
Abstract
Yb-Y inter-diffusion along a single grain boundary of a synthetic yttrium aluminium garnet (YAG) bicrystal has been studied using analytical transmission electron microscopy (ATEM). To investigate the diffusion, a thin-film containing Yb as the diffusant was deposited perpendicular to the bicrystal grain boundary by pulsed laser deposition (PLD). Structural properties and their change with time in both the diffusant source and the grain boundary are reported. The diffusion profiles are incorporated in a numerical diffusion model, which is applied to determine the grain boundary diffusion coefficient, D gb , at 1.723 K it is equal to 3 × 10−15 m2/s. We find that grain boundary diffusion is 4.85 orders of magnitude faster than volume diffusion, which was determined from the same diffusion experiment. This result is discussed in the context of special versus general grain boundaries. Finally, we successfully tested the capability of synchrotron-based nano-X-ray fluorescence analysis to map micro-chemical patterns in two dimensions with sub-micrometre resolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grain boundary diffusion governs numerous phenomena in solid materials, such as Coble creep, sintering properties, diffusion-induced grain boundary migration, different discontinuous reactions, recrystallization, grain growth and diffusive crack healing (Evans and Charles 1977; Gupta 1975; Hailong and Jun 2002; Wanamaker and Evans 1985). Grain boundary diffusion is the dominating atomic transport mechanism at low temperatures where volume diffusion is sluggish. Even if the rates of grain boundary and volume diffusion approach with increasing temperature, it is commonly assumed that grain boundary diffusion prevails over volume diffusion during most small-scale transport processes of elements in large portions of the solid Earth (e.g. grain boundary migration, recrystallization or cobble creep, Kaur et al. 1995; Mishin and Herzig 1999). Grain boundary diffusion is influenced by the structure of the grain boundary itself.
The most general classification of grain boundaries is based on their structure, and it is differentiated between special and general grain boundaries (Brandon 1966; Smith 1948). Special grain boundaries are those that display special properties, for example good atomic fit across the interface resulting low interface energy and slower element diffusion rates compared to general grain boundaries (Gleiter and Chalmers 1972; Kingery 1974). Special grain boundaries can be described using the Σ nomenclature based on the grain boundary geometry and relates the number of lattice points in the unit cells of the lattices of the two adjacent grains and is defined as Σ = 1/n, where n is the fraction of the lattice points of the two super-imposed lattices that coincide (Chadwick and Smith 1976).
Most solutions to the grain boundary diffusion problem relate to the product of D gb and the effective (or chemical) grain boundary width, δ (e.g. Fisher 1951). To obtain D gb , δ is assumed. The effective grain boundary width is defined as the zone of enhanced diffusion around a grain boundary (White 1973) and results from strain induced by misfit of the adjacent crystal lattices. In ionic crystals, it is accompanied by a space-charge layer (Kingery 1974; Kliewer and Koehler 1965; Lehovec 1953). The structural (or physical) grain boundary width, in contrast, is defined as the distance between two adjacent crystal lattices and is probably smaller than the effective grain boundary width.
In the geomaterial context, previous experimental attempts to exactly quantify grain boundary diffusion are hampered by a number of difficulties such as: (i) Grain boundary diffusion studies are mostly performed on polycrystals with randomly shaped grain boundaries where orientation is not well constrained. Thus, the diffusion geometry is very complex and often impossible to characterize adequately. (ii) The methods commonly used, for example secondary ion mass spectrometry (SIMS), give little or no information about the local defect structure. They also have rather poor lateral resolution. Therefore, grain boundary and volume diffusion are often measured simultaneously, and the extraction of the respective diffusion coefficients is based on numerous assumptions. Moreover, it is known from studies on metals and alloys that the diffusivity can vary by orders of magnitude with varying grain boundary orientation (Guan 2003; Herbeuval et al. 1973; Klugkist et al. 2001), which is a consequence of changing grain boundary energies. This cannot unequivocally be determined, and often an averaged diffusivity is obtained, as several grain boundaries with potentially different transport properties are analysed at the same time. (iii) Analytical or numerical solutions to the grain boundary diffusion problem are often oversimplified or they are not well adapted to the experimental set-up, sometimes simply because the samples cannot be characterized well enough.
It may be surprising that diffusion along a single grain boundary has not yet been studied on the nanometre scale, even though it has been proposed as early as 1986 (Clarke and Wolf 1986), and nanometre-resolved analyses of low-temperature volume diffusion were performed in 1983 (Nicholls and Jones 1983). However, site-specific sample preparation such as lamellae for transmission electron microscopy (TEM) using the focused ion beam (FIB) technique became available only in the last decade (Lee et al. 2003; Phaneuf 1999; Wirth 2004). Furthermore, the fabrication of grain boundaries with exactly defined orientations by wafer direct bonding was developed in the geosciences only lately (Heinemann et al. 2001; Heinemann et al. 2005).
Here, we report an alternative approach to quantify grain boundary diffusion by taking advantage of the above methods recently developed in different scientific disciplines. The main improvements in the present study over previous experimental and analytical procedures are the following:
-
1.
Simple geometry: We use bicrystal synthesis by direct wafer bonding to produce a perfectly straight and exactly oriented grain boundary. As the diffusant source, a thin-film is deposited perpendicular to the grain boundary using pulsed laser deposition. The resulting experimental geometry is simple and extremely well defined (Fig. 1).
Fig. 1 
Schematic sketch of the sample geometry. The grain boundary of the bicrystal is perpendicular to the thin-film (dark grey). The thickness of the thin-film is about 200 nm. The blow-up shows a sketch of the TEM lamella prepared with the focused ion beam (FIB) technique. The dimensions of the foils are usually 15 × 10 μm2 with a thickness of about 100 nm, whereas the bicrystal’s size is in the millimetre range
-
2.
