
Abstract
To constrain the melting phase relationships of phlogopite and magnesite in the presence of clino- and orthopyroxene, we performed experiments in the K2O–CaO–MgO–Al2O3–SiO2–H2O (KCMAS–H2O) and K2O–CaO–MgO–Al2O3–SiO2–H2O–CO2 (KCMAS–H2O–CO2) systems at pressures of 4–8 GPa and temperatures from 1100 to 1600 °C. We bracketed the carbonate-free solidus between 1250 and 1300 °C at 4 and 5 GPa, and between 1300 and 1350 °C at 6, 7 and 8 GPa. The carbonate-bearing solidus was bracketed between 1150 and 1200 °C at 4, 5 and 6 GPa, and between 1100 and 1150 °C at 7 and 8 GPa. Below the solidus in both systems at 4–6 GPa, phlogopite is in equilibrium with enstatite, diopside, garnet (plus magnesite in the carbonate-bearing system) and a fluid. At 7 GPa, phlogopite coexists with KK-richterite, enstatite, diopside, garnet (plus magnesite in the carbonate-bearing system) and a fluid. KK-richterite is the only stable K-bearing phase at 8 GPa and coexists with enstatite, diopside, garnet (plus magnesite in the carbonate-bearing system) and a fluid. In KCMAS–H2O, phlogopite is present to ~100 °C above the solidus. Olivine forms at the solidus and coexists with enstatite, diopside, garnet and melt. At depth in a subcontinental lithospheric mantle keel, phlogopite would be stable with orthopyroxene, clinopyroxene and magnesite to ~5 GPa along a 40 mW/m2 geotherm. A hydrous, potassic and CO2-bearing melt that intrudes the subcontinental mantle can react with olivine, enstatite and garnet, crystallizing phlogopite, magnesite and potentially liberating a hydrous fluid.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the Earth’s interior, volatiles such as H2O and CO2 play fundamental roles in processes such as metasomatism and melting. Water-bearing minerals, such as mica and amphibole, and carbonates play key roles in melting processes that depend on the stability of these OH- and CO2-bearing phases (cf. reviews by Frost 2006; Dasgupta and Hirschmann 2010; Luth 2014). It has been demonstrated that at low pressures, phlogopite-bearing mantle can be the source for potassium-rich melts (Elkins-Tanton and Grove 2003; Holbig and Grove 2008; Condamine and Médard 2014). Previous experimental studies involving phlogopite at high pressures mainly focus on subsolidus breakdown reactions (cf. review by Frost 2006), and only a few studies constrain the melting relations: pure phlogopite systems (Sato et al. 1997; Trønnes 2002), phlogopite with enstatite (Modreski and Boettcher 1972; Sato et al. 1997), phlogopite with diopside (Luth 1997), phlogopite with magnesite (Enggist et al. 2012) and phlogopite with enstatite and diopside (Modreski and Boettcher 1973). Although their primary focus was on studying the subsolidus reactions by which phlogopite broke down to form amphibole in natural and model (KNCMASH) lherzolite systems, Konzett and Ulmer (1999) also constrained the phlogopite + K-richterite-bearing solidus at 7 GPa between 1100 and 1300 °C and the K-richterite-bearing solidus at 8 GPa between 1300 and 1400 °C in their subalkaline KNCMASH lherzolitic composition. Wendlandt and Eggler (1980) determined the solidus of phlogopite-bearing natural lherzolite under both vapor-absent and H2O-saturated conditions from 1 to 3 GPa. These authors also bracketed the melting reaction phlogopite + enstatite + magnesite = forsterite + pyrope + liquid between 1200 and 1250 °C at 4 and 5 GPa. These brackets are indistinguishable from the brackets for the melting of phlogopite + magnesite at the same pressures determined by Enggist et al. (2012), which suggests minimal involvement of orthopyroxene in the melting reaction determined by Wendlandt and Eggler (1980). More information on phlogopite stability comes from multiple saturation experiments on orangeites (Yamashita et al. 1995; Ulmer and Sweeney 2002) and from experiments on lherzolite + H2O and lherzolite + H2O + CO2 systems (e.g., Mengel and Green 1989; Conceição and Green 2004; Fumagalli et al. 2009; Tumiati et al. 2013). The work of Modreski and Boettcher (1973) was extended to higher pressures by Sudo and Tatsumi (1990) for subsolidus reactions involving phlogopite with enstatite and diopside.
Melting relations of phlogopite coexisting with both clino- and orthopyroxene are unconstrained at P > 3 GPa, as is the effect of CO2 on such a phase assemblage. Previous authors suggested that the infiltration of a C–O–H fluid into metasomatized peridotite will trigger the breakdown of phlogopite or that a K–OH fluid will destabilize carbonates and will result in K–C–OH-rich melts at pressures >4 GPa (Wendlandt and Eggler 1980; Ulmer and Sweeney 2002).
Enggist et al. (2012) showed that phlogopite can be stable in the presence of carbonate to pressures greater than 7 GPa. The results of that simple system need to be extended into more complex systems. As a first step, in this study we constrain the melting relations of phlogopite and pyroxene with and without magnesite from 4 to 8 GPa. This study contributes to the understanding of potassium and water recycling into the Earth’s mantle, sources of alkali-rich melts or fluids that are potential metasomatic agents and the effect of CO2 on the stability of K–OH phases.
Experimental procedures
Starting materials
High-purity oxides (Al2O3, MgO and SiO2 of 99.99, 99.95 and 99.5 % purity, respectively) and carbonate (K2CO3 of 99.0 % purity) from Alfa Chemicals were used to prepare an anhydrous mixture with phlogopite stoichiometry. Phlogopite was synthesized using an end-loaded piston cylinder at the University of Alberta (for details, see Enggist et al. 2012). The products of the synthesis were checked by XRD to ensure that only phlogopite (phl) is present. Because of their small size (typically ≤1 μm), quantitative chemical analysis of the synthesized phlogopite by electron microprobe was difficult. Fragments of natural magnesite (mag) from Mt. Brussilof, British Columbia, Canada, that were free of visible inclusions were selected under a microscope and were ground in an agate mortar before being added to the starting material mixture. A synthetic enstatite–diopside (en–di) glass (en50di50; wt%) was prepared from high-purity oxides (MgO and SiO2; purity as above) and CaCO3 powder of 99.95 % purity from Alfa Chemicals. The mix, contained in a Pt crucible, was fused in a one atmosphere furnace at 1600 °C for 8 h. The melt was quenched to a glass by dipping the Pt crucible into distilled water. The resulting glass disk was wrapped into a plastic bag and crushed into small fragments using a hammer. The fragments then were ground in an agate mortar. The resulting glass powder was melted again at 1600 °C for 8 h and quenched as before. This crushing, melting and quenching cycle was repeated 3 times for each en–di glass batch in order to obtain a homogeneous glass composition. For the KCMAS–H2O and KCMAS–H2O–CO2 system, starting materials of phl33en33di33 and phl32en32di32mag4 (by weight) were prepared, respectively, and used in the experiments (Table 1). The composition of the synthetic phlogopite and natural magnesite and their relative molar proportion in the starting material (2:1) are the same as in Enggist et al. (2012).
