
Abstract
Late Alpine fissures and fractures in amphibolite-facies basement gneisses at Arvigo (Val Calanca, Swiss Alps) show distinct cm-sized reaction selvages parallel to the fracture walls that composed of subgreenschist facies assemblages produced by the interaction of water present in the fracture porosity with the old high-grade gneiss assemblages. The process of selvage or reaction-vein formation occurred in the brittle deformation regime and at temperatures characteristic of, first the prehnite-pumpellyite facies and then later of the zeolite facies. The vein formation occurred during uplift and cooling at very late stages of the Alpine orogeny. The reaction veins are composed of a selvage of altered gneiss on both sides of the central fracture and a central zone with fissure minerals that have been growing in the open fracture pore space. The central zone of the Arvigo veins contains an early assemblage with epidote, prehnite and chlorite and a late succession sequence of various species of zeolite. The veins of the Arvigo quarry are convincing evidence that fracture fluids in gneiss and granite have the potential to precipitate Ca–zeolite. This is an important find because many fluids recovered from deep continental drill holes and from geothermal energy exploration are found to be oversaturated in respect to a number of Ca–zeolite species. Vein formation during late uplift and cooling of the Alps occurred at continuously decreasing T and at hydrostatic pressure: (1) coexisting prehnite/epidote records temperatures of 330–380°C, (2) chlorite formation at temperature of 333 ± 32°C and (3) formation of zeolites <250°C. In the selvages the prime reaction is the replacement of plagioclase by albite along a sharp reaction front that separates the selvage from unaltered gneiss. In addition to albitisation, chloritisation of biotite is the second important reaction in the alteration process. The reactions release components for the formation of Ca–Al silicates. The water–rock interaction is associated with a depletion of Al, Si, Ca, Fe and K in the altered wall rock. The overall reaction is associated with an increase in porosity of up to 14.2 ± 2.2% in the selvage zone (altered wall rock), caused by the volume decrease during albitisation and the removal of biotite. The propagation of the sharp reaction front through the gneiss matrix occurred via a dissolution-reprecipitation mechanism. Zeolite formation is tied to the plagioclase alteration reaction in the rock matrix, which releases components for zeolite formation to a CO2-poor aqueous liquid.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Water is reaction and transport medium in fractured basement rocks of the upper continental crust. The aqueous liquid present in the fracture porosity drives re-equilibration by dissolution of unstable early minerals and precipitation of more stable product phases at low P and T (e.g. Bucher et al. 2009; Nordstrom et al. 1989; Stober and Bucher 1999). These secondary low-grade minerals of the subgreenschist facies give important information about the physical and chemical condition of re-equilibration.
Alpine-type orogenic mountain belts are characterised by a late stage of continental collision creating an isostatically unstable low-density root prone to uplift, erosion and cooling. Once continental basement rocks of the orogen have crossed the ductile–brittle transition zone during uplift, fracture porosity is generated and, because P–T conditions are subcritical, liquid water can freely circulate in the fracture system (Stober and Bucher 2005). Low-T alteration of gneiss and granite is caused by interaction with this circulating water. Alteration of crustal rocks first takes place along the fracture surfaces. Minerals may precipitate from the water at hydrostatic pressure in the open fracture pores space as fissure minerals (e.g. famous Alpine rock crystals). The alteration reactions attack gneiss and granite on both sides along the fractures and penetrate the high-grade basement rocks to various extents (mm–m). The resulting structures are alteration or reaction veins that, if fracture density is high, may completely replace high-grade rocks with their low-T hydrated counterparts. Very typical are cm-sized reaction veins that can be related to the described water–rock interaction processes (e.g. Bucher-Nurminen 1982; Ferry 1979; Mercolli et al. 1984; Parneix and Petit 1991). The product assemblages bear information on P–T and water composition during fluid-rock interaction (e.g. Cho et al. 1986; Bevins et al. 1991; Diegel and Ghent 1994; Faryad and Dianiska 2003; Gianelli et al. 1998; Liou 1979, 1985; Rose et al. 1992; Thompson 1971).
Highly soluble minerals such as calcite and grain boundary salt are first removed from the fracture surfaces. Later, the prime objects of the alteration damage in granite and gneiss are plagioclase and biotite. Products of the reactions are predominantly chlorite, K-feldspar and Ca–Al silicates such as epidote, prehnite or zeolite depending on temperature and water composition. Zeolites are formed in and limited to fractures. Zeolite formation typically affects only a few cm of the wall rock (Weisenberger and Bucher 2010). Zeolites are common in volcanic and volcanoclastic rocks of basaltic to acidic composition and in sedimentary environments, where elements necessary for the formation of secondary minerals mainly derive from dissolution of primary glass and feldspar (e.g. Gottardi 1989; Hay 1966, 1977; Hay and Sheppard 1977, 2001; Neuhoff et al. 1999; Walker 1960, 1963). Low-T Ca–Al-silicates have been reported from gneiss and granite as a result of feldspar alteration (e.g. Faryad and Dianiska 2003; Freiberger et al. 2001; Weisenberger and Bucher 2010). A detailed study of the fissure zeolite distribution in the Central Swiss Alps (Weisenberger and Bucher 2010) has shown that zeolites hosted in gneiss and granite are widespread and that they are an important product of water–rock interaction in the upper continental crust. Water sampled during deep continental drilling programs is mostly oversaturated with respect to a number of zeolite species (Bucher et al. 2009; Nordstrom et al. 1989, Stober and Bucher 1999, 2005).
This study focuses on the process of fracture related water–rock interaction that produces Ca–Al silicates, especially zeolites in gneiss and granite material at low-T conditions. We present a detailed mineralogical and petrological study of exceptionally well-developed alteration veins in granitic gneiss of the Simano nappe in the Central Alps. The samples were collected in an active quarry in Arvigo (Val Calanca, Switzerland; Fig. 1). They show classic effects of reaction of water traveling along fractures with the gneissic rock matrix and expose the details of the alteration process. Knowledge of this process is needed for an understanding of solute acquisition of water in deep drillholes, for basic research, for geothermal energy or nuclear waste disposal projects.

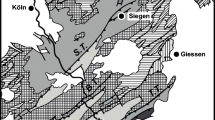
Geological maps: a Simplified tectonic map of the Simano nappe complex (modified after Spicher 1980); b Enlarged from (a): Simplified geological map of the studied area along the NS trending Val Calanca (modified after Berger et al. 2005). c Outline of Switzerland with the location of Fig. 1a indicated
Water produced alteration veins consist of two parallel bleached zones (alteration selvages) adjacent to the central fracture (open fissure, central zone), filled with low-T minerals. The selvages are separated from the unaltered gneiss by a sharp reaction front, which represents interface between plagioclase-bearing gneiss and completely albitised low-T rock. Mineral and chemical data from the veins are used in this contribution to reveal the mass transfer and the porosity evolution related to the water driven alteration of crystalline rocks of the upper continental crust during water–rock interaction.
Geological setting
The Arvigo locality is situated in the N–S striking Alpine valley Val Calanca, Grisons (Fig. 1). The rocks exposed in Arvigo belong to the Simano nappe, a Penninic basement nappe of the Central Alps and represents paleogeographically the southern passive margin of the European plate (Wenk 1955).
The Simano nappe is a metamorphic complex including several metagranitic bodies of Caledonian and Variscan age (Jenny et al. 1923; Keller 1968; Köppel and Grünenfelder 1975; Schaltegger et al. 2002), whereas the upper parts of the Simano nappe mainly consist of pre-Mesozoic gneisses and micaschists, intercalated with numerous amphibolite and calcsilicate lenses (Rütti et al. 2005; Schaltegger et al. 2002). The large Simano basement nappe is located between the underlying Leventina nappe and the Adula nappe as hanging wall (Berger et al. 2005), which is separated by thin Mesozoic metasediments (Fig. 1). Peak metamorphic conditions of Barrovian late Alpine metamorphism for the region around Arvigo yield a temperature range from 600–680°C and pressure of 550 to 600 MPa (Engi et al. 1995; Nagel et al. 2002; Todd and Engi 1997).
Simano granite gneiss is mined as building stones in a quarry south of the Arvigo (Fig. 1). The gneisses generally strikes in NNW-SSE orientation with a dip of ~30° in NE direction (146/30° NE to 158/31° E) that is parallel to the slope of the valley. Fissures and veins are generally perpendicular to the schistosity and structural analysis by Weiß and Forster (1997) yielded an orientation of 60/85°N to 70/90° for early fissures whereas later formed fissures tend to change orientation with time from 70/80°S to 20/90°. Both early and late fissures and veins contain hydrous Ca–Al silicates. Fissures are often brecciated due to late stage deformation. The Arvigo quarry is famous for a large number of Alpine fissure minerals, which occur in extension fractures and cavities of the granitic gneiss (Ruppe 1966; Simonetti 1971; Wagner 1968, 1980, 1981, 1983; Wagner et al. 2000a, b; Weiß and Forster 1997). Brittle structures, fissures, veins and gashes formed during exhumation and uplift of the Alpine orogen (Mullis et al. 1994). The Arvigo veins contain more than 40 different minerals (Armbruster 2000; Armbruster et al. 2000, Wagner et al. 2000a, b; Weiß and Forster 1997). The principal vein minerals are epidote, prehnite and zeolites (Fig. 2). The mineralisation of fissures and veins depends on the rock type including the vein (Table 1). The major fissure minerals quartz, adularia, epidote, prehnite and zeolite are present in all veins. However, different characteristic minerals that are present in small or trace amounts.