High-resolution analyses: We combine site-specific sample preparation by focused ion beam (FIB) with measurements by TEM. The latter has two main advantages: first, it has a very high spatial resolution, and second, it provides information on the local defect structure, without being destructive. The simple diffusion geometry allows for appropriate TEM analyses because it is possible to orient the grain boundary parallel to the axis of the incident electron beam and the energy-dispersive X-ray (EDX) detector, whereby artefacts from planar defects are excluded (e.g. Williams and Carter 1996). Furthermore, volume and grain boundary diffusion are distinguished and measured on the same sample from one experiment using exactly the same experimental and analytical settings.
-
3.
Numerical modelling: We perform numerical simulations to account for all our experimental observations. We will show that a quantitative evaluation of the diffusion coefficients in our system cannot be performed by applying conventional analytical solutions because none would meet the present experimental configuration and our observations on the nanometre scale.

We use synthetic yttrium aluminium garnet (YAG) bicrystals as model samples. YAG has several advantages over naturally occurring garnet, particularly the availability of large stoichiometric and highly pure single-crystal wafers and its resistance to electron beam damage. Moreover, grain boundary diffusion of rare earth elements in YAG has previously been studied using SIMS (Jiménez-Melendo et al. 2001), which allows for the comparison of different approaches.
Materials and methods
Bicrystal synthesis by direct bonding
Starting material for our experiments was high-purity Y3Al5O12 single crystals (Dobrzycki et al. 2004; Lupei et al. 2001; Weber and Abadie 2001; Yin et al. 1998) for the bicrystal synthesis (Hartmann et al. 2010) and Yb-doped YAG as precursor for the thin-film, i.e. the diffusant source. YAG bicrystal samples were synthesized by the wafer direct bonding method (Gösele et al. 1999; Heinemann et al. 2005; Pößl and Kräuter 1999; Reiche 2006; Tong and Gösele 1999; Tong et al. 1995). The highly polished and ultra clean crystal surfaces are saturated with pure adsorbed water and are brought into contact without force. When contact is established, hydrogen bonds of the opposing crystal surfaces are expected to form. The adsorbed water evaporates at elevated annealing temperatures leaving a grain boundary behind.
A detailed description of the bonding procedure, as well as the specific orientation relationship for the samples used, is given by Hartmann et al. (2010). The contact geometry is close (6.5° misorientation) to a tilt boundary of 36.9° about the bicrystals’ common [100] direction, this equals a twist boundary of 180° parallel to the (210) plane. The additional misorientation results in a twist component and relatively poor atomic fit across the interface and can thus be considered as a model for a general grain boundary. The bicrystals’ grain boundary can also be considered as a near Σ5 (210)/[100] grain boundary.
Diffusion experiments
Diffusion experiments are designed in thin-film geometry, where the grain boundary is covered with the thin-film perpendicular to the surface (Fig. 1). Pulsed laser deposition (PLD, Dohmen et al. 2002; Marquardt et al. 2010) was used to deposit Yb-doped YAG thin-films on the bicrystal. A general review of the method is given by Chrisey and Hubler (2003). As source material (target) for the deposition, we used a Czochralski-grown Yb:YAG homogeneous single crystal (Yb1.17Y1.83Al5O12, Marquardt et al. 2009). Prior to thin-film deposition, the polished bicrystal samples were heated to 893 K in order to desorb potential organic contamination. The diffusion anneals of different time ranges were performed in a gas mixing furnaces at 1,723 K and ambient atmosphere (air). The annealing durations were 5, 20, 40 min, 2, 24.1, 48 and 68 h. For the shortest experiments, the furnace was overheated by about 25°C to compensate for the drop in temperature related to the insertion of the sample holder that cooled during the positioning of the sample. After the diffusion anneal, the sample was quenched by placing it on a bloc of copper. Given the thermal diffusivity in YAG (Marquardt et al. 2009) and the thickness of the samples (~1 mm), the time to reach thermal equilibrium during heating and cooling is very small compared to the annealing times. Temperature was monitored in situ by a type-B thermocouple. For the determination of the grain boundary diffusion, the 24.1-h anneal was used, because the length of both the volume diffusion profile and the grain boundary diffusion profile was long enough for the analyses, but did not extend over the FIB-foil dimensions.
Transmission electron microscopy
TEM lamellae (15 × 8 × 0.1 μm3) were prepared using the FIB technique (Lee et al. 2003; Overwijk et al. 1993; Phaneuf 1999; Wirth 2004). Using this method, samples of constant thickness can be prepared, which is a prerequisite to calculate element ratios directly from energy-dispersive X-ray spectroscopy (EDS) data.
We used analytical and energy-filtered high-resolution transmission electron microscopy (ATEM, HRTEM) on a Tecnai F20 X-Twin TEM operated at 200 kV with a field-emission gun (FEG) as electron source. The TEM is equipped with a post-column Gatan imaging filter (GIF Tridiem). All of the TEM images presented are energy-filtered images, where a 10 eV window was applied to the zero loss peak. The Gatan DigitalMicrograph software was used to analyse the transmission electron micrographs.
ATEM was performed with an EDAX X-ray analyser equipped with an ultrathin window. The intensities were measured during a counting time of 100 s in scanning transmission mode (STEM). The incident electron beam was focused to a diameter of 3.8 nm. We measured in pre-selected windows of 20 × 40 nm2. The sample was inclined towards the detector by an angle of 15–20°. The thin-film–substrate interface of the sample was oriented approximately parallel (within 2–5°) to the detector axis and the incident beam. We avoided crystal orientations where strong diffraction occurs, such as two-beam (Bragg) or in-zone-axis orientations, to circumvent the potential bias of EDS analysis due to electron channelling.