Experimental setup
Approximately 1–3 mg of starting material was loaded into 1.5 mm outer diameter Pt capsules. The tops of the capsules were triple-crimped and the capsules were dried at 120 °C overnight. No water was added to the samples. The capsules were welded shut with a dampened tissue wrapped around the lower part of the capsule to keep the starting material cool. Sealed capsules were gently compressed into a cylindrical shape in a brass die.
The capsules were run in 18/11 assemblies (Walter et al. 1995) that were prepared as described in Enggist et al. (2012). Shorting of the thermocouple wires to the anvils, which can lead to erroneous emf readings, was reduced by replacing the solid graphite ring in the top part of the assembly by a split ring.
All experiments were performed using the UHP-2000 uniaxial split-sphere multi-anvil apparatus at the University of Alberta. Samples were pressurized first and then heated at a rate of ~60 °C min−1 at pressures from 4 to 7 GPa. At 8 GPa, the heating rate above 800 °C was reduced to ≤30 °C min−1, which decreased the number of thermocouple failures. Experiments were quenched by cutting off the power to the furnaces, causing the temperature to drop below 300 °C in 2–5 s, and then decompressed over 2.5–4 h. Experimental charges were mounted in epoxy (Petropoxy 154), ground open and impregnated with more epoxy under vacuum. Samples were polished using Al2O3 powder of 5, 1 and 0.05 μm grain size with oil, ethanol or acetone to preserve water-soluble phases. Polished samples were cleaned in an ultrasonic bath, dried at 60 °C and then carbon-coated for microprobe analysis.
Analytical methods
A JEOL 8900 electron microprobe at the University of Alberta was used for the analysis of the experimental products. The acceleration voltage was 15 kV and the cup beam current was 15 nA. Standards were albite (Na), diopside (Ca), Fo93 olivine (Mg), hematite (Fe), pyrope (Al, Si), orthoclase (K) and willemite (Mn). Counting times were 20 and 10 s on peak and background, respectively. A focused beam was used to analyze diopside, enstatite, garnet, magnesite and olivine, and beam diameters of up to 3 μm, depending on grain size, were chosen for phlogopite and KK-richterite grains. The Phi-Rho-Z correction (Armstrong 1988) provided by JEOL was applied to the raw data, and norm spreadsheets by P. Ulmer (personal communication, 2007) were used to calculate mineral compositions.
Results
General observations
Our run durations of 8–144 h at high and low temperatures, respectively, produced samples that appear texturally equilibrated with homogeneous phase compositions (Table 2). Shorter experiments, especially at 1100 and 1150 °C, showed textural evidence of disequilibrium, with zones within the capsule that resembled the starting material, and chemical evidence of disequilibrium, with high variability in the mineral compositions. The results of these experiments with demonstrable disequilibrium have been omitted from this manuscript.
Below the solidus, the grain size can be extremely small (<1–5 µm). In the carbonate-free system (KCMAS–H2O), the solidus is characterized by a change in texture with the occurrence of interstitial quench phlogopite and melt in the hot spot of the capsules. In the CO2-bearing system (KCMAS–H2O–CO2), the disappearance of magnesite together with the occurrence of quench phlogopite and melt marks the solidus. A small amount of fluid is present below the solidus, resulting from a breakdown reaction of the synthetic phlogopite in the starting material to phlogopite, garnet and fluid as observed in previous studies (Luth 1997; Sato et al. 1997; Enggist et al. 2012). The phlogopite that coexists with garnet, diopside, enstatite, plus magnesite in the carbonate-bearing system and a fluid below the solidus is referred to as “primary phlogopite” to distinguish it from the phlogopite in supersolidus experiments that crystallizes from the melt during the quenching of the experiment. At higher pressure, amphibole forms as a subsolidus breakdown product of phlogopite (Sudo and Tatsumi 1990; Luth 1997; Sato et al. 1997; Konzett and Ulmer 1999). In Na-bearing systems, this amphibole is a K-richterite, with K in the A-site and Na in the M2-site. In Na-free systems, K is present in both A- and M2-sites and the amphibole has been called KK-richterite (Konzett and Ulmer 1999; Yang et al. 1999). We adopt this terminology in this study.
The solidus in KCMAS–H2O was bracketed at 1250–1300 °C at 4–5 GPa, and at 1300–1350 °C at 6–8 GPa (Fig. 1). In KCMAS–H2O–CO2, the solidus is located at 1150–1200 °C at 4–6 GPa, and at 1100–1150 °C at 7–8 GPa (Fig. 2). Below the solidus at 4–6 GPa in KCMAS–H2O, primary phlogopite is in equilibrium with garnet, enstatite, diopside and fluid. At 7 GPa, primary phlogopite coexists with KK-richterite, garnet, enstatite, diopside and fluid (Fig. 3). At 8 GPa, the subsolidus assemblage is KK-richterite, garnet, enstatite, diopside and fluid. The same subsolidus assemblages, with the addition of magnesite, are present in the KCMAS–H2O–CO2 system. Primary phlogopite and KK-richterite coexist with melt in experiments at temperatures up to ~100 °C above the solidus at 4–6 GPa (phlogopite) and 7–8 GPa (KK-richterite).