Field relationships and schematic sketches of fracture related alteration of Arvigo gneiss. Mineral abbreviation after Bucher and Frey (2002). a Fissure mineral assemblages: Qtz-Kfs-Ep-Sco. b Assemblage: Cal-Sco-Lmt. Calcite shows corrosion. Coin for scale. c Photograph of a vein hosted in biotite-rich gneisses (Arvigo/Val Calanca). Hammer for scale. The vein is characterised by a ~1 cm leaching zone trending in vertical direction, which appears to be lighter, due to the destruction of biotite. The open space of the fissure is filled with secondary phases, which consists mainly on chlorite. d Schematic sketch of an Arvigo fissure
Previous work
Previous work is limited to fissure mineral descriptions (Brughera 1984; Graeser and Stalder 1976; Ruppe 1966; Simonetti 1971; Wagner, 1968, 1980, 1981, 1983; Wagner et al. 2000a, b; Weiß and Forster 1997). The chemical evolution of fluids during various stages of Alpine orogeny has been derived from fluid inclusion studies (e.g. Mullis et al. 1994; Poty et al. 1974). CO2 rich fluids have been characterised for the initial uplift and exhumation period (Mullis et al. 1994). The presence of epidote in Arvigo veins suggests that pCO2 was low during the later stages of vein and fissure formation. The evolution in fluid composition from early CO2-rich fluids towards late aqueous fluids was confirmed by fluid inclusion studies on quartz crystals (Stalder 2007; Wagner et al. 2000a, b). Fluid inclusions in the core of quartz crystals are rich in CO2 (up to 40 vol% CO2, Stalder 2007), whereas the aqueous fluid inclusions in quartz rims are CO2 free. Mineral inclusions in the quartz core zone, coexisting with CO2-rich fluid, are hornblende, ilmenite, biotite and carbonate (Wagner 2000a). Those minerals are often found as precursor phases of Ca–Al-silicates. The quartz rim zone contains the following sequence of minerals: fibrous actinolite, epidote, chlorite and calcite (Wagner 2000a). Zeolites were not found as inclusions. Homogenization temperatures of fluid inclusions in the quartz core are up to 365°C (Stalder 2007; Wagner et al. 2000a, b). In contrast, fluid inclusions in the rim yield homogenization temperatures of 160 to 230°C. NaCl content of the fluids in quartz increases from core to rim up to 5.9 wt% NaCl Stalder 2007; Wagner et al. 2000a, b).
Sampling and analytic methods
Representative samples of different veins and fissures were collected in the quarry in the years 2006, 2007 and 2008. A subset of samples was selected for petrographic, bulk-rock and electron-microprobe analysis.
Modal analyses were determined by point counting of more than 1,600 evenly spaced points in each thin section using a standard polarized microscope. Quantitative analysis of mineral composition was performed at the Institute of Geosciences, University of Freiburg, using a CAMECA SX 100 electron microprobe equipped with five WD spectrometers and one EDS detector with an internal PAP-correction program (Pouchou and Pichior 1991). Major and minor elements for zeolites were determined at 15 kV accelerating voltage and 10 nA beam current with a defocused electron beam of 20 μm in diameter and counting times up to 20 s. Na and K were measured first, to minimize the effect of Na and K loss during determination. Since the zeolite loses water when heated, the crystals were mounted in epoxy resin to minimize loss of water. Natural and synthetic standards were used for calibration. The charge balance of zeolite formulas is a reliable measure for the quality of the analysis. It correlates with the extent of thermal decomposition of zeolites during microprobe analysis. A useful test is based on the charge balance between the non-framework cations and the amount of tetrahedral Al (Passaglia 1970). Analyses are considered acceptable if the sum E% = ([100*((Al)-(Na + K) + 2(Mg + Ca + Sr + Ba)/(Na + K) + 2(Mg + Ca + Sr + Ba))]) of the charge of the extra-framework cations (Ca2+, Sr2+, Na+, and K+) is within 10% of the framework charge.
Identification of minerals was made with a BRUKER AXS D8 Advance X-ray powder diffractometer (XRD) and the DIFFRACplus v5.0 software. Whole rock analyses were performed by standard X-ray fluorescence (XRF) techniques at the Institute of Geosciences at the University Freiburg, using a Philips PW 2404 spectrometer. Pressed powder and Li-borate fused glass discs were prepared to measure contents of trace and major elements, respectively. The raw data were processed with the standard XR-55 software of Philips. Relative standard deviations are <1 and <4% for major and trace elements, respectively. Loss on ignition was determined by heating at 1,100°C for 2 h.
A slab, 7 mm thick, of sample Arvigo 12 was impregnated with fluorescent epoxy under high pressure conditions at the EMPA (Swiss Federal Institute for Materials Testing and Research) in Dübendorf, Switzerland to reveal sample porosity. The porosity was measured from digital analysis of photomicrographs. Images of the alteration profile Arvigo 12 were digitized using the software package ImageJ 1.38x (Wayne Rasband, National Institute of Health, USA) to calculate porosity.
Assemblage stability diagrams involving zeolites have been computed with the computer program Domino/Theriak (de Capitani and Brown 1987) using the thermodynamic data by Berman (1988), Frey et al. (1991) and Maeder and Berman (1991).
Results
Petrography
Fracture related alteration selvages extend a few centimeters into gneissic host rock of from discrete central fractures (Fig. 2). Macroscopically the alteration zone is characterised by a light and porous bleached selvage. Reaction product minerals are precipitated in the open central fissure but also, rarely, in the porosity of the alteration selvage.
Unaltered rock
The unaltered gneisses (G, Fig. 3c) in Arvigo are represented by biotite-gneiss, leucocratic plagioclase gneiss and dark biotite-hornblende gneiss. The dark-coloured rocks vary from fine to medium grained biotite gneiss with mafic and felsic layers parallel to the foliation to coarse-grained augen-gneiss with feldspar crystals up to 12 mm in size. The amphibolite facies assemblage includes the minerals: plagioclase, quartz, K-feldspar, biotite, muscovite and ±hornblende and the accessory apatite, zircon, rutile, ilmenite and titanite. The modal composition varies over a wide range from biotite, biotite-hornblende, biotite-poor, and biotite-muscovite to muscovite gneiss (Table 1).

Sample Arvigo 12 that shows a complete section from unaltered host rock gneiss trough the different alteration zone to the central open fissure. a Overview over hand specimen. Alteration increases in direction of arrow. Schistosity is perpendicular to the cut section. b Sawed slices showing the position of XRF analyses presented in Table 3. c Sketch of different alteration areas: unaltered gneiss (G) weakly altered zone (L) marked by incomplete biotite chloritisation, the central altered zone (M) (selvage, bleached vein) contains turbid albite (red dashed line). Boundary (M) represents the albitisation front, the outer limit of the bleached selvage and the sharp front of the reaction vein. Zone (M) grades into the highly altered part (H), where chlorite is absent. Hydrate low-T minerals precipitated along the fissure wall in the centre of the vein (V). A thin chlorite-vein in the weakly altered part (L) is shown. d Thin section photographs along the section. Red dashed line marks the sharp interface between albitised plagioclase and plagioclase. Thin section numbers corresponds to Table 2
Altered rock
The extent of alteration differs for different stages, rock types, fissure minerals and porosity. Generally, the altered rocks are lighter coloured and have a higher porosity.
The first sign of alteration of gneiss (L, Fig. 3c) is the chloritisation of biotite (Fig. 4e). The alteration of biotite increases gradually toward the central fissure (Fig. 3c). In medium altered gneiss (M, Figs. 3c, 4f), almost all biotite is replaced by chlorite. Microscopic observation shows the development of turbidity in plagioclase (Fig. 4b, c). In the outer zone (H, Fig. 3c) all chlorite is destroyed.

Representative photomicrographs of typical textures in gneiss and vein selvages (Sample Arvigo 12, Fig. 3): a Plagioclase crystal in gneiss showing polysynthetic twins. b Albitized plagioclase grains. Albite crystals are turbid due to reaction-created porosity. Red dashed line marks the grain boundary between two albite grains. The orientation of pores is crystallographically controlled. c Alteration front (red dashed line) between the selvage and highly altered zone close to the central fissure. d Typical gneiss assemblage not affected by late hydration and alteration. e Biotite alteration to chlorite and K-feldspar in the weakly altered zone. K-feldspar is etched by HF and stained by Na3Co(NO2)6 to distinguish it from plagioclase. f Total replacement of biotite by chlorite, K-feldspar and ilmenite in weakly altered gneiss
Fissure minerals
More than 40 different fissure minerals are known from the Arvigo locality. Most of them are rare and it is not the purpose of this paper to explain their mode of formation. Epidote, prehnite, chlorite and zeolites are the dominant fissure minerals in addition to the ubiquitous quartz, adularia and calcite. Quartz and adularia represent early fissure minerals overgrown by Ca–Al silicates, chlorite and calcite. Chlorite appears in two generations. The mineral time sequence is as follows (deduced from consistent overgrowth relationships and listed from old and high-T to late and low-T): quartz, adularia, chlorite I, epidote, prehnite, chlorite II, calcite and zeolites. Scolecite and laumontite are by far the dominant zeolite species, whereas heulandite, chabazite, stilbite and epistilbite were only found sporadically.
Changes in modal mineralogy
The modal mineral composition changes along the alteration profile (Fig. 3) are given in Table 2. Plagioclase is the dominant mineral in gneiss and the modal content decreases with alteration. Quartz decreases in the highly altered zone, suggesting a quartz consuming reaction during alteration from the unaltered to the slightly altered sample (Fig. 3d, Table 2). Considering potassium bearing phases, an increase of the modal contents of K-feldspar and muscovite is accompanied by biotite decrease to the ultimate disappearance of biotite in the highly altered zone (M, Figs. 3c, 4). This decrease in biotite is associated with the increase in chlorite in the rock matrix. Calcite, epidote, prehnite and laumontite occur as accessory minerals only in TS 12.1. Petrographic data show that titanite and ilmenite are inversely correlated with biotite alteration. The Ti-minerals ilmenite and titanite resulted from the breakdown of biotite (Fig. 4f).
Mineralogy and mineral chemistry
Modal mineral inventory data and chemical composition of high-T and alteration minerals was obtained in detail from sample Arvigo 12 (Fig. 3). This sample was chosen, because it contains unaltered and altered zones on a hand-specimen scale.