The background was subtracted from each TEM–EDS spectrum using a second-order polynomial, where the parameters were adjusted to obtain the best least-squares fit to the background before and after each relevant peak. Furthermore, we measured zero-Yb spectra to obtain an average value for the background noise in our EDS measurements. The noise was then subtracted from each spectrum. Integrated X-ray intensities of Yb (L α,β ) and Y (L α,β ) were used to calculate the intensity ratio I Yb /I Y. The intensity ratios were converted to concentration ratios using the Cliff–Lorimer equation (Cliff and Lorimer 1975) to finally calculate the mole fraction of Yb.
A more detailed description of the data processing is given by Marquardt et al. (2010). They also provide more details on the error propagation using the Cliff–Lorimer equation and on the estimated excitation volume as well as the resolution due to window measurements.
Nano-X-ray fluorescence analysis at the ESRF
Qualitative elemental maps of the diffusion zone in the bicrystal have been obtained at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. Experiments were performed at the nanoprobe beamline ID13, where a hard X-ray nanobeam generated with nanofocusing X-ray lenses (NFLs, Boye 2009) is used to run fluorescence analyses with a spatial resolution ranging from 200 to 50 nm (Hanke et al. 2008; Schroer et al. 2005; Schropp et al. 2010). A FIB-cut lamella (10 × 8 × 0.25 μm3) was mounted on a perforated carbon-film on a 3-mm copper grid, which is a standard procedure to mount TEM-FIB-lamellae. The energy of the X-rays was 24.3 keV. The spatial resolution is basically defined by the spot size, which was determined by gold knife-edge scans to be 165 × 171 nm2 in our experiment. The sample is oriented 45° with respect to both the energy-dispersive detector and the incident beam to record maximum fluorescence signals.
Experimental results
Thin-film crystallisation
The grain boundary produced by direct wafer bonding is thoroughly described elsewhere (Hartmann et al. 2010) and is used for our grain boundary diffusion experiment. The thin-film, which serves as diffusant source, is oriented perpendicular to the grain boundary (Fig. 1). It was initially amorphous (Fig. 2a), but crystallized during the diffusion anneal (Fig. 2b) and adapted the structure and orientation of the bicrystal. As a result, the grain boundary extends into the homoepitaxially grown thin-film (Fig. 2c).

Bright field (BF) images of the bicrystal with the thin-film prior to and after annealing. For FIB sample preparation, a protection layer of platinum was deposited on the carbon coating (upper part of the images). a The thin-film is initially amorphous and has an excellent contact with the bicrystal. b Thin-film after 20-min annealing at 1,723 K is partially recrystallized. Some parts of the thin-film are epitactic with the bicrystal, indicated by broken lines. Still, some large crystals are present. The right bicrystal grain is in a strongly diffracting orientation and the lower region of the thin-film has the same orientation. The outermost ~2% of the thin-film are amorphous, framed with red broken lines. The slightly curved contrast changes below the thin-film–bicrystal interface on the left part are caused by a hole in the sample supporting perforated carbon film. c Thin-film after 2-h annealing at 1,723 K. Diffraction contrast causes a different shading of the opposing crystals. The thin-film grew epitaxially on the bicrystal; thus, the grain boundary continues into the thin-film. The thin-film of this specific sample is 380 nm thick; at the surface of the left grain, some portions (up to 13%) are not crystalline, indicated by red broken lines. Note that the grain boundary produced in the former thin-film is slightly curved. Close to the surface, it is inclined with respect to the incident beam. A groove formed during annealing at the surface of the thin-film
The observed progress of the thin-film crystallization was as follows: After 20 min, the films are polycrystalline with polyhedral crystals ranging from about 100 to 200 nm in size, their diameter exceeds the thin-film thickness (Fig. 2b). Some amorphous material still remained between the crystals, and at the thin-film–bicrystal interface, local epitaxial growth is present. After 40 min, epitaxy and crystallisation are highly advanced (Fig. 3). Thin-film and bicrystal substrate are completely homoepitactic after 2 h (Fig. 2c). Diffraction patterns designate only the presence of garnet phases, and the grain boundaries are now fully crystalline. In contrast, amorphous material at the surface of the thin-film was still present, even after annealing for 24 h. The thickness of the remaining amorphous layer differs with space, mostly it is in the range of 2% of the entire thin-film thickness, but at one position, it reached 13% of the thin-film thickness (Figs. 2, 3).

BF images of the grain boundary in the thin-film after annealing at 1,723 K for a period of 40 min. Images a and b are acquired using different defocus settings on the same sample; thus, different planes of depth were imaged. The thin-film was about 180 nm thick. a The grain boundary of the bicrystal substrate starts to curve towards the thin-film and joins a grain boundary between two grains of the polycrystalline thin-film. b At a different image plane of the same sample, the grain boundary of the bicrystal continues straight into the thin-film. The thin-film is close to full epitaxy. The pitted appearance of the images arises from the relatively thick sample
Grain boundary characteristics in the bicrystal and thin-film
We characterised the grain boundary of the original bicrystal after each anneal. Structural changes, such as orientation relationships between the adjacent grains, rigid body displacements, grain boundary migration, dislocation or step formation, were only rarely observed. For example, in one experiment, we observed that the orientation and placement of the grain boundary of the bicrystal changed as it became curved towards the thin-film–bicrystal interface (Fig. 4). Samples where such phenomena occurred were excluded from further investigation. Lattice fringe images reveal no defects in the lattice planes imaged. Based on high-resolution transmission electron micrographs published elsewhere (Fig. 5 in Hartmann et al. 2010), we estimate the structural grain boundary width to be 2 nm as it alternates periodically between 0.5 and 3 nm. The alternation is related to the periodic strain contrast that arises from the additional (6.5°) misorientation with respect to the perfect Σ5 grain boundary (Hartmann et al. 2010). It is surrounded by a substantially strained lattice in both of the adjacent grains.