Phase relations of phlogopite with pyroxene in the KCMAS–H2O system. Filled quadrants indicate phase presence as noted in legend. Light gray K-phase quadrant indicates primary phlogopite coexisting with melt. Long-dashed line above the solidus shows the maximum stability of phlogopite coexisting with melt. Di diopside, En enstatite, f/m fluid present if quadrant dark gray/dark gray quadrant with m to M for increasing melt portion, Gr garnet, K primary potassium-bearing phase (phlogopite or KK-richterite), Kr KK-richterite, Ol olivine and Phl phlogopite

Phase relations of phlogopite and pyroxene with magnesite in the KCMAS–H2O–CO2 system. Light gray K-phase quadrant indicates primary phlogopite coexisting with melt. Diagram omits experiments at >1400 °C (cf. Table 2). Di diopside, En enstatite, f/m fluid present if quadrant dark gray/dark gray quadrant with m to M for increasing melt portion, Gr garnet, K primary potassium-bearing phase (phlogopite or KK-richterite), Kr KK-richterite; Mag magnesite, Ol olivine and Phl phlogopite

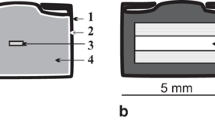
a Subsolidus assemblage: p-phl in equilibrium with gr, mag, en, di and kr. Mag is homogeneously distributed throughout the capsule. b Supersolidus: at high pressures en grows to ≤200 µm and encloses di and gr. Phases present are en, di, gr, ol and melt. c Supersolidus: p-phl reacted out, and a melt phase is present with q-phl needles, di, en, gr and ol grains. d Supersolidus: closer to solidus, p-phl is coexisting with melt, en, di, gr, ol. Q-phl present besides melt. e Temperature on sample was too high (thermocouple shorting) and is estimated to be ~1600 °C. Ol coexists with melt. A fluid exsolved upon quench (voids). f Temperature on sample was too high (thermocouple shorting) and is estimated by using 2-pyroxene thermometers to be ~1500 °C. Ol, en and di coexist with melt. A fluid exsolved from the melt upon quench (voids). di diopside, en enstatite, gr garnet, kr KK-richterite, mag magnesite, p-phl primary phlogopite and q-phl quench phlogopite. Scale bar in upper left of each image is 100 µm
In both systems, after quench, a hydrous solution is present in experiments at the solidus, which was observed to exit the capsule after it was breached during polishing. The amount is very small and no precipitates on the capsule surface were observed, in contrast to the observations in the phlogopite + magnesite system described in Enggist et al. (2012). At ≥50 °C above the solidus, no hydrous solution was seen to escape the charge upon breach.
Diopside
The size of diopside crystals below the solidus is ≤5 µm. At and above the solidus, diopside crystals are up to 25 µm in size, sub- to euhedral and of equant habit. Diopside is free of mineral inclusions, and the grain size decreases (1–10 µm) with increasing pressure. With increasing temperature, the Mg content increases by up to ~2 wt% and the Ca content decreases by up to ~5 wt%. The measured K2O and Al2O3 contents range from 0.1 to 1.4 and 0.4–2.4 wt%, respectively, with higher values at higher temperatures (Table 3). This trend probably results from the dissolution of phlogopite or KK-richterite into the melt, which increases the activity of K2O and Al2O3 in the melt, thereby increasing the K and Al content of the pyroxene (Modreski and Boettcher 1972; Enggist et al. 2012).
Enstatite
At 4–6 GPa, enstatite crystals are 5–20 µm in diameter below the solidus and up to 30 µm above the solidus. Enstatite is sub- to euhedral and prismatic and often contains inclusions of euhedral garnet and anhedral diopside grains. At >6 GPa, grains measure 100–200 µm in length below and above the solidus, respectively, and contain numerous small (1–10 µm) inclusions (Fig. 3c). The concentrations of Al2O3 (0.3–2.4 wt%) and CaO (0.4–2.4 wt%) increase with increasing temperature (Table 4). SiO2 contents (~57–60 wt%) are close to stoichiometric with 2 Si apfu. As in the case of diopside (see above), the increase in Al can be attributed to the melting of phlogopite or KK-richterite. The increasing Ca content above the solidus reflects the increasing temperature and the associated incorporation of Ca in the orthopyroxene.
Garnet
Garnet grains are 1–10 µm in diameter below the solidus, up to 20 µm at the solidus and ~1–3 µm above the solidus. Grains are sub- to euhedral and are free of mineral inclusions. SiO2 contents range from ~43 to 46 wt% (Table 5), with no obvious systematic relationship to pressure or temperature. Al2O3 varies from ~20 to 25 wt%, with lower Al contents at higher pressures. The garnets contain 4–6 wt % CaO, consistent with previous studies (Brey et al. 1990; Sudo and Tatsumi 1990; Luth 1997; Konzett and Ulmer 1999).
KK-richterite
Grains of KK-richterite are 10–80 µm long and are sub- to euhedral. Larger grains often enclose garnet (Fig. 3a). KK-richterite occurs at 7 GPa, where it coexists with primary phlogopite, and at 8 GPa as the only K-bearing phase below the solidus (Table 6). Residual KK-richterite above the solidus contains up to 2.7 wt% Al2O3, compared to 1.3–1.9 wt% at subsolidus conditions. Relative to the ideal composition (K2CaMg5Si8O22(OH)2), the present KK-richterite is deficient in Si (7.85–7.88 apfu), as the site is shared with Al. The mineral, furthermore, is deficient in K (1.72–1.87 apfu) and Mg (4.75–4.95 apfu), contains extra Ca (1.10–1.18 apfu) and contains 0.22–0.45 Al apfu, similar to the compositions reported by Sudo and Tatsumi (1990), Konzett and Ulmer (1999), and Konzett and Fei (2000).
Magnesite
Magnesite is sub- to euhedral, prismatic and only occurs below the solidus. Grains are up to 20 µm in size and are free of mineral inclusions. Magnesite is the first phase to disappear at the solidus. All magnesite grains contain Ca, and CaO decreases with increasing pressure from 1.9 to 0.4 wt% at 4–8 GPa, respectively. The MgO content of the magnesite at 4 GPa is low (44.4 wt%) compared to those from higher pressures (Table 7). The low MgO content relative to stoichiometric MgCO3 (47.80 wt% MgO) is similar to that reported in previous studies (Luth 1997; Keshav and Gudfinnsson 2010; Enggist et al. 2012). This may be a result of the higher Ca content of this carbonate relative to the others in our experiments and the effect of that on the composition calculated with the Phi-Rho-Z correction scheme. Alternatively, one of the reviewers suggested the possibility that the 4-GPa carbonate might contain some OH. To our knowledge, such an occurrence has not be documented in experimental studies at high pressure to date.
Olivine
Olivine was found in experiments above the solidus and in one subsolidus experiment in KCMAS–H2O at 8 GPa. Grains have a prismatic to equant habit, are subhedral, up to 30 µm in size, contain no mineral inclusions and are stoichiometric features that are consistent with previous studies (Luth 1997; Trønnes 2002; Enggist et al. 2012).
Phlogopite
Primary phlogopite grains are 5–30 µm in length, are sub- to euhedral of prismatic habit and do not contain mineral inclusions. Quench phlogopite are 5–50 µm in length and exhibit a dendritic habit.
In both KCMAS–H2O and KCMAS–H2O–CO2, the Al2O3 content of the primary phlogopite decreases with increasing pressure (Table 8). This trend is similar to that observed in our earlier study (Enggist et al. 2012).