Plagioclase-albite
The prime mineralogical difference between high-T and altered rock is the complete albitisation of plagioclase (Electronic supplementary material 1; Fig. 5). During albitisation plagioclase of amphibolite facies gneiss of oligoclase composition (An15–19) was replaced by albite (An0.5–2; Electronic supplementary material 1; Fig. 5). Figure 5 shows a profile of the composition of plagioclase across the alteration profile (Fig. 3). The profile shows a very sharp decrease in anorthite component at about 25 mm from the fissure wall. This sharp change in plagioclase composition defines the reaction front of the alteration process and coincides with the outer limit of the reaction vein. This reaction front also coincides with the appearance of turbidity in plagioclase (Fig. 3d, 4b, c). The development of turbidity in albite grains in thin section is related to a porosity increase (Fig. 6, Electronic supplementary material 2). The intra-granular pores have an angular to elongated shape of up to 10 μm in length (Fig. 6, Electronic supplementary material 2). The slight decrease of anorthite component at around 70 mm from the fissure wall coincides with a thin chlorite vein (Fig. 3c). A slight decrease in the Or content of plagioclase along the profile (Electronic supplementary material 1) has been observed. Additionally a depletion of Sr in albite can be observed (Electronic supplementary material 1; Fig. 5). Saussuritisation and sericitisation of plagioclase, processes that are characterised by the replacement of plagioclase by fine-grained epidote and muscovite (e.g. Sandström et al. 2010) are not observed in our samples. Although the plagioclase is totally albitised, the optical expression of original albite law twinning in plagioclase has been preserved in many of the altered grains.


Images of TS 12.1 showing products of the albitisation process (Pl + Qtz + H2O = Ab + Lmt). a BSE-image showing the porosity (black dots) in albite following albitisation of gneiss plagioclase. Laumontite is visible due to the perfect cleavage (~90°). Using image analyses methods a porosity of ~15% was determined. b K element map. c Ca element map that point out the complex grain boundary of laumontite. d Na element map. e Al element map. f Si element map
Biotite-chlorite
Chloritisation of biotite increases gradually from the gneiss, where biotite is unaltered, into the light altered zone (Fig. 3c, 4d–f). In the medium and highly altered zone biotite is completely replaced by chlorite. Chlorite also occurs as spherulitic aggregates in the central zone of the veins. The zone of chloritised biotite extends beyond the visibly bleached selvage (Fig. 3d, red-dashed line).
Chloritisation of biotite is accompanied by the formation of K-feldspar and the Ti-minerals ilmenite (Fig. 4f) or titanite. Chloritisation progresses preferentially along cleavage planes of biotite. Representative analysis of biotite and chlorite are given in the Electronic supplementary material 3. Biotite has an XMg of 0.21–0.24, XMg of chlorite ranges from 0.23 to 0.42. Ti is minor element in biotite with an average concentration of 3.12 wt% (Electronic supplementary material 3). Ti is present in chlorite in traces only thus Ti of former Bt is present in the reaction products Ilm or Ttn.
Muscovite
Modal muscovite was preserved during alteration and its composition varies in a limited range with no systematic pattern over the profile (Table 2). The celadonite component is 7–10 mol%. The paragonite content is 4–7 mol% and significantly higher than the margarite component (<1 mol%; Electronic supplementary material 3).
K-feldspar
Original K-feldspar of the gneiss is preserved during alteration. Additional K-feldspar formed by chloritisation of biotite. The composition of K-feldspar ranges from Or85–94 and Ab15–06, with An-component less than 0.5 mol% (Electronic supplementary material 4). Euhedral adularia in fissures and central zones of veins is very pure Kfs (Or93–99, Ab07-01, An00-01, Electronic supplementary material 4).
Quartz
Quartz is abundant in Arvigo gneiss. Modal quartz decreases during alteration (Table 2). However, fissure quartz is the earliest mineral that precipitated in the central zones of the veins. Fluid and solid inclusions in these early quartzes retain important information about fluid and mineral composition/evolution during quartz growth (cf. Previous work).
Epidote
Epidote occurs as dominant mineral in veins and fissures overgrowing the early alteration minerals quartz and adularia (Fig. 2a). Epidote is itself overgrown by prehnite and zeolites. Epidote forms green to dark-green sheaf-like aggregates up to 20 mm in length, but common size ranges between 5 and 10 mm. Analyzed epidote (Electronic supplementary material 5) has (Fe3+/(Fe3++Al)) in the range from 0.15 to 0.30. Minor components are MnO and SrO (up to 0.64 wt% and 0.30 wt%, respectively). No chemical zoning in epidote was observed.
Prehnite
Prehnite in fissures and vein centres forms colourless to pale-green fan-shaped radiating aggregates, so-called bow-tie structures (Phillips and Rickwood 1975), or sheaf like aggregates (Electronic supplementary material 6). The aggregates are up to 10 mm in diameter overgrowing quartz and epidote. Prehnite served as a substrate mineral for the later growth of zeolites. Prehnite composition lies close to its ideal endmember composition Ca2(Fe3+,Al)2Si3O10(F,OH)2 (Electronic supplementary material 7). A small content of fluorine due to the OH vs. F substitution is detectable. Significant Al/Fe3+ variation occurs within prehnite on the octahedral site (Electronic supplementary materials 6, 7). The Fe3+ content controls the colour of prehnite: colourless to pale-green prehnite is low in Fe, whereas pale-green prehnite is Fe-rich. Prh aggregates often show a chemical zoning (Electronic supplementary materials 6, 7), suggesting an Fe3+ vs. Al substitution during growth, with Fe decreasing towards the rim.
Zeolite minerals
In vein centres and fissures at the Arvigo quarry, scolecite and laumontite are the absolutely dominant zeolite species, heulandite, chabazite, stilbite and epistilbite are found sporadically. All but one sample from Arvigo consistently shows that laumontite is later than scolecite (Fig. 2). For Alpine fissures following zeolite chronology can be obtained: scolecite, laumontite, heulandite, chabazite and stilbite (Weisenberger and Bucher 2010).
Laumontite (Ca4(Al8Si16O48)·18 H2O) typically forms radiating aggregates, thin fibres or prisms elongated along the c-axis with a square cross-section. Laumontite forms as {110} prism and commonly be twinned on {100} to form “swallow tail” or “V” twins. It is white with a length between <1 and 15 mm. Ca is the dominant extra-framework cation (average value of 97%), with minor amounts of Na and K (Electronic supplementary materials 8, 9). Maximum values for K and Na are 0.7 and 0.1 wt%, respectively. Other elements occur only in trace amounts. The extra-framework cation K increases during growth from core to the rim. The Si/(Si + Al) ratio varies slightly between 0.67 and 0.69 (Electronic supplementary material 9). The content of alkalies increases with increasing Si and decreasing Ca and Al during growth, which can be explained by the coupled substitution Si4+ + (Na+, K+) vs. Al3+ + Ca2+.
Scolecite (Ca8(Al16Si24O80)·24 H2O) occur in veins and fissures as white fibrous crystals with vitreous or slightly silky luster, forming characteristic radiating sprays (Fig. 2) either as radiating aggregates as well as loose fibres. Scolecite ranges between 1 and 20 mm in length, but the common length is between 3 and 6 mm. 23 microprobe analyses from 3 different samples indicate no major chemical variations (Electronic supplementary material 9). Ca is the dominant extra-framework cation (Electronic supplementary materials 8, 9), which on average occupies 98% of the extra-framework cation sites. Minor amounts of Na up to 4%, but on an average 2% is observed. K and Sr are not incorporated into the framework structure of scolecite (Electronic supplementary material 8) and occur only in trace amounts (Electronic supplementary material 8). The substitution Si/(Si + Al) shows a small range between 0.61 and 0.62 (Electronic supplementary material 9).
Heulandite ((Na,K)Ca4(Al9Si27O72)·24 H2O) in veins and fissures forms crystals up to 5 mm in length. The monoclinic crystals occur in tabular habit parallel {010} and elongated in its typical coffin-shaped appearance. Crystals are transparent to translucent and colourless. Representative analyses for heulandite are given in the electronic supplementary material 8. The average composition of heulandite from Arvigo is Ca3.27Na0.19K1.45Sr0.30(Al8.84Si27.16O72)·22 H2O, which is nearly identical to the heulandite composition (Ca3.37Na0.07K0.88Sr0.55(Al8.42Si27.49O72)·22 H2O) determined by Armbruster et al. (1996) for Gibelsbach heulandites in the western Aar Massif and from other Alpine fissures (Weisenberger and Bucher 2010). Heulandite can be classified by its geochemistry as heulandite-Ca (Coombs et al. 1998), with higher amounts of Sr and K (Electronic supplementary materials 8, 9). Ca (average of 63% of all extra-framework cations) is the important extra-framework cation. Besides Ca, K is a major element in heulandite, with an average of 28% (maximum to 31%) of K-endmember. Sr content is also significant between 5 and 7% Sr-heulandite endmember. Thus distinguishes heulandite from the other zeolites found in Arvigo and in Alpine fissure, except chabazite, which also shows elevated Sr (Weisenberger and Bucher 2010). Si/(Si + Al) ranges between 0.74 and 0.76 (Electronic supplementary material 9), which is in agreement with the definition of heulandite (Coombs et al. 1998). It is distinguished from clinoptilolite, which has a Si/(Si + Al) ratio larger than 0.8.
In general, all zeolites at the Arvigo quarry, as well as in fissures hosted in granite and gneiss in the Central Alps (Weisenberger and Bucher 2010), are Ca-dominated and the Si/Al ratio increases with decreasing temperature (time along cooling path).
Whole rock geochemistry and mass changes
The alteration process reacts water in the central fissure with host rock gneiss. The losses and gains of components to form the bleached selvage from the original gneiss is an important issue for understanding the overall process. Excess components derived from the gneiss can be invested spent in fissure mineral deposition (minerals in the central zone). A method for mass-balance analyses was described by Gresens (1967) based on the assumption that one or more elements are immobile and conserved during the alteration process. The ratio of mobile elements in the fresh and altered rock is then compared to the ratio of the immobile elements, in order to calculate the mass or volume change during alteration.