STEM image of an experiment where grain boundary migration was observed and which was therefore discarded from further investigation. Grain boundary migration is rather the exception than the rule in these experiments. The image is partially influenced by diffraction contrast, visible due to the chosen camera length

STEM image of the sample investigated with nano-XRF (1,723 K and 24.1 h). The camera length is chosen to probe a mixture of Z-contrast and diffraction contrast; the thin-film is fully epitactic. The platinum layer is the brightest grey in the image, the thin-film appears in light grey and the two parts of the bicrystal are dark grey. The region mapped using the YbLα peak from synchrotron X-ray fluorescence at the ESRF is shown in the enlargement
Furthermore, we investigated the grain boundary formed within the thin-film. During the initial stages of annealing (5–40 min), the grain boundary within the thin-film meandered between the crystals of the polycrystalline thin-film and was often inclined with respect to the bicrystal grain boundary (Fig. 3a, b). Figure 3b, taken after a 40-min anneal, shows that at some parts, the grain boundary cut already straight through the thin-film.
After longer annealings, the grain boundary of the thin-film has the same orientation as in the bicrystal (Figs. 2c, 5). In some experiments, however, minor changes in orientation occur. Such minor changes are, for example, the absence of steps that are normally present at the grain boundary in intervals of 40 nm (Hartmann et al. 2010) or slight curvatures in the newly generated grain boundary. The grain boundary in the thin-film of the sample annealed for 24.1 h displays the same orientation as the grain boundary in the bicrystal (Fig. 5 STEM image). Lattice fringe images reveal no defects in the lattice planes imaged.
Diffusion profiles
Inter-diffusion profiles of Yb-Y in the volume and in the grain boundary are plotted as mole fractions of Yb versus distance from the thin-film–bicrystal interface (Fig. 6). In the next section the procedure to extract the grain boundary diffusion coefficient by numerical fitting is explained. An overview map of the Yb distribution along and across the grain boundary is shown in Fig. 5.

Comparison of the experimental measurements (points) and numerical solutions (solid lines) for the volume (blue, data from Marquardt et al. 2010) and grain-boundary (red) diffusion. Experimental points were measured by EDX-based ATEM on the same lamella. The interface between thin-film and substrate is at zero. The blue line was obtained by adjusting the analytical solution for the volume diffusion profile. The red line results from the direct modeling of the grain boundary as described in the text. The calculated grain boundary diffusion coefficient is 4.85 orders of magnitude higher than the volume diffusion coefficient
Numerical modelling of grain boundary diffusion
Fast diffusion along an isolated grain boundary, accompanied by relatively slow diffusion into the volume of the adjacent crystals, has been considered theoretically in a number of studies, starting with the pioneering work of Fisher (1951). The Whipple–LeClaire analytical solution cannot be used for the present study as it presupposes a constant concentration in the thin-film and applies to averaged concentration with depth, where the spatial average is taken over a series of surfaces parallel to the thin-film (Le Claire 1963). In contrast, the concentrations in our experiments are integrated over small windows across a single isolated grain boundary. The thin-film, which serves as the diffusant source, was, however, only treated very approximately, either it was considered as a constant source (e.g., Fisher 1951; Whipple 1954) or as an instantaneous source (Suzuoka 1961; Suzuoka 1964). In our case, the thin-film undergoes a structural change during the diffusion anneal that must be taken into account (see Marquardt et al. 2010). Therefore, the diffusion profiles were modelled with the following working assumptions:
-
1.
The final diffusant distribution inside the thin-film is time-dependent and non-uniform; the Yb mole fraction decreases towards the thin-film–substrate interface. Therefore, a detailed analysis of diffusion inside the thin-film is necessary.
-
2.
The diffusion coefficient in the thin-film decreases by several orders of magnitude during annealing, since it is initially amorphous and subsequently crystallized.
-
3.
After crystallization, diffusion is similarly slow in both the thin-film and the bicrystal.
-
4.
As the narrow outer thin-film layer remains amorphous, diffusion therein and at the surface of the thin-film is still fast.
-
5.
After crystallization, the grain boundary cuts through the thin-film, thus, it is connected to the outermost layer of the thin-film, where diffusion is fast at all times.
A numerical model that accounts for these characteristics of the diffusion assembly was developed using the COMSOL Multiphysics software package for the implementation of the finite elements approach. The system geometry is shown in Fig. 7. The OX-axis lies along the grain boundary, the thin-film region corresponds to negative values of x. The OY-axis traces the initial interface between the thin-film and the bicrystal. In our modelling, we use a two-dimensional setting; the dependence of the diffusant distribution on z is ignored.

System geometry for the numerical model. b Initial stage of the system geometry used for modelling. c System geometry after t = t cr
The transport of Yb in the YAG bicrystal (blue region in Fig. 7) is governed by a linear diffusion equation
for the Yb mole fraction
where C Y,Yb are the Y and Yb concentrations, respectively. The parameter D v denotes the volume inter-diffusion coefficient between Yb and Y in the bicrystal. Material transport along the grain boundary is described by a reduced diffusion equation (see Fisher 1951)
for the Yb mole fraction u(x, t) along the boundary
The parameter D gb in Eq. (2) is the grain boundary diffusion coefficient, δ is the effective grain boundary width. The last term in Eq. (2) describes particle loss due to transversal (y-parallel) diffusion into the volume of the bicrystal. Equation (3) ensures continuity of the mole fraction.
