling of potassium from subducted oceanic crust. Science 272:1927–1930
Schmidt MW, Poli S (1998) Experimentally based water budgets for dehydrating slabs and consequences for arc magma generation. Earth Planet Sci Lett 163:361–379
Seifert F, Schreyer W (1971) Synthesis and stability of micas in the system K2O–MgO-SiO2–H2O and their relations to phlogopite. Contrib Mineral Petrol 30:196–215
Sobolev NV, Kaminsky FV, Griffin WL, Yefimova ES, Win TT, Ryan CG, Botkunov AI (1997) Mineral inclusions in diamonds from the Sputnik kimberlite pipe, Yakutia. Lithos 39:135–157
Sobolev NV, Logvinova AM, Efimova ES (2009) Syngenetic phlogopite inclusions in kimberlite-hosted diamonds: implications for role of volatiles in diamond formation. Russ Geol Geophys 50(12):1234–1248
Sudo A, Tatsumi Y (1990) Phlogopite and K-amphibole in the upper mantle: implication for magma genesis in subduction zones. Geophys Res Lett 17(1):29–32
Syracuse EM, van Keken PE, Abers GA (2010) The global range of subduction zone thermal models. Phys Earth Planet Inter 183:73–90
Thompson AB (1992) Water in the earth’s upper mantle. Nature 358:295–302
Thomsen TB, Schmidt MW (2008) Melting of carbonated pelites at 2.5–5.0 GPa, silicate–carbonatite liquid immiscibility, and potassium–carbon metasomatism of the mantle. Earth Planet Sci Lett 267:17–31
Trønnes RG (2002) Stability range and decomposition of potassic richterite and phlogopite end members at 5–15 GPa. Mineral Petrol 74:129–148
Ulmer P, Sweeney RJ (2002) Generation and differentiation of group II kimberlites: constraints from a high-pressure experimental study to 10 GPa. Geochim Cosmochim Acta 66:2139–2153
van Achterbergh E, Griffin WL, Stiefenhofer J (2001) Metasomatism in mantle xenoliths from the Letlhakane kimberlites: estimation of element fluxes. Contrib Mineral Petrol 141:397–414
van Keken PE, Kiefer B, Peacock SM (2002) High-resolution models of subduction zones: implications for mineral dehydration reactions and the transport of water into the deep mantle. Geochem Geophys Geosyst 3:1056–1076
Vedder W, Wilkins RWT (1969) Dehydroxylation and rehydroxylation, oxidation and reduction of micas. Am Mineral 54:482–509
Vielzeuf D, Schmidt MW (2001) Melting relations in hydrous systems revisited: application to metapelites, metagreywackes and metabasalts. Contrib Mineral Petrol 141:251–267
Vigouroux N, Wallace PJ, Kent AJR (2008) Volatiles in high-K magmas from the western trans-Mexican volcanic belt: evidence for fluid fluxing and extreme enrichment of the mantle wedge by subduction processes. J Petrol 49(9):1589–1618
Walter MJ, Thibault Y, Wei K, Luth RW (1995) Characterizing experimental pressure and temperature conditions in multianvil apparatus. Can J Phys 73:273–286
Wendlandt RF, Eggler DH (1980) The origins of potassic magmas: 2. Stability of phlogopite in natural spinel lherzolite and in the system KAlSiO4–MgO–SiO2–H2O-CO2 at high pressures and high temperatures. Am J Sci 280:421–458
Wyllie PJ (1980) The origin of kimberlite. J Geophys Res 85:6902–6910
Wyllie PJ, Sekine T (1982) The formation of mantle phlogopite in subduction zone hybridization. Contrib Mineral Petrol 4:375–380
Wyllie PJ, Huang W-L, Otto J, Byrnes AP (1983) Carbonation of peridotites and decarbonation of siliceous dolomites represented in the system CaO–MgO–SiO2–CO2 to 30 kbar. Tectonophysics 100:359–388
Yamashita H, Arima M, Ohtani E (1995) High pressure melting experiments on group II kimberlite up to 8 GPa; implications for mantle metasomatism. Proc Int Kimberlite Conf 6:669–671
Yoder HS, Kushiro I (1969) Melting of a hydrous phase: phlogopite. Am J Sci 267(A):558–582
Acknowledgments
This research was funded by a Discovery Grant from the National Science and Engineering Research Council of Canada to Luth. Many thanks are addressed to D. Caird for instructing the first author on the multi-anvil apparatus and to S. Matveev for the help with the electron microprobe. Comments by D. Canil and an anonymous reviewer improved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. L. Grove.
Rights and permissions
About this article
Cite this article
Enggist, A., Chu, L. & Luth, R.W. Phase relations of phlogopite with magnesite from 4 to 8 GPa. Contrib Mineral Petrol 163, 467–481 (2012). https://doi.org/10.1007/s00410-011-0681-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-011-0681-9