Bulk rock chemistry and density measurements along the alteration profile (Fig. 3) are presented in Table 3. Figure 7a represents the isocon diagram. It has been computed from the simplified graphical method after Grant (1986) for the Gresens (1967) equation and assuming that TiO2 is conserved during the alteration process. Conservation of TiO2 is justified by the rarity or absents of titanite or other Ti-bearing minerals in the central fissure (Wagner et al. 2000a, b; Weiß and Forster 1997).

Isocon diagram and bar diagram for chemical mass loss and gain during alteration. Gneiss is based on sample Arvigo 12 VIII, whereas the selvage composition is based on analyses Arvigo 12 II (Fig. 3, Table 3). a Isocon diagram showing constant mass (CM), constant volume (CV) and Isocon line. Elements below the lines are depleted in the selvage. b Bar diagram showing oxide and element mass changes compared to their respective mass in gneiss. Mfi weight concentration of component i in selvage, Moi weight concentration of component i in gneiss, Mo mass of the gneiss, (Mfi-Moi)/Moi mass change in relation to element mass in gneiss, (Mfi-Moi)/Mo mass change in relation to gneiss mass. Diagrams were constructed by using the program GEOISO (Coelho 2006). c Mass change for major elements along the Arvigo12 profile in relation to original element mass of sample Arvigo 12 VIII
Components above the isocon have been enriched in the process relative to gneiss, elements below the isocon have been depleted during the alteration process. The slope of the obtained isocon is 1.068 (Fig. 7), equivalent to a mass loss of 6.8%. Changes of the rock volume during alteration can be calculated using the mass ratio of immobile elements and the rock densities of gneiss and selvage. Using the density and the mass ratio of immobile elements of 0.932 (inverted slope of the isocon), a volume loss of 3.9% is obtained. Isocons representing constant mass and constant volume instead of constant TiO2 are included in Fig. 7a for comparison.
Ca, Sr and Rb are the elements that show the largest depletion (Fig. 7). They decreased by 64, 63 and 25%, respectively. K, Si, Mg, Fe Mn and Al are lost in minor amounts, 3, 3, 4, 6, 7 and 13%, respectively. Based on a Ti-conserving alteration process, also Na was conserved (Fig. 7). Ba, Zr and P are gained during alteration, 13%, 10% and 9%, respectively. Al, Si, Ca Fe and K are the significant elements that are lost during alteration (Fig. 7b). The total mass loss of 6.8% and the losses of the individual components can be summarized in a mass-balance equation for the process that alters gneiss and leaves behind the bleached selvage (Eq. 1):
Mass change of major elements along the profile (Fig. 3; Table 3) reveals that Ca, Si and Al depletion is limited to the zone of albitisation (Fig. 7c). K is slightly depleted over most parts of the alteration profile. In the alteration zone on the gneiss side of the sharp reaction front, K is the main element that has been depleted. Fe is enriched in Arvigo 12 VI (Fig. 3, 7), the slice containing the small chlorite veinlet. Mg shows depletion in the zone where biotite is removed (Fig. 7c).
Porosity
Porosity of the altered rock increased as a result of volume loss during albitisation (Eqs. 2, 3) and due to the dissolution of gneiss minerals such as biotite and the dissolution of low-T reaction products. The porosity of gneiss varies between 1.0 and 1.9 vol%. The porosity in the alteration selvage varies from 3.8% at the alteration front of albitisation to 6.2% in zone M of the profile (Fig. 3c). The highly altered zone H (Fig. 3c) characterised by the removal of chlorite has a porosity of up to 14.2 ± 2.2%. On a small scale (microscope), the porosity generated by albitisation appears not to be connected (isolated pores). The isolated pores are not filled with epoxy during impregnation, thus digital analysis methods may underestimate the porosity (uncertainty 15%). The albitisation reaction predicts a volume change of ~16% (Eqs. 2, 3). This is in good agreement with measured porosity in albite using image analysis of 12.7%.
with the following mineral compositions: oligoclase = Na0.84Ca0.16Al1.16Si2.84O8; albite = Na0.99Ca0.01Al1.01Si2.99O8.
Using the volume of plagioclase (Table 2) a porosity increase of 8.5% can be assigned to the albitisation process on a whole rock scale. Electronic supplementary material 2 shows a relic grain of plagioclase that was not completely replaced by albite. The texture shows the porosity development during replacement reaction. The measured porosity in albite is 5 vol% and in the porous zone around the relic plagioclase porosity increases up to 34 vol% (Electronic supplementary material 2).
Discussion
Mineral reactions
The most significant alteration reactions are albitisation of plagioclase (Figs. 3, 4, 5, 6, Electronic supplementary material 2) and chloritisation of biotite (Figs. 3, 4). Chloritisation of biotite extends into the gneiss beyond the sharp reaction front caused by the albitisation process and so forming bleached zone of the vein (Fig. 3). This observation suggests that water preferentially migrates along connected sheet silicate clusters (Sandström et al. 2010).
The chloritisation reaction (Eq. 4) conserves Mg and Ti (Ferry 1979; Parry and Downey 1982; Tulloch 1979). Equation 4 depicts observed mineral changes and average biotite and chlorite composition. Because Fe3+/Fetotal in biotite and chlorite is unknown, all Fe has been assumed to be Fe2+.
with the mineral compositions: biotite = K1.8(Fe3.0Mg1.6)(Al0.8Ti0.4)(Si5.4Al2.6O20)(OH)4; chlorite = (Mg4.2Fe5.2Al2.6)(Si5.6Al2.4O20)(OH)16; K-feldspar = KAlSi3O8; ilmenite = FeTiO3.
Chloritisation by Eq. 4 is confirmed by petrographic observations (Fig. 4f). Using the volumes of the solids in Eq. 4 the reaction results in a volume increase of 11%. However, the textures suggest that the chloritisation process conserves volume. Therefore, dissolved components were lost from the reaction sites to achieve volume conserving pseudomorphic replacement. Suggested by the measured the mass changes along the alteration profile (Fig. 7c; Table 3), volume conservation was attained by removal of K and Fe. Reaction 5 expresses the iso-volume chloritisation of biotite, where K-feldspar component is removed to achieve volume conservation.
Volume-conserving chloritisation (Eq. 5) consumes H2O and Si and releases Fe, Al and K.
Albitisation (Eq. 2) is a common process under H2O saturated conditions over a wide P–T range from diagenesis (Lee et al. 2003; Saigal et al. 1988) to greenschist (Leichmann et al. 2003) and even amphibolite facies metamorphism (Clark et al. 2005). During the albitisation of plagioclase, dissolution of oligoclase occurs with coeval formation of albite. The depletion in Ca, Al and Si is characteristic for the albitisation zone (Fig. 7c) and it shows that it is not reprecipitated as low-T Ca–Al silicate in the selvage. The dissolving An-component migrates to the central zone of the vein and precipitates there as Ca–Al-silicates, epidote, prehnite and zeolite. Mass-change along the section of Arvigo12 (Fig. 7) shows an enrichment of An-component in the weakly altered zone ahead of the reaction front. This surprising computed mass-change is caused by the used Ti-conserved reference frame. Assuming Ti conservation could be incorrect because Ti minerals are occasionally part of the fissure assemblage. Alternatively, the distribution of Ti minerals along the section (Fig. 4) is very irregular, which, together with the small sample slices, assigns large uncertainties to bulk Ti.
Oligoclase in the Arvigo samples was replaced by albite (Electronic supplementary material 1; Fig. 5). The albitisation reaction produced porosity due to the differences in molar volume between the solid phases (Eq. 2). Microporosity in plagioclase/albite increases with alteration (Fig. 6, Electronic supplementary material 2) and the orientation of the elongated pores is crystallographically controlled. Albitised plagioclase preserved twinning after the albite-twinning law of the original oligoclase. This implies epitactic overgrowth of product albite on reactant oligoclase. Similar replacement textures have been previously described from plagioclase (Engvik et al. 2008) and from K-feldspar (Cole et al. 2004; Walker et al. 1995). This implies that the albitisation process is controlled by a dissolution-reprecipitation mechanism along a moving interface (e.g. Engvik et al. 2008; Putnis and Putnis 2007), resulting in porosity generation.
Muscovite and K-feldspar of the Arvigo gneiss do not show alteration textures. The two passive minerals have not been included in the general alteration scheme in Fig. 8.

Flow chart illustrating component transfer during alteration of Arvigo gneiss. Gneiss assemblage is Pl + Bt transformed to selvage assemblage Ab + Chl. Ms and Kfs are excess minerals. Mineral symbols in the gneiss zone represent gneiss minerals (Pl, Bt and Qtz) and their alteration products (Ab, Kfs, Chl, Ttn and Ilm) that remain in the wall rock during alteration. Black arrows represent element migration paths, which are based on alteration reactions and average mineral composition discussed in the text. Breakdown of biotite is associated with a volume increase that is balanced by the removal Kfs
Mass changes and element mobility
Significant mass changes and element mobility at the whole rock scale is limited to few elements (Fig. 7). Significant loss is marked by the major elements Ca, Al, Si, Fe, Mg and K (Fig. 7). In contrast, the relative loss of trace elements Rb and Sr and gain of Ba, Zr and P is high. However, the absolute mass changes are very small (Fig. 7b), whereas the distribution of those trace element bearing minerals (e.g. apatite, zircon) along the section (Fig 4) is very irregular, which, together with the small sample slices, assigns large uncertainties to the bulk of the trace elements. The computed mass changes represent dissolving biotite and An-component of plagioclase and can be linked to the two major alteration reactions of chloritisation of biotite and albitisation of plagioclase (Eqs. 1, 5). Element mobility is summarised in Fig. 8 representing an alteration scheme of redistribution of elements during low-T alteration between gneiss minerals, alteration minerals and water.
The loss of Ca is compatible with albitisation of plagioclase (Eq. 2), during which Ca is mobilised (Fig. 7, Table 3). The change in Sr geochemically follows Ca (Sun et al. 1974) and is also mobilised during plagioclase dissolution. Most of the Ca was lost from the wall rock gneiss (Fig. 7; Table 3). Consequently it was transferred to the fissure or vein centre where it precipitated as Ca–Al-silicates (epidote, prehnite and zeolites) and calcite, depending on water composition and temperature. Approximately 1 wt% CaO remained in the altered rock as An-component of newly formed albite (Electronic supplementary material 1) and some rare titanite, which formed during chloritisation but also as Ca–Al minerals in reaction created porosity (Fig. 6). Strontium is strongly enriched in heulandite (Electronic supplementary material 8).