Diffusive transport inside the thin-film is governed by a time-inhomogeneous diffusion equation
where the volume diffusion coefficient in the thin-film, D f (t), decreases from some (large) initial value D ∞ to D v in the course of the structural change of the thin-film from amorphous to crystalline. For simplicity, we assume that D f = D ∞ for 0 < t < t cr (crystallization time) and D f = D v afterwards. Initially, Yb is only present in the thin-film, such that
where h and L denote the extension of the thin-film and of the bicrystal along the x-axis, respectively. Both mole fraction and diffusive flux are continuous at the thin-film–bicrystal interface, such that
The numerical solution was calculated in two steps. First, Eqs. (1–4) were solved for 0 < t < t cr for the system shown in Fig. 7a. A no-flux boundary condition was applied at the outer border of the thin-film. During the first stage, the large parameter D ∞ creates a practically uniform mole fraction distribution in the thin-film, which slowly decreases due to loss of Yb into the bicrystal.
Instantaneous crystallization is assumed to occur at t = t cr , the corresponding value of the mole fraction for the thin-film is denoted by c cr . For t > t cr , the grain boundary extends into the thin-film as shown in Fig. 7b and we replace D f = D ∞ with D f = D v in Eq. (4). Fast diffusion still occurs at the outer border of the thin-film, an assumption based on the observations described in the section on thin-film crystallization. In contrast, inside the thin-film, the mole fraction distribution is now almost “frozen”, changing only slowly, because D v « D ∞ . Therefore, we assume that for t > t cr , the outer border distribution of c is uniform and time independent, and we applied a Dirichlet boundary condition c = c cr for x = −h accordingly. In a second stage, Eqs. (1–4) were solved for t cr < t < t to , where tto is the total annealing time.
The parameter values were chosen to represent the experimental settings. Specifically, t to = 24.1 h, t c = 40 min, h = 185 nm, δ = 2 nm (which is a mean value of the observed variations in the structural grain boundary width), c 0 = 0.403. The Yb-Y inter-diffusion coefficient in the YAG crystal is D v = 4.3 × 10−20 m2 /s as determined by Marquardt et al. (2010), D ∞ = 105 × D v . For the numerical solution, the bicrystal domain was restricted to |y| < 2,000 nm and L = 8,000 nm. Performing calculations for 0 < t < t cr , we have found that c cr /c 0 = 0.92. This value was applied to the Dirichlet boundary condition at the outer border of the thin-film for t cr < t < t to .
Diffusion profiles were simulated for different values of D gb . For each simulation, the resulting mole fraction was evaluated along the grain boundary by averaging c over 20 × 40 nm2 boxes, in order to be comparable with the ATEM measurement procedure (see section material and methods). The best fit between the experimental and numerical results (Fig. 6) yields the grain boundary diffusion coefficient
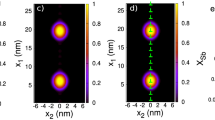
The space distribution of the mole fraction c(x, y, t) (normalized over c 0) is shown in Fig. 8 for t = 0, t = t cr , and t = t to . As expected, the Yb distribution in the thin-film is uniform at t = t cr but differs from that at t = 0. For t = t to , we see a scenario of fast diffusion along the grain boundary accompanied by comparatively slow volume diffusion into the bicrystal from both the grain boundary and the thin-film. Note that within the thin-film, the Yb mole fraction is now decreasing towards the thin-film–bicrystal interface in agreement with the experimental observations.

Density plots for the mole fraction c(x, y, t) normalized over the initial value in the thin-film c 0 at different times. Left the initial distribution, t = 0; middle the distribution at the time of crystallization, t = t cr , right the final distribution at t = T, it corresponds to the ATEM measurements
Figure 6 shows the calculated compositions (integrated over 20 × 40 nm2 windows) along the grain boundary versus experimental data. The close agreement suggests that the model (1–4) and the grain boundary diffusion coefficient (5) correctly describe the experimental observations. A better fit for the first 500 nm of the diffusion profile may only be obtained by a further refinement of the thin-film model.
Also note that the crystallization of the thin-film results in a reduction of the diffusant supply that influences the distribution of the diffusant itself along the grain boundary and thus D gb . But as the value of D gb is the logarithmic derivative of the concentration, it is not very sensitive. However, the measured concentrations that are compared to numerics in Fig. 6 are strongly affected by D f .
Discussion
Our study was performed on YAG at 1,723 K and ambient atmosphere; the experimental conditions of other studies used for comparison are similar. For the sake of readability, we will not repeat the conditions continuously.
Thin-film and grain boundary properties
The observed change of the crystals’ orientation within the thin-film is related to a grain growth process, which is dominated by the largest grains, i.e. the bicrystal substrate (Humphreys and Hatherly 1996; Kaur et al. 1995). It has repeatedly been observed that grain growth in thin-films stagnates when a grain diameter of a size approximately equal to the thin-film thickness is reached (e.g. in gold thin-films, Harris et al. 1998, olivine and orthopyroxene, Milke et al. 2007). This is not observed in the present study, probably because epitaxial growth dominates the final thin-film structure.
The nature of the amorphous surface layer is not completely clear. We suspect that it formed during crystallisation of the thin-film and that it is a non-stoichiometric residue that accumulated at the thin-film surface, because the thin-film crystallisation started at the interface with the substrate. In our case, minor non-stoichiometry cannot be detected using TEM–EDS as the composition in the outermost layer of the sample is influenced by gallium implantation during sample preparation (Lee et al. 2003; Lee et al. 2007). Alteration of the thin-film’s surface caused by sample preparation cannot be excluded (Lee et al. 2007), but is improbable because the thickness of the amorphous layer varies randomly on a single FIB-foil.