As illustrated in Fig. 4, the compositions of the 7-GPa phlogopite in KCMAS–H2O, and all phlogopites in KCMAS–H2O–CO2, show Si enrichment and K depletion relative to ideal phlogopite. This suggests that there is a small amount of the talc/10 Å component as discussed by Fumagalli et al. (2009), as well as some of either a celadonite (K2Mg2Al2Si8O20(OH)4) or a montdorite (K2Mg5Si8O20(OH)4) component to account for the observed offset from the talc/10 Å exchange vector in Fig. 4. In comparison with previous work in either simple (Fig. 4a) or natural (Fig. 4b) systems, the phlogopite in our study has less of the talc/10 Å component and does not have the Tschermak substitution toward eastonite (K2Mg4Al2(Si4Al4)O20(OH)4).

a Compositional trends of phlogopite compared to previous simple system experimental studies. This study: gray circles KCMAS–H2O primary phlogopite; open squares KCMAS–H2O–CO2 primary phlogopite. Black crosses compositions from previous studies of Enggist et al. (2012), Konzett and Ulmer (1999), Trønnes (2002), Luth (1997), Modreski and Boettcher (1973). b Compositional trends of phlogopite compared to previous experimental studies in natural systems. This study: gray circles KCMAS–H2O primary phlogopite; open squares KCMAS–H2O–CO2 primary phlogopite. Black crosses compositions from previous studies of Wendlandt and Eggler (1980), Mengel and Green (1989), Thibault et al. (1992), Sato et al. (1997), Konzett and Ulmer (1999), Hermann (2002), Conceição and Green (2004), Holbig and Grove (2008), Fumagalli et al. (2009), Tumiati et al. (2013), Condamine and Médard (2014). For reference, the composition of ideal phlogopite is shown by gray star in both a, b. Vectors emanating from ideal phlogopite illustrate the effect of different exchange components as labeled. The AlVIAlIVMg−1Si−1 vector is the Tschermak exchange pointing toward eastonite (K2Mg4Al2(Si4Al4)O20(OH)4). The exchanges SiAlVIAl IV−1 Mg−2 and Si2Mg−1Al IV−2 , which point toward celadonite (K2Mg2Al2Si8O20(OH)4) and montdorite (K2Mg5Si8O20(OH)4) components, respectively, are superimposed in this (K + Na) versus Si compositional space. The [v]SiK−1Al IV−1 vector points toward the “talc/10 Å” end member (Mg6Si8O20(OH)4) (cf. discussion in Fumagalli et al. 2009). See text for discussion
Melt
Melt compositions were problematic to quantify for three reasons. First, the amount of melt close to the solidus is very low and the melt is located either in very small interstitial pockets or occurs as overgrowths on minerals, making quantitative analysis difficult or impossible even with a focused beam. Second, the presence of quench phlogopite, and the hydrous and alkali-rich nature of this system, makes it possible (likely?) that there was additional crystallization on other phases during the quench that would further obscure the melt composition. Finally, in near-solidus experiments, the quenched melt consists of a hydrous solution that exits the capsule upon breach, quench phlogopite, plus an unidentifiable, non-phlogopite quench material. Only at very high melt fractions did the melt quench to a glass (Fig. 3e, f).
The compositions of the quench material and the glass are given in Table 9. Because of the uncertainties induced by the presence of the hydrous quench solution, the variability in the composition of quench phlogopite and the quench phase, we were unable to reconstruct reliable melt compositions.
Discussion
Assessment of equilibrium
All of the experiments in this study were synthesis experiments, and we believe that they approached, or attained, equilibrium based on the following reasoning. For each experiment, textural and chemical equilibrium was assessed. For well-equilibrated experiments, two main textures were observed: a relatively dense aggregate of subhedral to euhedral crystals of similar size for each type throughout the capsule, and loose, interstitial quench crystals of varying size that quenched from the melt. The former texture was observed below the solidus, and both textures were found in experiments above the solidus. In poorly equilibrated experiments, such as some low-temperature experiments with short run durations, textures varied from patches containing anhedral to subhedral crystals of random size to areas that still resembled the starting material with a very fine-grained texture, where no crystals could be identified.
In addition to the textural assessment, chemical analyses of the crystalline phases were evaluated to determine whether equilibrium was achieved. In well-equilibrated experiments, crystals of the same phase had similar compositions throughout the charge. If crystals of the same phase showed a lot of compositional variation, this was taken as evidence for disequilibrium. Experiments showing random textures, and/or widely varying chemical compositions, or experiments that were not reproducible were considered not at equilibrium and, therefore, were not included in this study.
A reviewer of an early version of this manuscript suggested using Ca-in-opx geothermometry to calculate temperatures, which could be compared to experimental temperatures as a test of equilibrium. We undertook this exercise and expanded it to consider two-pyroxene geothermometers as well. Based on our results for both our experiments and previously published “simple system” studies, we suggest that a precise geothermometer for these “simple” systems remains to be calibrated at >4 GPa. In contrast, the agreement between calculated and experimental temperatures for experiments, both ours and previously published ones, at <4 GPa was much better. Full details of this exercise and its results are given in the online supplementary material (Appendix 1).
Subsolidus formation of garnet
In our experiments, the original synthetic phlogopite in the starting material reacts at subsolidus conditions to form primary phlogopite, garnet and an inferred small amount of fluid. A similar reaction was observed in previous studies at pressures >5 GPa (Sato et al. 1997), >6 GPa (Luth 1997) and at 4–8 GPa (Enggist et al. 2012).
In the present study, diopside must be involved in this reaction to provide the Ca present in the garnets. Using the phase compositions from ae-151 (KCMAS–H2O–CO2, 4 GPa, 1150 °C) as an example, a mass-balanced reaction with reactants pure diopside and the starting material phlogopite (Table 1) can be written as:
This reaction produces ~1.9 wt% H2O, and given that the starting materials contained ~1/3 phlogopite, there would be ~0.6 wt% H2O in the experiment. Mass balance calculations using the compositions of the phases present and the starting materials (Table 10) confirm that less phlogopite is present in subsolidus experiments than was present in the starting materials. These calculations tend to underestimate the K2O content of the bulk compositions, consistent with some K2O dissolving into the fluid phase.
Formation of amphibole
At >6 GPa, the primary phlogopite reacts to produce KK-richterite at subsolidus conditions, such that phlogopite coexists with KK-richterite at 7 GPa but is absent at 8 GPa. Similar behavior, but at higher pressure, was observed by Sudo and Tatsumi (1990) in their study of phlogopite + diopside. They proposed a divariant reaction to explain the breakdown:
They suggested that in the presence of orthopyroxene, a different reaction would be responsible for the breakdown of phlogopite and the formation of KK-richterite:
A variant of this reaction that produces more KK-richterite and does not require K to dissolve into the fluid was proposed by Konzett and Ulmer (1999):
This reaction consumes more enstatite and produces more olivine as well as KK-richterite relative to reaction (3).