Loss of Al (Fig. 7) is notably high considering that Al is generally relatively immobile compared to other elements during water–rock interaction (Carmichael 1969; Ragnarsdottir and Walther 1985; Verdes et al. 1992). Al has been used as a reference frame for mass balance calculations in many studies of allochemical processes (e.g. Grant 1986; Thompson 1975). Nevertheless, field evidence (Fig. 2), mass changes (Fig. 7; Table 3) and deduced mineral reactions (Eq. 2) suggest loss of Al during water–rock alteration and consequently Al transport via the aqueous liquid. In the albitisation process, by which porosity is generated, Al migrated to the fissure at the vein centre and precipitated there as Ca–Al–silicate. Volume conserved chloritisation is an additional mechanism for Al loss, where Al was removed (Eq. 5) to achieve fully iso-volumetric replacement, without textural evidence for volume expansion.
The decrease of Si during alteration is linked to the leaching of silica during chloritisation and albitisation (Eqs. 1, 5). According to model reaction Eq. 5, chloritisation released Fe in accordance with the mass balance calculations, which indicate a loss in Fe (Fig. 7). Iron (Eq. 5) migrates to the central fissure, where it is oxidized and precipitates in epidote and prehnite (Electronic supplementary material 5, 6). The decrease in K is consistent with the chloritisation process (Eq. 5) during which K leached from the gneissic wall rock. The increase of volatiles (LOI, Table 3) is a consequence of hydration and formation of hydrous Ca–Al-silicates during albitisation. No changes are observed in Na concentration (Fig. 7a) during alteration. This agrees well with volume calculations during albitisation and measured microporosity in altered plagioclase.
Components released during albitisation and chloritisation were transported to the central fissure and deposited as fissure minerals in the open water-filled fracture porosity. It is a remarkable observation that the An-component dissolved at the reaction front does not precipitate as zeolite in the created porosity but rather leaves the reaction site and migrates a few cm and finally precipitates as Ca–Al mineral e.g. zeolite in the central fissure. We do not have an immediate and plausible explanation for this observation. However, we suggest that the observed unexpected system behaviour is related to cross-effects associated with fluxes caused by dissimilar driving forces such as the coupling between heat and matter fluxes (by diffusion or advection).
In general, solute transport in porous material is accomplished by three principal mechanism: advection, aqueous diffusion and hydrodynamic dispersion (Steefel 2008). The solute transport from the reaction front in the selvage to the fissure could have been driven by a hydraulic gradient caused by lithostatic pressure at the reaction front and hydrostatic pressure in the fissure (Yardley and Lloyd 1995). However a temperature gradient between advecting water in the central fissure and water at the reaction front could also the driving force for element migration (thermodiffusion, Soret effect). Solubility differences associated with the potential P–T-μ gradients could explain precipitation in the central fissure and not in the porosity of the selvage.
Mineral stability and mineral equilibria
Prehnite and epidote
Prehnite and epidote are common minerals in low-grade metamorphic rocks (e.g. Bevins et al. 1991; Cho et al. 1986; Freiberger et al. 2001; Kuniyoshi and Liou 1976; Liou et al. 1983; Liou 1985; Rose and Bird 1987; Tulloch 1979). Temperature and/or fO2 conditions, during formation of prehnite and epidote, are reflected in their chemical composition. Pumpellyite, which is often associated with prehnite and epidote, was not found in Arvigo rocks. The observed heterogeneity of the Al2O3/Fe2O3 ratio in prehnite may be due to (1) partial re-equilibration during changing P–T-fO2 conditions, where iron in prehnite increases with decreasing temperature and increasing fO2 (Kuniyoshi and Liou 1976; Liou et al. 1983), (2) succeeding discrete events of water–rock interaction (Freiberger et al. 2001), or (3) local chemical influence of host minerals. For the Arvigo samples, the Fe3+ enrichment in the core of prehnite (Electronic supplementary materials 6, 7) suggests that fO2 decreased during the vein forming.
Prehnite in metabasites can be stable up to 400°C and up to 300 MPa (Frey et al. 1991; Liou 1985). However the absence of pumpellyite, which is stable between 100 and 800 MPa, suggests that pressure conditions during the formation of Ca–Al-silicates were below 100 MPa (Frey et al. 1991; Kuniyoshi and Liou 1976; Liou 1985).
Sample Arvigo 1 (Electronic supplementary material 5) contains coexisting prehnite-epidote. The Prh-Ep pair could be co-genetic and formation temperatures from 330 to 380°C can be derived from the thermometer of Rose and Bird (1987) that uses the iron partitioning between coexisting prehnite and epidote (Fig. 9). The compositional limits on the stability of coexisting prehnite and epidote are represented by the two dashed lines (Fig. 9). The data from Arvigo (Fig. 9) suggest that Ep-Prh pairs may not have coexisted at equilibrium because they are metastable with respect to the Zo-Grs pair. Also the estimated temperature of 350°C appears to be high for prehnite in Arvigo veins, although the temperature is within the range of reported prehnite stability in active geothermal systems (275–350°C; Bird et al. 1984) and in hydrothermal experiments (376°C, Liou et al. 1983).

Temperature of formation of coexisting prehnite and epidote, using the Fe3+-Al partitioning (Rose and Bird 1987). Distribution of Fe3+ between coexisting epidote and prehnite is expressed as mole fraction of Ca2FeAlSi3O10(OH)2 in prehnite and as the pistacite component Ca2Fe3Si3O12(OH) in epidote. Dashed lines represents limits on the compositional range for coexisting prehnite and epidote (I: prehnite ⇒ zoisite + grossular + quartz; II: laumontite + prehnite ⇒ clinozoisite + quartz). Solid lines are isotherms, based on constant log K by using thermodynamic properties of the reaction: Al-prehnite + epidote ⇒ Fe-prehnite + clinozoisite (adapted from Rose and Bird 1987)
Chlorite
Several chlorite thermometers based on structural and chemical parameters are available from the literature (e.g. De Caritat et al. 1993). The empirical calibration based on AlIV content (Cathelineau and Nieva 1985; Cathelineau 1988) was tested for low-grade basic rocks within a regional metamorphic context (Bevins et al. 1991). However, De Caritat et al. (1993) have shown that the content of AlIV is not dependent on the geochemical composition of the host rock and therefore the Cathelineau (1988) thermometer is frequently used for chlorite hosted in granite and gneiss (e.g. Orvosová et al. 1998; Rahn et al. 1994). Although the reliability of the thermometer is difficult to assess, Vidal et al. (2001) showed that the variation of AlIV in chlorite with temperature is thermodynamically sound.
Chemical composition of chlorite in Arvigo was measured in 40 grains. The calculated formation temperature varies from 275°C to 380°C, with an average value of 333°C (Electronic supplementary material 3). Two distinct groups of chlorite are present resulting in two different groups of computed T. The first group shows a formation temperature of around 310°C and the higher temperature group varies from 330 to 380°C. Chlorite, which occurs as pseudomorphs after biotite, trends to record higher formation temperatures, in contrast to spherulitic chlorite precipitated in the central fissure and in open porosity, which records lower temperatures.
Chlorite temperatures are similar to those derived from epidote and prehnite and suggest that the early phase of water–rock interaction occurred at about 300 to 350°C.
Zeolites
Zeolites along with apophyllite are the youngest minerals formed in Alpine fissures in Arvigo. The general chronology of the Arvigo zeolites is (from early high-T to late low-T): scolecite, laumontite, heulandite, and stilbite. This succession is comparable with zeolite sequences found in open fissures elsewhere in the Alps and consistent with thermodynamic phase modelling in the system CaAl2Si2O8–SiO2–H2O (Weisenberger and Bucher 2010).
Reactions involving Ca–zeolites have been studied experimentally (e.g. Cho et al. 1987; Frey et al. 1991; Liou 1971; Thompson 1970). In general, P–T limits of zeolite stability are in agreement with observations in geothermal systems (Frey et al. 1991; Kristmannsdóttir and Tómasson 1978). However, observed T at some zeolite isograd reactions do not agree well with computed phase equilibria (Frey et al. 1991; Kristmannsdóttir and Tómasson 1978; Neuhoff et al. 2000). This shows that experimental data may not adequately reflect natural zeolite-facies rocks. The discrepancies may be related to the presence of additional extra-framework cations like Sr, Na, K and Mg in natural aqueous solutions and other water composition parameters that are not modelled by experimental studies. The neglected parameters may result in different amounts of H2O incorporated in the zeolite channel structure. Also the order–disorder state of experimental and natural zeolites may be considerably different (e.g. Cho et al. 1987; Frey et al. 1991; Liou 1971; Neuhoff et al. 1999; Thompson 1970). Lastly, water pressure may be hydrostatic in natural fractured environments rather than lithostatic as in experimental studies with the consequence that T estimates should be derived from computed low-P models.
Mineral and porosity evolution during alteration and vein formation were modelled by using computed assemblages stability diagrams with the Theriak/Domino software (de Capitani and Brown 1987). The formation of zeolites at Arvigo occur at T < 300°C in the subgreenschist facies given by P–T estimates for the Chl–Prh–Ep assemblage (see above). This limit corresponds to 10 km depth assuming a geotherm of 30°C/km during exhumation and a related lithostatic pressure of 270 MPa. Since the assemblage has been precipitated in the open fissure and vein centre, pressure may have been hydrostatic and as low as 100 MPa. During the zeolite forming stage pressure in the central fissure may have been much lower than 100 MPa. The occurrences of scolecite and laumontite indicate maximum formation temperatures of 280–300°C using equilibrium phase modelling in the system CaO-Al2O3-SiO2-H2O-CO2 (Fig. 10).