The change in grain boundary orientation and placement as shown in Fig. 3 suggests that grain rotation, grain boundary migration and/or chemical induced grain boundary migration takes place at the chosen annealing temperatures. It probably influences the kinetics of crystallisation of the thin-film, even though its influence on the original bicrystal grain boundary is only secondary. We believe that grain rotation and grain boundary migration inside the recrystallizing thin-film do not significantly affect the diffusion process inside the grain boundary of the bicrystal, at least not so as long as the grain boundary diffusion lengths in the bicrystal are significantly higher than that in the thin-film, i.e. at sufficiently long run durations. We therefore excluded both mechanisms for the evaluation of the diffusion processes.
Diffusion
The Yb distribution map (Fig. 5) obtained with nano-XRF has a relatively poor resolution compared to TEM and cannot be interpreted quantitatively. Nevertheless, it is consistent with the numerical simulations (compare Figs. 5, 8). The resolution of about 165 nm is revolutionary for synchrotron XRF measurements and is very promising for diffusion studies with profile lengths in the range of a few micrometres where it could provide an outstanding spatial data coverage.
Our measured diffusion profile yield a grain boundary diffusion coefficient of 3 × 10−15 m2/s at 1,723 K and significantly differs from the grain boundary diffusion coefficient of about 1.6 × 10−13 m2/s as reported by Jiménez-Melendo et al. (2001) using SIMS. A similar discrepancy is found for the determined volume diffusion coefficients as determined by Jiménez-Melendo et al. (2001) and Marquardt et al. (2010), possible reasons including different experimental set-ups and analyses were discussed therein. For the detailed comparison, it is disadvantageous that the characteristics of the thin-film diffusant source are not known in the study of Jiménez-Melendo et al. (2001). This lead to the use of a diffusion model not precisely adapted to the experimental conditions. In addition, diffusion profiles measured by SIMS on polycrystals yield average profiles that contain information on volume diffusion and on grain boundary diffusion in many differently oriented grain boundaries. Both studies extracted the grain boundary diffusion coefficients from the product of D gb and δ. For δ, both use a directly measured structural grain boundary width, because there is no information about the effective grain boundary width. Jiménez-Melendo et al. (2001) used a value of 1 nm, whereas we used a value of 2 nm as determined on the specific grain boundary investigated. However, structural and effective grain boundary widths are not necessarily the same (Farver and Yund 1991).
We have shown that our clearly defined experimental geometry has several advantages for the study of grain boundary diffusion. First, it allows for the measurements of grain boundary diffusion on a single grain boundary and the investigation of its orientation at the same time. Second, a thorough monitoring of the thin-film’s evolution and associated changes of the local defect structure is feasible using TEM. These investigative improvements are the basis for the development of our 2D numerical model, which is well adapted to the experimental observations.
In our study, we find the grain boundary diffusion coefficient to be 4.85 orders of magnitude higher than the determined volume diffusion coefficient. Jiménez-Melendo et al. (2001) report a respective difference of about 5.38 orders of magnitude. Assuming that the two studies are internally consistent, this indicates that the near Σ5 grain boundary studied in this work exhibits a lower grain boundary diffusion coefficient than an average over many different grain boundaries. This finding is consistent with the common assumption that small angle and Σ grain boundaries have the lowest grain boundary diffusivities (Crank 1975; Joesten 1991; Kaur et al. 1995; Mishin and Herzig 1999; Schwarz et al. 2002; Smoluchowski 1952; Turnbull and Hoffman 1954). As the investigated grain boundary has properties of general grain boundaries due to its additional 6.5° misorientation with respect to the perfect Σ5 grain boundary, our finding likely represents a diffusion coefficient closer to the lower bound; in between the lowest grain boundary diffusivities occurring in special grain boundaries and the highest diffusivities occurring in most-disturbed general grain boundaries. We suggest that the presented combination of methods is applied to systematically study the effect of grain boundary orientation on the diffusion coefficient.
Conclusion and outlook
We performed diffusion experiments on a single, well-characterized grain boundary. In addition, we carefully characterized the time-dependent properties of the diffusant source in our experiments. Based on the observed structural changes of the thin-film and the associated changes of its transport properties, we developed a numerical model to extract the grain boundary diffusion coefficient from the measured diffusion profile. We find that grain boundary diffusion in our system is 4.85 orders of magnitude faster than volume diffusion and conclude that this value is at the lower end of differences between grain boundary and volume diffusion.
We show that used combination of miniaturized diffusion experiments, high-resolution analytical techniques and numerical simulations is exceptionally suited to study volume and grain boundary diffusion simultaneously.
Finally, we note that synchrotron-based nano-XRF to map diffusion in two dimensions, which we explored in this study, has high potential for systematic diffusion studies, where grain boundary diffusion extends over few micrometre lengths. Furthermore, direct measurements of the effective grain boundary width are necessary for a better understanding of its influence on D gb .