These reactions all produce olivine, which was only observed in the 8-GPa subsolidus experiment in KCMAS–H2O, where it coexists with KK-richterite. Olivine was not observed in the 7-GPa subsolidus experiment that had coexisting phlogopite and KK-richterite, nor was it found in the KK-richterite-bearing subsolidus experiments in KCMAS–H2O–CO2. This may be the result of either low modal abundance of olivine, such that it was not present in the cross section of the capsule imaged and analyzed, or because phlogopite broke down via a different reaction entirely. For example, Luth et al. (1993) and Trønnes (2002) wrote a balanced and fluid-absent reaction for KNCMASH that does not produce olivine:
In the Na-free KCMASH system, the analogous reaction would be:
The low K content of the clinopyroxenes in our experiments (Table 3) argues against this reaction being significant. There are other possible reactions involving fluid constituents as reactants, such as
or
(Trønnes, pers. comm.), but the small amount of fluid coexisting with phlogopite in our experiments makes these unlikely to be significant.
Mass balance calculations (Table 10) that provide estimates of modal abundances of the phases present can provide some insight into which reaction may be most realistic. Comparing the 5- and 8-GPa experiments in KCMAS–H2O for which all phase compositions were available, the abundance of both enstatite and diopside decreases by approximately the same amount upon the formation of KK-richterite, whereas garnet increases in abundance and olivine appears. These observations are most consistent with the original reaction [(3) above] proposed by Sudo and Tatsumi (1990).
In contrast, in the KCMAS–H2O–CO2 system, the formation of KK-richterite does not change the abundance of enstatite, but that of diopside does decrease and that of garnet increases. Olivine did not appear. A different KK-richterite-forming reaction in the presence of magnesite seems unlikely, but further work is needed to investigate this issue. It would be most worthwhile to see whether these differences were also present in the KNCMAS systems as an initial effort.
In the KNCMASH system, the appearance of K-richterite was bracketed by Konzett and Ulmer (1999) at 6–6.5 GPa at 800 °C, and at 6.5–7 GPa at 1100 °C. In the natural lherzolite bulk composition they studied, K-richterite became stable between 6 and 6.5 GPa at 1100 °C. Comparing these to our results for KK-richterite (6–7 GPa at 1100–1300 °C), we suggest that the addition of Na or Fe does not stabilize K-amphibole relative to phlogopite at significantly lower pressures.
Melting phase relationships
The solidi found in the present study involve the K-bearing phases as reactants, in agreement with the results of previous studies (Luth 1997; Sato et al. 1997; Trønnes 2002; Enggist et al. 2012). Sato et al. (1997) found that a natural, Mg-rich phlogopite melted to olivine + liquid at 4 GPa and to olivine + garnet + liquid at 5–8 GPa. At 7.5 GPa, Trønnes (2002) observed that KMASH phlogopite reacted to olivine, garnet and “fluid” and drew the stability curve for phlogopite shown as “Phl” in Fig. 5. At ~10 GPa, he found that phlogopite was breaking down to a higher-pressure potassic phase (phase “x”—see his Fig. 2 for details). Because there was no Ca in his experiments, KK-richterite did not form. Luth (1997) found phlogopite + diopside melted incongruently to olivine and liquid at 3 and 5 GPa and to olivine, garnet and liquid at 7.5 and 9 GPa (“Phl–Di” curve in Fig. 5). The apparent stability of phlogopite + diopside to higher temperatures above 6 GPa than phlogopite alone may be less contradictory than it appears in Fig. 5 and may have more to do with the limited data available for both systems. For example, a 7.4-GPa, 1400 °C experiment on synthetic phlogopite by Trønnes (2002) produced phlogopite + garnet + olivine + fluid, despite it lying above the reaction boundary as shown.

Comparison of different melting reactions involving phlogopite with subcontinental geotherms from Hasterok and Chapman (2011) (dashed lines with numbers that are the heat flow in mW/m2) and with a mantle adiabat (“AMA,” also from Hasterok and Chapman 2011). The hottest portion of the Northern Cascadia subduction model of Syracuse et al. (2010) is shown as the shaded region. Curves from this study are shown in red: Phl–En–Di–Mag: KCMAS–H2O–CO2 phlogopite + enstatite + diopside + magnesite, Phl–En–Di KCMAS–H2O phlogopite + enstatite + diopside (this study). The short-dashed lines on each of these solidi represent the change in K-phase from phlogopite to phlogopite + KK-richterite to KK-richterite with increasing pressure. Previous studies: Phl–Mag phlogopite + magnesite (Enggist et al. 2012), Phl–En phlogopite + enstatite (Sato et al. 1997), Phl synthetic phlogopite breakdown curve (Trønnes 2002), Phl–Di phlogopite + diopside (Luth 1997) and dry carb peridotite nominally anhydrous, carbonated peridotite (Dasgupta and Hirschmann 2006, 2007). See text for discussion
Sato et al. (1997) report melting reactions phlogopite + enstatite to olivine and melt at 4 GPa, to olivine, garnet and melt at pressures of 5 and 6 GPa, and to garnet and melt at 8 GPa (“Phl–En” curve in Fig. 5). Enggist et al. (2012) determined a melting reaction in the phlogopite and magnesite system between 4 and 8 GPa of the form phlogopite + magnesite + fluid = olivine + enstatite + garnet + melt (“Phl–Mag” curve in Fig. 5).
Adding both pyroxenes to the end-member system decreases the temperature of the solidus involving phlogopite in both the carbonate-free and carbonate-bearing systems (Fig. 5). The slope of the solidus in KCMAS–H2O (“Phl–En–Di” curve in Fig. 5) remains positive in P–T space at least to 8 GPa, paralleling that for phlogopite + diopside (“Phl–Di”, Fig. 5) but at lower temperature. The appearance of KK-richterite above 6 GPa in our KCMAS–H2O experiments, and its absence in the Ca-free phlogopite + enstatite system, explains the crossover of the “Phl–En–Di” and “Phl–En” curves in Fig. 5.
The solidus in our KCMAS–H2O–CO2 system has a negative slope (“Phl–En–Di–Mag” curve, Fig. 5) from 4 to 8 GPa, similar to that for phlogopite + magnesite (“Phl–Mag,” Fig. 5). These two curves converge in P–T space at higher pressures, but the K-bearing phase differs, because of the formation of KK-richterite in KCMAS–H2O–CO2 system but not in the Ca-absent phlogopite + magnesite study.