Equilibrium T-X CO2 diagrams at P = 10 MPa, 50 MPa and 100 MPa, for the CaO-Al2O3-SiO2-H2O-CO2 system and different Ca/Al ratios
Important for the formation of zeolite is the composition of water from which they precipitated. Rare occurrences of zeolite that formed during uplift and cooling in the amphibolite-facies Lepontine Alps can be related to absence of CO2 dominated fluids that are typical in this part of the Alps (Mullis et al. 1994; Poty et al. 1974; Stalder 2007). Zen (1961) noticed that zeolite mineral assemblages could be experimentally obtained by increasing the chemical potential of H2O relative to that of CO2, at constant temperature and pressure. In relatively low CO2 water, calcium zeolites are stabilised relative to assemblages that contain calcite, quartz and clay minerals (Senderov 1973; Zen 1961), an observation supported by the presence of CO2 free fluid inclusions at zeolite occurrences. However, the lack of zeolite inclusions in quartz, suggests that quartz growth was finished before zeolite formation started and no information about water composition at the time of zeolite formation is available. This is in agreement with the models (Fig. 10) showing the effect of CO2 on the stability of zeolites and other Ca–Al-silicates for different Ca/Al bulk ratios and pressure conditions in the system CaO-Al2O3-SiO2-H2O-CO2. Zeolites are stable in water with low CO2 concentrations. With increasing CO2, zeolites are replaced at low-T by kaolinite (e.g. Val Bedretto, Stalder et al. 1998) and other Ca–Al silicates, calcite and quartz at higher temperature (Fig. 10). Stilbite is stable only at a very low CO2 pressure in the water (<0.4 MPa at 10 MPa) and low temperature, whereas heulandite, laumontite and scolecite are stable at higher CO2 pressure (Fig. 10). The appearance of scolecite is controlled by pressure and it occurs only at lower pressures. The Ca/Al ratio in water can also affect the stability of zeolites. However the occurrences of heulandite may also be controlled by additional extra-framework cations like Sr, Na and K, which are preferentially incorporated in heulandite in Alpine fissures (e.g. Weisenberger and Bucher 2010). Generally, zeolite stability increases with decreasing pressures. At a pressure of 100 MPa, zeolites are stable at XCO2 below 0.05 (Fig. 10). However, in hydrothermal systems the fluid pressure is unlikely equal to the total pressure. Very low CO2 at Arvigo is supported by the absence of kaolinite, which would be present at lower temperatures with XCO2 > 0.04.
Mineral evolution
Major element bulk rock composition of the unaltered gneiss of sample Arvigo 12 VIII (Table 3) has been recalculated to atomic proportions. To simplify the calculations, Ti and Mn were ignored. The presence of chlorite, epidote and prehnite indicate that Fe occurs in di- and trivalent state and that some provision for the redox state must be made. The models use Fe3+/Fe2+ = 2/3 for the relatively oxidised selvages. Since alteration and vein formation is a process of hydration and consequently excess H2O was assumed.
The modelled assemblage distribution (Fig. 11) and the corresponding predicted assemblage evolution (Fig. 12) suggests that epidote starts to form at T ~ 420°C, which is higher than the T estimate from the iron distribution between Ep and Prh. At Arvigo initial (early) water was relatively rich in CO2 and consequently epidote may not appear until a much lower T has been reached.


Predicted assemblage evolution during the alteration process, calculated along an exhumation path (Fig. 11)
According to the model (Figs. 11, 12), chloritisation occurred at T of 350°C. This is in good agreement with the empirical chlorite thermometers of Cathelineau (1988) that yields an average chlorite formation temperature of 333°C.
Water–rock reactions depend on the composition of the reactive water entering and leaving the system, and can not be thermodynamically treated by merely considering the solid phases in the same way like isochemical reactions are used to determine P–T conditions. The model of mineral evolution (Fig. 12) predicts the appearance of prehnite and the zeolite-in reaction correctly and reflects a plausible mineral evolution. The precise prediction of the zeolite mineral succession is difficult because their formation is also related to minor cations such as Sr, which controls the formation of heulandite.
The porosity evolution can be calculated by removing the molar volume portion of the low-T alteration minerals from the initial volume of gneiss (Fig. 13) using the predicted assemblage evolution along the cooling path (Figs. 11, 12) and assuming that the components for the formed Ca–Al-silicates in the fissure were locally derived from the adjacent wall rock. Fig. 13 therefore shows the temporal porosity evolution along the P–T path (Fig. 11).

Porosity evolution during gneiss alteration and low-T hydration. Minerals precipitated during alteration have been precipitated in the central open fissure
One path is modelled by assuming that all chlorite remains in the wall rock, whereas the second path requires biotite dissolution and chlorite precipitation in the central fissure. The computed total porosities of the bleached selvage are 10.6 and 17.6 vol%. That is in agreement with the porosity estimated from digital image analysis of ~14%.
Fluid accessibility and composition
The alteration process that formed hydrous Ca–Al-silicates required a significant amount of H2O. Water from the central open fissure (extension fracture) entered the gneissic wall rocks and followed the progressing reaction front through the process-created porosity (e.g. Austrheim 1987; Bons 2001; Bucher-Nurminen 1982).
Fracturing is caused by brittle deformation that is much younger than the main Alpine ductile deformation and folding. It is related to the uplift of the Central Alps 10–20 Ma ago (Purdy and Stalder 1973; Steck 1968). Two distinct fracture directions can be observed in Arvigo, which suggest a change in the stress field with time. Recent dating of the latest fissure minerals in the Central Alps (Weisenberger and Bucher 2008) suggests a young age (~2 Ma) of zeolite formation in the Central Alps.
Biotite and chlorite occur as connected clusters across the foliation and provide migration pathways for the water throughout the altered zone. The sheet silicate packages modally increase toward the central fissure. In contrast, albitisation produces porosity that is constant across the alteration selvage (Fig. 3). Porosity produced by albitisation is generated at the reaction interface (reaction front) and dramatically changes the permeability of the rock. The porosity due to albitisation is concentrated at the grain boundaries (Electronic supplementary material 2) and it represents a prominent fluid migration path. Intragranular porosity may only have limited effects on the permeability due the poorly connected pores. Both alteration reactions, chloritisation of biotite and albitisation, enhance the permeability. This has the consequence that both main processes are self-accelerating (positive feed back loop).
If a part of the created porosity of the selvage has been filled by late alteration minerals the mass change calculation results in an apparent lower porosity increase. Precipitation of low-T minerals in an area adjacent to wall rock gneiss can clog earlier formed porosity. This decrease in permeability stops water migration and the ultimately the entire alteration process. These is illustrated by the chlorite vein in Fig. 3, where the gneiss surround the little vein is depleted in Ca (Fig. 5), but depleted to a clearly smaller extent compared with the bleached selvage. Ca remaining in albite can be related to clogging of water transport channels by secondary chlorite.
The source of the water in the fracture porosity that caused the alteration process and the vein formation is certainly meteoric surface related water (Bucher and Stober 2010; Seelig and Bucher 2010). This also means the components for mineral precipitates in the central vein are wall rock (gneiss) derived.
Summary and conclusions
Subgreenschist facies mineral assemblages in granite and gneiss formed by hydration of high-T assemblages are the key to understanding water–rock interaction in hydrothermal and geothermal systems. The Arvigo locality is an example of a crystalline basement unit consisting of granites and gneisses that has been significantly affected by late hydration and alteration, thereby forming a succession of zeolite minerals in fissures. Fissures and gashes formed by brittle deformation were generated at very shallow depth during exhumation and uplift of the Alpine orogen. The fractures and cavities were filled with water and low-T minerals crystallized in the open space.
-
I.
The fissures contain the early assemblage epidote and prehnite, then chlorite and finally various species of zeolites. In general succession is (from old high-T to young low-T): quartz ⇒ adularia ⇒ chlorite I ⇒ epidote ⇒ prehnite ⇒ chlorite II ⇒ zeolites. Calcium is the dominant extra-framework cation in all zeolites, whereas the specific zeolite formed in the fissures depends on temperature and/or water composition.
-
II.
The composition of coexisting prehnite and epidote records temperature conditions between 330 and 380°C for the pre-zeolite assemblage using the Rose and Bird (1987) calibration. The iron zoning pattern in prehnite suggest a redox change during growth of prehnite. AlIV on the tetrahedral site in chlorite (Cathelineau 1988) suggest T of 333 ± 32°C. Zeolite formation took place at temperatures below 250°C.
-
III.
Water induced mineral reactions occurred during the alteration of gneiss. Albitisation of plagioclase accompanied by chloritisation of biotite have been the key processes. The sharp reaction front propagating from the central fracture into the gneiss matrix is related to albitisation reaction. The first replacement reaction transforms biotite to chlorite within a 3 to 7 cm thick zone in the host rock ahead of the sharp reaction front related to albitisation. The plagioclase replacement reaction releases components for zeolite formation and forms a sharp reaction front in the gneiss at about 2 to 2.5 cm from the central fracture.
-
IV.
Albitisation is associated with a volume decrease. Albite remains in the wall rock, whereas the anorthite component dissolved in water, producing a porosity increase of ~ 16% in albite. We conclude that much of the material once present in the produced volume is transferred to the central extension fracture and deposited their as hydrous low-T Ca–Al. The porous product albite suggests that the propagation of the reaction front through the gneiss matrix occurred via a dissolution-precipitation mechanism. Chloritisation of biotite in gneiss was accompanied by the release of K, Fe, and Al in a volume conserving process.
-
V.
The transport of dissolved components towards the central open fissure must have been driven by chemical potential gradients (hydrodynamic dispersion). Water moved to the reaction front driven by the metastability of the oligoclase + H2O pair at the low-T conditions. Precipitation of Ca–Al hydrate silicates in the central fissure (vein centre) was caused by either a pressure drop from lithostatic to hydrostatic or by a dilution of high-pH Al-rich water by surface water.
-
VI.