References
Boye P (2009) Nanofocusing Refractive X-Ray Lenses. Institut für Strukturphysik, Doktor, Technische Universität Dresden, Dresden, p 145
Brandon DG (1966) The structure of high-angle grain boundaries. Acta Metall 14:1479–1484
Chadwick GA, Smith DA (1976) Grain boundary structure and properties. Academic Press, London
Chrisey DB, Hubler GK (2003) Pulsed laser deposition of thin films. Wiley, VCH, London, p 648
Clarke DR, Wolf D (1986) Chapter 3 grain boundaries in ceramics and at ceramic-metal interfaces. Mater Sci Eng 83(2):197–204
Cliff G, Lorimer GW (1975) The quantitative analysis of thin specimens. J Microsc 103:203–207
Crank J (1975) The mathematics of diffusion. Oxford University Press, New York
Dobrzycki L, Bulska E, Pawlak DA, Frukacz Z, Wozniak K (2004) Structure of YAG crystals doped/substituted with erbium and ytterbium. Inorg Chem 43:7656–7664
Dohmen R, Becker H-W, Meißner E, Etzel T, Chakraborty S (2002) Production of silicate thin films using pulsed laser deposition (PLD) and applications to studies in mineral kinetics. Eur J Miner 14:1155–1168
Evans AG, Charles EA (1977) Strength recovery by diffusive crack healing. Acta Metall 25(8):919–927
Farver JR, Yund RA (1991) Measurement of oxygen grain boundary diffusion in natural, fine-grained, quartz aggregates. Geochim Cosmochim Acta 55(6):1597–1607
Fisher JC (1951) Calculations of diffusion penetration curves for surface and grain boundary diffusion. J Appl Phys 22(1):74–77
Gleiter H, Chalmers B (1972) High-angle grain boundaries. Pergamon Press, Oxford
Gösele U, Tong QY, Schumacher A, Kräuter G, Reiche M, Plößl A, Kopperschmidt P, Lee TH, Kim WJ (1999) Wafer bonding for microsystems technologies. Sens Actuators A Phys 74(1–3):161–168
Guan Z (2003) Korngrenzstrukturen, Fremdatomdiffusion und Homogenität in nanokristallinen Metallen und Legierungen. Naturwissenschaftlich-Technische Fakultät II—Physik und Elektrotechnik -, Doctor, Universität des Saarlandes, Saarbrücken, p 140
Gupta TK (1975) Crack healing in thermally shocked MgO. J Am Ceram Soc 58(3–4):143
Hailong Z, Jun S (2002) Morphological evolution during diffusive healing of internal cracks within grains of α-iron. Acta Mech Sin 18(5):516–527
Hanke M, Dubslaff M, Schmidbauer M, Boeck T, Schoder S, Burghammer M, Riekel C, Patommel J, Schroer CG (2008) Scanning x-ray diffraction with 200 nm spatial resolution. Appl Phys Lett 92(19):193109-3
Harris KE, Singh VV, King AH (1998) Grain rotation in thin films of gold. Acta Mater 46(8):2623–2633
Hartmann K, Wirth R, Heinrich W (2010) Synthetic near Σ5 (210)/[100] grain boundary in YAG fabricated by direct bonding: structure and stability. Phys Chem Miner 37(5):291–300
Heinemann S, Wirth R, Dresen G (2001) Synthesis of feldspar bicrystals by direct bonding. Phys Chem Miner 28(10):685–692
Heinemann S, Wirth R, Gottschalk M, Dresen G (2005) Synthetic [100] tilt grain boundaries in forsterite: 9.9 to 21.5°. Phys Chem Miner 32(4):229
Herbeuval I, Biscondi M, Goux C (1973) Influence of Intercrystalline Structure on the Diffusion of Zinc in Symmetrical Bending Boundaries of Aluminum Mem. Sci Rev Met 70(1):39–46
Humphreys FJ, Hatherly M (1996) Recrystallization and related annealing phenomena. Pergamon, Oxford
Jiménez-Melendo M, Haneda H, Nozawa H (2001) Ytterbium cation diffusion in Yttrium Aluminum Garnet (YAG)—implications for creep mechanisms. J Am Ceram Soc 84(10):2356–2360
Joesten R (1991) Grain-boundary diffusion kinetics in silicate and oxide minerals. Diffusion, atomic ordering, and mass transport; selected topics in geochemistry, vol 8. Springer, New York, pp 345–395
Kaur I, Mishin Y, Gust W (1995) Fundamentals of grain and interphase boundary diffusion. Wiley, Chichester
Kingery WD (1974) Plausible Concepts Necessary and Sufficient for Interpretation of Ceramic Grain-Boundary Phenomena: II, Solute Segregation, Grain-Boundary Diffusion, and General Discussion*. J Am Ceram Soc 57(2):74–83
Kliewer KL, Koehler JS (1965) Space charge in ionic crystals. I. General approach with application to NaCl. Phys Rev 140(4A):A1226
Klugkist P, Aleshin AN, Lojkowski W, Shvindlerman LS, Gust W, Mittemeijer EJ (2001) Diffusion of Zn along tilt grain boundaries in Al: pressure and orientation dependence. Acta Mater 49(15):2941–2949
Le Claire AD (1963) The analysis of grain boundary diffusion measurements. British J Appl Phys 14(6):351–356
Lee MR, Bland PA, Graham G (2003) Preparation of TEM samples by focused ion beam (FIB) techniques; applications to the study of clays and phyllosilicates in meteorites. Miner Mag 67(3):581–592
Lee MR, Brown DJ, Smith CL, Hodson ME, MacKenzie M, Hellmann R (2007) Characterization of mineral surfaces using FIB and TEM: a case study of naturally weathered alkali feldspars. Am Miner 92:1383–1394
Lehovec K (1953) Space-Charge Layer and Distribution of Lattice Defects at the Surface of Ionic Crystals. J Chem Phys 21(7):1123–1128
Lupei V, Lupei A, Pavel N, Taira T, Ikesue A (2001) Comparative investigation of spectroscopic and laser emission characteristics under direct 885-nm pump of concentrated Nd:YAG ceramics and crystals. Appl Phys B Lasers Opt 73(7):757–762