In the present study, olivine appears at the solidus, and qualitatively the amount of enstatite and garnet increases as well. We infer that melting takes place via the following reactions, where the “K-bearing phase” is phlogopite at pressures of 4–6 GPa, phlogopite and KK-richterite at 7 GPa and KK-richterite at 8 GPa:
for KCMAS–H2O and KCMAS–H2O–CO2, respectively. Unfortunately, our inability to derive liquid compositions prohibits a more quantitative analysis. Mass balance calculations were attempted using the compositions of the quench material and the quench phlogopite, but for the reasons detailed above (“Melt” section of Results), the results were deemed unreliable.
Finally, it is interesting to note that phlogopite persists above the solidus in the KCMAS–H2O system, but does not do so to the same extent in the carbonate-bearing one. A similar persistence of phlogopite above the solidus was observed in the study of Condamine and Médard (2014) at 1 GPa. If this persists in more complex lherzolitic systems at 4–6 GPa, it would have implications for the behavior of trace elements hosted by mica during partial melting in the mantle.
Implications for the Earth
Direct application of the results of this study to the Earth is limited by the compositional simplicity of the KCMAS–H2O and KCMAS–H2O–CO2 systems relative to natural lherzolite. As models for melting of a peridotite containing phlogopite with or without magnesite, however, these systems may provide some useful insights. Given the effectiveness of magnesite in lowering the solidus in both systems, it is clear that melting of magnesite + phlogopite peridotite will occur at lower temperatures than will phlogopite (or KK-richterite) peridotite, consistent with conclusions reached by previous workers at lower pressures.
Comparison of our results with previous work provides some insight into the effects of the presence of both phlogopite and carbonate in lherzolitic mantle. An obvious initial comparison is to the melting in the lherzolite + H2O system. This system has been studied at high pressure for ~50 years, and there is still vigorous debate about the location of the water-saturated solidus. At 4–6 GPa, the solidus has been located at 800–820 °C by Till et al. (2012) (see also Grove et al. (2006) for lower-pressure data) but at significantly higher temperatures by Green et al. (2010) and Green et al. (2014): ~1225 °C at 4 GPa and ~1375 °C at 6 GPa. A thorough recent discussion of these results is provided by Kessel et al. (2015), who also provide new experimental data on the solidus of “K-rich” lherzolite + H2O, using a rocking multi-anvil and the cryogenic LA–ICP–MS technique (Kessel et al. 2004) to analyze solute compositions. As the effect of alkalis on influencing solidi in mafic and ultramafic systems is well documented (e.g., Foley et al. 2009; Brey et al. 2011; Kessel et al. 2015), it seems most appropriate to compare our results with those for K-enriched lherzolite + H2O from Kessel et al. (2015). These authors determined the water-saturated solidus to be between 900 and 1000 °C at 4 GPa, and between 1000 and 1100 °C at 5 and 6 GPa. They did not observe phlogopite as part of the subsolidus assemblage, which they attributed to preferential dissolution of K2O into the coexisting fluid phase (their experiments contained ~16 wt% H2O). Relative to their solidus, our dehydration melting solidus of Phl + En + Di is displaced to higher temperatures by >250 °C.
Comparing our results to the solidus of “anhydrous” carbonate-bearing peridotite (Dasgupta and Hirschmann 2006, 2007), we see that our solidus in the KCMAS–H2O–CO2 system is at lower temperatures at 5 GPa and above (“Phl–En–Di–Mag” curve compared to “carb peridotite solidus” curve in Fig. 5). A more realistic comparison would be to previous work on phlogopite + carbonate lherzolite. Thibault et al. (1992) determined the solidus of phlogopite + carbonate lherzolite to be ~1060 °C at 3 GPa. Foley et al. (2009) constrained the solidus of a phlogopite lherzolite with ~3 wt % CO2 to be ~1040 °C at 4 GPa, 1060–1070 °C at 5 GPa, and ~1090 °C at 6 GPa, consistent with a displacement to lower temperatures relative to our model system results. It is worth noting that the solidus of Foley et al. (2009) has a steep positive slope in P–T space, in contrast to the steep negative slope we observed in the KCMAS–H2O–CO2 system (“Phl–En–Di–Mag” curve in Fig. 5).
Another way to contextualize our results is to relate them to geothermal conditions in different environments in the Earth’s mantle. Both of our solidi (red lines in Fig. 5) lie at lower temperatures than a mantle adiabat (“AMA” in Fig. 5, from Hasterok and Chapman 2011), so a K-bearing melt, rather than phlogopite or phlogopite + magnesite, would be stable in the convecting mantle.
In subcratonic lithospheric mantle, the stability of phlogopite-bearing assemblages would depend on the ambient geothermal gradient; for example, our carbonate-bearing solidus (“Phl–En–Di–Mag,” Fig. 5) intersects the 40 mW/m2 geotherm of Hasterok and Chapman (2011) at ~5 GPa, marking the maximum stability depth of phlogopite + magnesite. Given the lower temperatures of the phlogopite + carbonate solidus in the natural system outlined above, the maximum stability depth would be offset to lower pressures. For comparison, the solidus of Foley et al. (2009) would cross the 40 mW/m2 geotherm at ~4.4 GPa. For cratons with cooler geotherms (e.g., 35 mW/m2 illustrated on Fig. 5), the maximum stability depth for phlogopite or phlogopite + magnesite would be correspondingly greater.
What happens when melt forms? The negative slope in P–T space that we found for the carbonate + phlogopite solidus in KCMAS–H2O–CO2 would provide a mechanism for trapping melts once they form—as soon as they start to rise, they would freeze—but available data for more realistic systems at these pressures (e.g., Foley et al. 2009) indicate that the solidus has a steep positive slope, steeper than the geotherm. The behavior of an ascending melt would thus depend on whether it rises and cools along the geotherm, which causes it to freeze. More rapid ascent would be required to let melts survive.
In cooler conditions, such as subduction-related environments, phlogopite with magnesite would be stable to depth in the subducting slab, allowing recycling into the mantle to at least ~7 GPa even in the warmest subduction zones (northern Cascadia model of Syracuse et al. 2010, gray region in Fig. 5). If a slab becomes imbricated to the bottom of the subcontinental lithosphere to “grow” that cratonic root from the bottom up (Helmstaedt and Schulze 1989) (see also Stachel and Harris 2008), the slab would heat up to the ambient geothermal gradient and—depending on the depth—could cross the solidus and begin to melt if it contains phlogopite and magnesite. If the melt rises into cooler lithospheric mantle, it would react with olivine, orthopyroxene and garnet (e.g., harzburgite) to crystallize phlogopite, magnesite and clinopyroxene (e.g., magnesite–phlogopite lherzolite) in the inverse of our melting reaction proposed above.