The remarkable and astonishing lack of zeolites in late fissures in the Lepontine Alps, compared to the exceptional Arvigo locality can be related to pCO2. In most areas of the Lepontine Alps pCO2 in the water is inherited from the main phase of metamorphism and is thus above a critical threshold value that makes zeolite formation possible. Computed assemblage stabilities suggest that water decreased in CO2 with time, which is observed in fluid inclusions in quartz, that formed prior to the zeolite formation.
-
VII.
Mass balance suggest a mass loss of 6.8 wt% and depletion of Al, Si, Ca, Fe and K in the selvage relative to the gneiss. These elements are subsequently found as major components in epidote, prehnite, calcite, adularia, chlorite and zeolites as fracture filling minerals. The mass transfer is associated with an increase in porosity, caused by the volume decrease during albitisation and the removal of chlorite in the wall rock.
References
Armbruster T (2000) Cation distribution in Mg, Mn-bearing babingtonite from Arvigo, Val Calanca, Grisons, Switzerland. Schweiz Mineral Petrogr Mitt 80:279–284
Armbruster T, Kohler T, Meisel T, Nägler TF, Götzinger MA, Stalder HA (1996) The zeolite, fluorite, quartz assemblage of the fissure at Gibelsbach, Fiesch (Valais, Switzerland): crystal chemistry, REE patterns, and genetic speculations. Schweiz Mineral Petrogr Mitt 76:131–146
Armbruster T, Stalder HA, Gnos E, Hofmann BA, Herwegh M (2000) Epitaxy of hedenbergite whiskers on babingtonite in Alpine fissures at Arvigo, Val Calanca, Grisons, Switzerland. Schweiz Mineral Petrogr Mitt 80:285–290
Austrheim H (1987) Eclogitization of the lower crustal granulites by fluid migration through shear zones. Earth Planet Sci Lett 81:221–232
Berger A, Mercolli I, Engi M (2005) Tectonic and petrographic map of the Central Lepontine Alps, 1:100’000. Schweiz Mineral Petrogr Mitt 85:109–146
Berman RG (1988) Internally-consistent thermodynamic data for minerals in the system Na2O–K2O-CaO-MgO-FeO-Fe2O3-Al2O3-SiO2-TiO2–H2O-CO2. J Petrol 29:445–522
Bevins RE, Rowbotham G, Robinson D (1991) Zeolite to prehnite-pumpellyite facies metamorphism of the late Proterozoic Zig-Zag Dal Basalt Formation, eastern North Greenland. Lithos 27:155–165
Bird DK, Schiffman P, Elders WA, Williams AE, McDowell SD (1984) Calcsilicate mineralization in active geothermal systems. Econ Geol 79:671–695
Bons PD (2001) The formation of large quartz veins by rapid ascent of fluid in mobile hydrofractures. Tectonophysics 336:1–17
Brughera F (1984) Aquamarin aus dem Steinbruch von Arvigo (Calancatal). Schweizer Strahler 6:498–501
Bucher K, Frey M (2002) Petrogenesis of metamorphic rocks. Springer, Berlin
Bucher K, Stober I (2010) Fluids in the upper continental crust. Geofluids 10:241–253
Bucher K, Zhu Y, Stober I (2009) Groundwater in fractured crystalline rock, the Clara mine, Black Forest, Germany. Int J Earth Sci 98:1727–1739
Bucher-Nurminen K (1982) Mechanism of mineral reactions inferred from textures of impure dolomitic marbles from East Greenland. J Petrol 23:325–343
Carmichael DM (1969) On the mechanism of prograde metamorphic reactions in quartz bearing pelitic rocks. Contrib Mineral Petrol 20:244–267
Cathelineau M (1988) Cation site occupancy in chlorites and illites as a function of temperature. Clay Minerals 23:471–485
Cathelineau M, Nieva D (1985) A chlorite solid solution geothermometer. The Los Azufres (Mexico) geothermal system. Contrib Mineral Petrol 91:235–244
Cho M, Liou JG, Maruyama S (1986) Transition from the zeolite to prehnite-pumpellyite facies in the Karmutsen Metabasites, Vancouver Island, British Columbia. J Petrol 27:467–494
Cho M, Maruyama S, Liou JG (1987) An experimental investigation of heulandite-laumontite equilibrium at 1000 to 2000 bar Pfluid. Contrib Mineral Petrol 97:43–50
Clark C, Schmist Mumm A, Faure K (2005) Timing and nature of fluid flow and alteration during Mesoproterozoic shear zone formation, Olary Domain, South Australia. J Metamorph Geol 23:147–164
Coelho J (2006) GEOISO—a WindowsTM program to calculate and plot mass balances and volume changes occurring in a wide variety of geologic processes. Comput Geosci 32:1523–1528
Cole DR, Larson PB, Riciputi LR, Mora CI (2004) Oxygen isotope zoning profiles in hydrothermally altered feldspars; estimating the duration of water-rock interaction. Geology 32:29–32
Coombs DS, Alberti A, Artioli A, Armbruster T, Colella C, Galli E, Grice JD, Liebau F, Mandarino JA, Minato H, Nickel EH, Passaglia E, Peacor DR, Quartieri S, Rinaldi R, Ross M, Sheppard RA, Tillmanns E, Vezzalini G (1998) Recommended nomenclature for zeolite minerals: report of the subcommittee on zeolites of the international mineralogical association, commission on new minerals and mineral names. Mineral Mag 62:533–571
de Capitani C, Brown TH (1987) The computation of chemical equilibrium in complex systems containing non-ideal solutions. Geochim Cosmochim Acta 51:2639–2652
De Caritat P, Hutcheon I, Walshe JL (1993) Chlorite geothermometry: a review. Clays and Clay Miner 41:219–239
Diegel S, Ghent ED (1994) Fluid-mineral equilibria in prehnite-pumpellyite to greenschist facies metabasites near Flin Flon, Manitoba, Canada: implications for petrogenetic grids. J Metamorph Geol 12:467–477
Engi M, Todd CS, Schmatz D (1995) Tertiary metamorphic conditions in the eastern Lepontine Alps. Schweiz Mineral Petrogr Mitt 75:347–369
Engvik A, Putnis A, Fitz Gerald JD, Austrheim H (2008) Albitisation of granitoid: the mechanism of plagioclase replacement by albite. Can Mineral 46:1401–1415
Faryad SW, Dianiska I (2003) Ti-bearing andradite-prehnite-epidote assemblage from the Malá Fatra granodiorite and tonalite (Western Carpathians). Schweiz Mineral Petrogr Mitt 82:47–56
Ferry JM (1979) Reactions mechanism, physical conditions, and mass transfer during hydrothermal alteration of mica and feldspar in granitic rocks from South-central Maine, USA. Contrib Mineral Petrol 68:125–139
Freiberger R, Hecht L, Cuney M, Morteani G (2001) Secondary Ca-Al silicates in plutonic rocks: implications for their cooling history. Contrib Mineral Petrol 141:415–429
Frey M, de Capitani C, Liou JG (1991) A new petrogenetic grid for low-grade metabasites. J Metamorph Geol 9:497–509
Gianelli G, Mekuria N, Battaglia S, Cheriscla A, Garofalo P, Ruggieri G, Manganelli M, Gebregziabher Z (1998) Water-rock interaction and hydrothermal mineral equilibria in the Tendaho geothermal system. J Volc Geothermal Res 86:253–276
Gottardi G (1989) The genesis of zeolites. Eur J Mineral 1:479–487
Graeser S, Stalder HA (1976) Mineral-Neufunde aus der Schweiz und angrenzenden Gebieten. Schweizer Strahler 4:158–171
Grant JA (1986) The isocon diagram—a simple solution the Gresens’ equation for metasomatic alteration. Econ Geol 81:1976–1982
Gresens RL (1967) Composition-volume relationships of metasomatism. Chem Geol 2:47–65
Hay RL (1966) Zeolites and zeolitic reactions in sedimentary rocks. Geol Soc Amer Special, Paper No 85, pp 1–130
Hay RL (1977) Geology of zeolites in sedimentary rocks. In: Mumpton FA (ed) Mineralogy and geology of natural zeolites, Mineralogical Society of America, Short Course Notes, Washington, DC, pp 53–64
Hay RL, Sheppard RA (1977) Zeolites in open hydrologic systems. In: Mumpton FA (ed) Mineralogy and geology of natural zeolites. Mineralogical Society of America, Short Course Notes, Washington, DC, pp 93–102
Hay RL, Sheppard RA (2001) Occurrences of zeolites in sedimentary rocks. In: Bish DL, Ming DW (eds) Natural zeolites: occurrence, properties; applications, Reviews in Mineralogy & Geochemistry, vol 45. Mineralogical Society of America, Washington, DC, pp 217–234
Jenny H, Frischknecht G, Knopp J (1923) Geologie der Adula. Beitr Geol Karte Schweiz. Schweizerische Geologische Kommision, Bern
Keller F (1968) Mineralparagenesen und Geologie der Campo Tencia-Pizzo Forno-Gebirgsgruppe. Beitr Geol Karte Schweiz. Schweizerische Geologische Kommision, Bern
Köppel V, Grünenfelder M (1975) Concordant U-Pb ages of monazite and xenotime from the Central Alps and the timing of the high temperature Alpine metamorphism, a preliminary report. Schweiz Mineral Petrogr Mitt 55:129–132
Kristmannsdóttir H, Tómasson J (1978) Zeolites zones in geothermal areas in Iceland. In: Sand LB, Mumpton FA (eds) Natural zeolites: occurrence Properties use. Pergamon Press, New York, pp 277–284
Kuniyoshi S, Liou JG (1976) Contact metamorphism of the Karmutsen Volcanics, Vancouver Islands, British Columbia. J Petrol 17:73–99
Lee MR, Thompson P, Poeml P, Parsons L (2003) Peristeritic plagioclase in North Sea hydrocarbon reservoir rocks: Implications for diagenesis, provenance and stratigraphic correlation. Am Mineral 88:866–875