Marquardt H, Ganschow S, Schilling F (2009) Thermal diffusivity of natural and synthetic garnet solid solution series. Phys Chem Miner 36(2):107–118
Marquardt K, Petrishcheva E, Abart R, Gardés E, Wirth R, Dohmen R, Becker H-W, Heinrich W (2010) Volume diffusion of Ytterbium in YAG: thin-film experiments and combined TEM–RBS analysis. Phys Chem Miner 37(10):751–760
Milke R, Dohmen R, Becker H-W, Wirth R (2007) Growth kinetics of enstatite reaction rims studied on nano-scale, Part I: methodology, microscopic observations and the role of water. Contrib Miner Petrol 154(5):519–533
Mishin Y, Herzig C (1999) Grain boundary diffusion: recent progress and future research. Mater Sci Eng A 260(1–2):55–71
Nicholls AW, Jones IP (1983) Determination of low temperature volume diffusion coefficients in an Al-Zn alloy. J Phys Chem Solids 44(7):671–676
Overwijk MHF, van den Heuvel FC, Bulle-Lieuwma CWT (1993) Novel scheme for the preparation of transmission electron microscopy specimens with a focused ion beam. J Vac Sci Technol B 11(6):2021–2024
Phaneuf MW (1999) Applications of focused ion beam microscopy to materials science specimens. Micron 30(3):277–288
Pößl A, Kräuter G (1999) Wafer direct bonding: tailoring adhesion between brittle materials. Materials Science and Engineering. Reports(25): 1–88
Reiche M (2006) Semiconductor wafer bonding. Phys Status Solidi a 203(4):747–759
Schroer CG, Kurapova O, Patommel J, Boye P, Feldkamp J, Lengeler B, Burghammer M, Riekel C, Vincze L, van der Hart A, Kuchler M (2005) Hard x-ray nanoprobe based on refractive x-ray lenses. Appl Phys Lett 87(12):124103-3
Schropp A, Boye P, Feldkamp JM, Hoppe R, Patommel J, Samberg D, Stephan S, Giewekemeyer K, Wilke RN, Salditt T, Gulden J, Mancuso AP, Vartanyants IA, Weckert E, Schoder S, Burghammer M, Schroer CG (2010) Hard x-ray nanobeam characterization by coherent diffraction microscopy. Appl Phys Lett 96(9):091102-3
Schwarz SM, Kempshall BW, Giannuzzi LA, Stevie FA (2002) Utilizing the SIMS technique in the study of grain boundary diffusion along twist grain boundaries in the Cu(Ni) system. Acta Mater 50(20):5079–5084
Smith CS (1948) Introduction to grains, phases, and interfaces-an interpretation of microstructure. Trans AIME 175:15–51
Smoluchowski R (1952) Theory of grain boundary diffusion. Phys Rev 87(3):482
Suzuoka T (1961) Lattice and grain boundary diffusion in polycrystals. Trans Jpn Inst Met 2:25–33
Suzuoka T (1964) Exact Solutions of Two Ideal Cases in Grain Boundary Diffusion Problem and the Application to Sectioning Method. J Phys Soc Jpn 19:839
Tong QY, Gösele U (1999) Semiconductor wafer bonding : science and technology. Wiley, New York
Tong QY, Gösele U, Martini T, Reiche M (1995) Ultrathin single-crystalline silicon on quartz (SOQ) by 150°C wafer bonding. Sens Actuators A 48(2):117–123
Turnbull D, Hoffman RE (1954) The effect of relative crystal and boundary orientations on grain boundary diffusion rates. Acta Metall 2(3):419–426
Wanamaker BJ, Evans B (1985) Experimental diffusional crack healing in olivine. In: Schock RN (ed) Point defects in minerals, vol 31. American Geophysical Union, Washington, pp 194–210
Weber R, Abadie J (2001) Processing and optical properties of YAG- and Rare-Earth-Aluminum Oxide-Composition Glass Fibers. Mater Res Soc 702:193–204
Whipple RTP (1954) Concentration contours in grain boundary diffusion. Philos Mag 45(371):1225–1236
White S (1973) Syntectonic recrystallization and texture development in quartz. Nature 244(5414):276–278
Williams DB, Carter BC (1996) Transmission electron microscopy. Springer, New York
Wirth R (2004) Focused ion beam (FIB): a novel technology for advanced application of micro- and nanoanalysis in geosciences and applied mineralogy. Eur J Miner 16(6):863–876
Yin H, Deng P, Gan F (1998) Defects in YAG:Yb crystals. J Appl Phys 83(7):3825–3828
Acknowledgments
We would like to express our appreciation to the group of Christian Schroer from the Universität Dresden for measuring the Yb distribution in our sample using nano-XRF at the European Synchrotron Radiation Facility and we would like to thank Manfred Burghammer and Sebastian Schoeder for assistance in using beam line ID13. Furthermore, the financial support from the German GeoForschungsZentrum Potsdam, GFZ. Finally, we thank Ralf Dohmen for his support during PLD thin-film production. K. M. thanks the CNV foundation for financial support and Hauke Marquardt fort the manifold discussions. Finally, we thank the reviewers Daniele Cherniak and Bruce Watson for their thorough reading that improved the manuscript and reduced sources of misunderstanding.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Hoefs.
Rights and permissions
About this article
Cite this article
Marquardt (née Hartmann), K., Petrishcheva, E., Gardés, E. et al. Grain boundary and volume diffusion experiments in yttrium aluminium garnet bicrystals at 1,723 K: a miniaturized study. Contrib Mineral Petrol 162, 739–749 (2011). https://doi.org/10.1007/s00410-011-0622-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-011-0622-7