This potentially offers a mechanism to explain situations in which (re)fertilized lherzolite underlies depleted harzburgite in subcratonic lithospheric mantle (e.g., Griffin et al. 1999, 2008). With continued heating of the slab from below, the melting/re-freezing could migrate to shallower depths.
Summary and conclusions
In the KCMAS–H2O system, the subsolidus assemblage phlogopite, enstatite, diopside, garnet and fluid are stable to 1250 °C at 4, 5 and 5.5 GPa, and to 1300 °C at 6 GPa. Phlogopite, KK-richterite, enstatite, diopside, garnet and a fluid are the subsolidus assemblage that coexists at 1300 °C and 7 GPa. At 8 GPa, the stable subsolidus assemblage to 1300 °C is KK-richterite, enstatite, diopside, garnet and a fluid.
In the KCMAS–H2O–CO2 system, phlogopite, enstatite, diopside, garnet with magnesite and a fluid are the stable subsolidus assemblage to 1150 °C at 4, 5 and 6 GPa. Phlogopite, KK-richterite, enstatite, diopside, garnet with magnesite and a fluid coexist to 1100 °C and 7 GPa. At 8 GPa, the stable assemblage to 1100 °C is KK-richterite, enstatite, diopside, garnet, magnesite and a fluid.
The solidus in the carbonate-bearing system is lower by 100–200 °C at 4 and 8 GPa, respectively, relative to the carbonate-free system. Above the solidus, olivine, enstatite, diopside and garnet coexist with melt in both systems.
We infer that only in hot subduction zones will the carbonate and K-bearing phase trigger melting, so that return of water and carbonate to the deeper mantle takes place in cooler subducting slabs. In subcontinental lithospheric mantle, ascending hydrous, potassic and carbonate-bearing melts will crystallize at depth to phlogopite, magnesite and diopside. The depth at which this will occur in the mantle requires calibration by experiments in more complex systems.
References
Armstrong JT (1988) Quantitative analysis of silicate and oxide materials: comparison of Monte Carlo, ZAF, and phi(rho Z) procedures. In: Newbury DE (ed) Microbeam analysis. San Francisco Press, San Francisco, pp 239–246
Brey GP, Kohler T, Nickel KG (1990) Geothermobarometry in 4-phase lherzolites. 1. Experimental results from 10 to 60 Kb. J Petrol 31(6):1313–1352
Brey GP, Bulatov VK, Girnis AV (2011) Melting of K-rich carbonated peridotite at 6–10 GPa and the stability of K-phases in the upper mantle. Chem Geol 281(3–4):333–342. doi:10.1016/j.chemgeo.2010.12.019
Conceição RV, Green DH (2004) Derivation of potassic (shoshonitic) magmas by decompression melting of phlogopite plus pargasite lherzolite. Lithos 72(3–4):209–229
Condamine P, Médard E (2014) Experimental melting of phlogopite-bearing mantle at 1 GPa: implications for potassic magmatism. Earth Planet Sci Lett 397:80–92
Dasgupta R, Hirschmann MM (2006) Melting in the earth’s deep upper mantle caused by carbon dioxide. Nature 440(7084):659–662. doi:10.1038/nature04612
Dasgupta R, Hirschmann MM (2007) Effect of variable carbonate concentration on the solidus of mantle peridotite. Am Mineral 92(2–3):370–379. doi:10.2138/am.2007.2201
Dasgupta R, Hirschmann MM (2010) The deep carbon cycle and melting in earth’s interior. Earth Planet Sci Lett 298(1–2):1–13. doi:10.1016/j.epsl.2010.06.039
Elkins-Tanton LT, Grove TL (2003) Evidence for deep melting of hydrous metasomatized mantle: pliocene high-potassium magmas from the Sierra Nevadas. J Geophys Res 108(B7):2350. doi:10.1029/2002JB002168
Enggist A, Chu L, Luth RW (2012) Phase relations of phlogopite with magnesite from 4 to 8 GPa. Contrib Mineral Petrol 163:467–481. doi:10.1007/s00410-011-0681-9
Foley SF, Yaxley GM, Rosenthal A, Buhre S, Kiseeva ES, Rapp RP, Jacob DE (2009) The composition of near-solidus melts of peridotite in the presence of CO2 and H2O between 40 and 60 kbar. Lithos 112(S1):274–283. doi:10.1016/j.lithos.2009.03.020
Frost DJ (2006) The stability of hydrous mantle phases. Rev Mineral Geochem 62(1):243–271. doi:10.2138/rmg.2006.62.11
Fumagalli P, Zanchetta S, Poli S (2009) Alkali in phlogopite and amphibole and their effects on phase relations in metasomatized peridotites: a high-pressure study. Contrib Mineral Petrol 158(6):723–737. doi:10.1007/s00410-009-0407-4
Green DH, Hibberson WO, Kovacs I, Rosenthal A (2010) Water and its influence on the lithosphere-asthenosphere boundary. Nature 467(7314):448–451. doi:10.1038/nature09369
Green DH, Hibberson WO, Rosenthal A, Kovács I, Yaxley GM, Falloon TJ, Brink F (2014) Experimental study of the influence of water on melting and phase assemblages in the upper mantle. J Petrol 55(10):2067–2096
Griffin WL, Doyle BJ, Ryan CG, Pearson NJ, O’Reilly SY, Davies R, Kivi K, Van Achterbergh E, Natapov LM (1999) Layered mantle lithosphere in the Lac de Gras area, Slave Craton: composition, structure and origin. J Petrol 40(5):705–727
Griffin WL, O’Reilly SY, Afonso JC, Begg GC (2008) The composition and evolution of lithospheric mantle: a re-evaluation and its tectonic implications. J Petrol 50(7):1185–1204. doi:10.1093/petrology/egn033
Grove TL, Chatterjee N, Parman SW, Medard E (2006) The influence of H2O on mantle wedge melting. Earth Planet Sci Lett 249(1–2):74–89. doi:10.1016/j.epsl.2006.06.043
Hasterok D, Chapman DS (2011) Heat production and geotherms for the continental lithosphere. Earth Planet Sci Lett 307(1–2):59–70. doi:10.1016/j.epsl.2011.04.034
Helmstaedt HH, Schulze DJ (1989) Southern African kimberlites and their mantle sample: implications for Archean tectonics and lithosphere evolution. In: Ross J (ed) Kimberlites and related rocks: their composition, occurrence, origin and emplacement GSA Spec Publ No 14, vol 1. Blackwell, Carlton, pp 358–368
Hermann J (2002) Experimental constraints on phase relations in subducted continental crust. Contrib Mineral Petrol 143(2):219–235. doi:10.1007/s00410-001-0336-3
Holbig ES, Grove TL (2008) Mantle melting beneath the Tibetan Plateau: experimental constraints on ultrapotassic magmatism. J Geophys Res. doi:10.1029/2007JB005149
Keshav S, Gudfinnsson GH (2010) Experimentally dictated stability of carbonated oceanic crust to moderately great depths in the earth: results from the solidus determination in the system CaO–MgO–Al2O3–SiO2–CO2. J Geophys Res 115:B05205. doi:10.1029/2009JB006457
Kessel R, Ulmer P, Pettke T, Schmidt MW, Thompson AB (2004) A novel approach to determine high-pressure high-temperature fluid and melt compositions using diamond-trap experiments. Am Mineral 89(7):1078–1086
Kessel R, Pettke T, Fumagalli P (2015) Melting of metasomatized peridotite at 4–6 GPa and up to 1200 °C: an experimental approach. Contrib Mineral Petrol 169(4):1–19. doi:10.1007/s00410-015-1132-9
Konzett J, Fei YW (2000) Transport and storage of potassium in the Earth’s upper mantle and transition zone: an experimental study to 23 GPa in simplified and natural bulk compositions. J Petrol 41(4):583–603. doi:10.1093/petrology/41.4.583
Konzett J, Ulmer P (1999) The stability of hydrous potassic phases in lherzolitic mantle—an experimental study to 9.5 GPa in simplified and natural bulk compositions. J Petrol 40(4):629–652
Luth RW (1997) Experimental study of the system phlogopite-diopside from 3.5 to 17 GPa. Am Mineral 82(11–12):1198–1209
Luth RW (2014) 3.9—volatiles in earth’s mantle. In: Holland HD, Turekian KK (eds) Treatise on geochemistry, 2nd edn. Elsevier, Oxford, pp 355–391
Luth RW, Trønnes R, Canil D (1993) Volatile-bearing phases in the Earth’s mantle. In: Luth RW (ed) Short course handbook on experiments at high pressure and applications to the earth’s mantle, vol SC-21. Mineralogical Association of Canada, Edmonton, pp 445–485
Mengel K, Green DH (1989) Stability of amphibole and phlogopite in metasomatized peridotite under water-saturated and water-undersaturated conditions. In: Ross J, Jacques AL, Ferguson J, Green DH, O’Reilly SY, Danchin RV, Janse AJA (eds) Kimberlites and related rocks: their composition, occurrence, origin and emplacement GSA Spec Publ No 14. Blackwell, Carlton, pp 571–581
Modreski PJ, Boettcher AL (1972) Stability of phlogopite + enstatite at high-pressures—model for micas in interior of earth. Am J Sci 272(9):852–869
Modreski PJ, Boettcher AL (1973) Phase relationships of phlogopite in the system K2O–MgO–CaO–Al2O3–SiO2–H2O to 35 kilobars: a better model for micas in the interior of the Earth. Am J Sci 273(5):385–414
Sato K, Katsura T, Ito E (1997) Phase relations of natural phlogopite with and without enstatite up to 8 GPa: implication for mantle metasomatism. Earth Planet Sci Lett 146(3–4):511–526
Stachel T, Harris JW (2008) The origin of cratonic diamonds—constraints from mineral inclusions. Ore Geol Rev 34(1–2):5–32. doi:10.1016/j.oregeorev.2007.05.002
Sudo A, Tatsumi Y (1990) Phlogopite and K-amphibole in the upper mantle: implication for magma genesis in subduction zones. Geophys Res Lett 17(1):29–32. doi:10.1029/GL017i001p00029
Syracuse EM, van Keken PE, Abers GA (2010) The global range of subduction zone thermal models. Phys Earth Planet Inter 183(1–2):73–90. doi:10.1016/j.pepi.2010.02.004
Thibault Y, Edgar AD, Lloyd FE (1992) Experimental investigation of melts from a carbonated phlogopite lherzolite—implications for metasomatism in the continental lithospheric mantle. Am Mineral 77(7–8):784–794
Till CB, Grove TL, Withers AC (2012) The beginnings of hydrous mantle wedge melting. Contrib Mineral Petrol 163(4):669–688
Trønnes RG (2002) Stability range and decomposition of potassic richterite and phlogopite end members at 5–15 GPa. Mineral Petrol 74(2–4):129–148. doi:10.1007/s007100200001
Tumiati S, Fumagalli P, Tiraboschi C, Poli S (2013) An experimental study on COH-bearing peridotite up to 3.2 GPa and implications for crust–mantle recycling. J Petrol 54(3):453–479. doi:10.1093/petrology/egs074
Ulmer P, Sweeney RJ (2002) Generation and differentiation of group II kimberlites: constraints from a high-pressure experimental study to 10 GPa. Geochim Cosmochim Acta 66(12):2139–2153
Walter MJ, Thibault Y, Wei K, Luth RW (1995) Characterizing experimental pressure and temperature conditions in multi-anvil apparatus. Can J Phys 73(5–6):273–286
Wendlandt RF, Eggler DH (1980) The origins of potassic magmas: 2, stability of phlogopite in natural spinel lherzolite and in the system KAlSiO4–MgO–SiO2–H2O–CO2 at high pressures and high temperatures. Am J Sci 280(5):421–458. doi:10.2475/ajs.280.5.421
Yamashita H, Arima M, Ohtani E (1995) High pressure melting experiments on group II kimberlite up to 8 GPa; implications for mantle metasomatism. In: Proceedings of the 6th international Kimberlite conference, pp 259–270
Yang HX, Konzett J, Prewitt CT, Fei YW (1999) Single-crystal structure refinement of synthetic K–M4–substituted potassic richterite, K(KCa)Mg5Si8O22(OH)2. Am Mineral 84(4):681–684
Acknowledgments
We gratefully acknowledge the training provided by D. Caird to the first author for assembling experiments and operating the multi-anvil apparatus. We thank S. Matveev for the help with the electron microprobe. Comments by R. Trønnes and three anonymous reviewers considerably improved iterations of this manuscript. This research was funded by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada to RWL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Timothy L. Grove.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Enggist, A., Luth, R.W. Phase relations of phlogopite and pyroxene with magnesite from 4 to 8 GPa: KCMAS–H2O and KCMAS–H2O–CO2 . Contrib Mineral Petrol 171, 88 (2016). https://doi.org/10.1007/s00410-016-1304-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-016-1304-2