Leichmann J, Broska I, Zachovalova K (2003) Low-grade metamorphic alteration of feldspar minerals: a CL study. Terra Nova 15:104–108
Liou JG (1971) P-T stabilities of laumontite, wairakite, lawsonite, and related minerals in the system CaAl2Si2O8-SiO2–H2O. J Petrol 12:379–411
Liou JG (1979) Zeolite facies metamorphism of basaltic rocks from the East Taiwan Ophiolite. Am Mineral 64:1–14
Liou JG (1985) Phase equilibria and mineral parageneses of metabasites in low-grade metamorphism. Mineral Mag 49:321–333
Liou JG, Kim HS, Maruyama S (1983) Prehnite-epidote equilibria and their petrologic applications. J Petrol 24:321–342
Maeder UK, Berman RG (1991) An equation of state for carbon dioxide to high pressure and temperature. Am Mineral 76:1547–1559
Mercolli I, Schenker F, Stalder HA (1984) Geochemie der Veränderungen von Granit durch hydrothermale Lösungen. Schweiz Mineral Petrogr Mitt 64:67–82
Mullis J, Dubessy J, Poty B, O’Neil J (1994) Fluid regimes during late stages of a continental collision: physical, chemical and stabel isotope measurements of fluid inclusions in fissure quartz from a geotraverse through the Central Alps, Switzerland. Geochim Cosmochim Acta 58:2239–2267
Nagel T, de Capitani C, Frey M (2002) Isograds and P-T evolution in the eastern Lepontine Alps. J Metamorph Geol 20:309–324
Neuhoff PS, Fridriksson T, Arnórsson S (1999) Porosity evolution and mineral paragenesis during low-grade metamorphism of basaltic lavas at Teigarhorn, Eastern Iceland. Am J Sci 299:467–501
Neuhoff PS, Fridriksson T, Bird DK (2000) Zeolite parageneses in the North Atlantic Igneous Provinces: implications for geotectonics and groundwater quality of basaltic crust. Int Geol Rev 42:15–44
Nordstrom DK, Ball JW, Donahoe RJ, Whittemore d (1989) Groundwater chemistry and water-rock interactions at Stripa. Geochim Cosmochim Acta 53:1727–1740
Orvosová M, Majzlan J, Chovan M (1998) Hydrothermal alteration of granitoid rocks and gneisses in the Sb-Au Dúbrava deposit, Western Carpathians. Geol Carp 49:377–387
Parneix JC, Petit JC (1991) Hydrothermal alteration of an old geothermal system in the Auriat Granite (Massif Central, France); petrological study and modelling. Chem Geol 89:329–351
Parry WT, Downey LM (1982) Geochemistry of hydrothermal chlorite replacing igneous biotite. Clays Clay Miner 30:81–90
Passaglia E (1970) The crystal chemistry of chabazite. Am Mineral 55:1278–1301
Phillips ER, Rickwood PC (1975) The biotite-prehnite association. Lithos 8:275–281
Poty BP, Stalder HA, Weisbrod AM (1974) Fluid inclusions studies in quartz from fissures of Western and Central Alps. Schweiz Mineral Petrogr Mitt 54:717–752
Pouchou G, Pichior F (1991) Quantitative analysis of homogeneous or stratified microvolumes applying the model of “PAP”. In: Heinrich KFJ, Newbiry DE (eds) Electron probe quantitation. Plenum Press, New York, pp 31–75
Purdy JW, Stalder HA (1973) K-Ar ages of fissure minerals from the Swiss Alps. Schweiz Mineral Petrogr Mitt 53:79–98
Putnis A, Putnis CV (2007) The mechanism of reequilibration of solids in the presence of a fluid phase. J Solid State Chem 180:1783–1786
Ragnarsdottir KV, Walther JV (1985) Experimental determination of corundum solubilities in pure water between 400–700°C and 1–3 kbars. Geochim Cosmochim Acta 49:2109–2115
Rahn M, Mullis J, Erdelbrock K, Frey M (1994) Very low-grade metamorphism of the Taveyanne greywacke, Glarus Alps Switzerland. J Metamorph Geol 12:625–641
Rose NM, Bird DK (1987) Prehnite-epidote phase relations in the Nordre Aputiteq and Kruuse Fjord Layered Gabbros, East Greenland. J Petrol 28:1193–1218
Rose NM, Bird DK, Liou JG (1992) Experimental investigation of mass transfer—albite, Ca-Al-silicates, and aqueous solutions. Am J Sci 292:21–57
Ruppe H (1966) Val Calanca—Graubünden. Aufschluss 17:105–109
Rütti R, Maxelon M, Mancktelow NS (2005) Structure and kinematics of the northern Simano Nappe, Central Alps, Switzerland. Eclogae Geol Helv 98:63–81
Saigal GC, Morad S, Bjørlykke K, Egeberg PK, Aagaard P (1988) Diagenetic albitization of detrital K-feldspar in Jurassic, Lower, Cretaceous, and tertiary clastic reservoir rocks from offshore Norway; I textures and origin. J Sed Petr 58:1003–1013
Sandström B, Annersten H, Tullborg E-L (2010) Fracture related hydrothermal alteration of metagranitic rock and associated changes in mineralogy, geochemistry and degree of oxidation: a case study at Forsmark, central Sweden. Int J Earth Sci 99:1–25
Schaltegger U, Gebauer D, von Quadt A (2002) The mafic and ultramafic rock association of Loderio-Biasca (lower Pennine nappes, Ticino, Switzerland); Cambrian oceanic magmatism and its bearing on early Paleozoic paleogeography. Chem Geol 186:265–279
Seelig U, Bucher K (2010) Halogens in water from the crystalline basement of the Gotthard rail base tunnel (central Alps). Geochim Cosmochim Acta 74:2581–2595
Senderov EE (1973) Effect of CO2 on the stability of laumontite. Geochem Int 10:114–139
Simonetti A (1971) Le zeoliti a le loro paragenesi nelle fessure delle rocce del canton Ticino, della Val Calanca e della Val Mesolcina. Boll Soc Ticinese Sci Nat 62
Spicher A (1980) Tektonische Karte der Schweiz 1:500’000, 2nd edn. Schweizerische Geologische Kommision, Bern
Stalder HA (2007) Kluft-Mineralien aus dem Steinbruch von Arvigo im Calancatal. Schweizer Strahler 1:5–7
Stalder HA, Wagner A, Graser S, Stuker P (1998) Mineralienlexikon der Schweiz. Wepf Verlag, Basel
Steck A (1968) Junge Bruchsysteme in den Zentralalpen. Eclogae Geol Helv 61:387–393
Steefel CI (2008) Geochemical kinetics and transport. In: Brantley SL, Kubicki JD, White AF (eds) Kinetics of water-rock interaction. Springer, New York, pp 545–589
Stober I, Bucher K (1999) Deep groundwater in the crystalline basement of the Black Forest region. Appl Geochem 14:237–254
Stober I, Bucher K (2005) The upper continental crust, an aquifer and its fluid: Hydaulic and chemical data from 4 km depth in fractured crystalline basement rocks at the KTB test site. Geofluids 5:8–19
Sun C-O, Williams RJ, Sun S-S (1974) Distribution coefficients of Eu and Sr for plagioclase-fluid and clinopyroxene-liquid equilibria in oceanic ridge basalt: an experimental study. Geochim Cosmochim Acta 38:1415–1433
Thompson AB (1970) Laumontite equilibria and the zeolite facies. Am J Sci 269:267–275
Thompson AB (1971) PCO2 in low-grade metamorphism; zeolite, carbonate, clay mineral, prehnite relations in the system CaO-Al2O3-SiO2-CO2–H2O. Contrib Mineral Petrol 33:145–161
Thompson AB (1975) Calc-silicate diffussion zones between marble and pelitic schist. J Petrol 16:314–346
Todd CS, Engi M (1997) Metamorphic field gradients in the Central Alps. J Metamorph Geol 15:513–530
Tulloch AJ (1979) Secondary Ca-Al silicates as low-grade alteration products of granitoid biotite. Contrib Mineral Petrol 69:105–117
Verdes G, Gout R, Castet S (1992) Thermodynamic properties of the aluminate ion and bayrite, boemite, diaspore and gibbsite. Eur J Mineral 4:767–792
Vidal O, Parra T, Trotet F (2001) A thermodynamic model for Fe-Mg aluminous chlorite using data from phase equilibrium experiment and natural pelitic assemblages in the 100–600°C, 1–5 kbar range. Am J Sci 6:557–592
Wagner A (1968) Mineralien aus den Stenbrüchen von Arvigo. Schweizer Strahler 1:128–131
Wagner A (1980) Die Mineralien aus den Gesteinsbrüchen von Arvigo im Bild (1 Teil). Mineralienfreund 18:137–141
Wagner A (1981) Die Mineralien aus den Gesteinsbrüchen von Arvigo im Bild (2 Teil). Mineralienfreund 19:56–64
Wagner A (1983) Die Mineralien aus dem Val Calanca und den Steinbrüchen von Arvigo. Schweizer Strahler 6:336–355
Wagner A, Stalder HA, Stuker P, Offermann E (2000a) Arvigo—eine der bekanntesten Mineralfundstellen der Schweiz. Schweizer Strahler 12:41–70
Wagner A, Stalder HA, Stuker P, Offermann E (2000b) Arvigo—eine der bekanntesten Mineralfundstellen der Schweiz. Schweizer Strahler 12:118–154
Walker GPL (1960) Zeolite zones and dike distribution in relation to the structure of the basalts of Eastern Iceland. J Geol 68:515–528
Walker GPL (1963) The Breiddalur central volcano, Eastern Iceland. Quart J Geol Soc Lond 119:29–63
Walker FDL, Lee MR, Parsons L (1995) Micropores and micropermeable texture in alkali feldspars; geochemical and geophysical implications. Mineral Mag 59:505–534
Weisenberger T, Bucher K (2008) Porosity evolution and mass transfer during low-grade metamorphism in crystalline rocks of the upper continental crust. In: 33rd IGC International Geological Congress, Oslo MPN03710L
Weisenberger T, Bucher K (2010) Zeolite in fissure of granites and gneisses of the Central Alps. J Metam Geol 28:825–847
Weiß S, Forster O (1997) Arvigo, Val Calanca: Kluftminerale aus dem Süden Graubündens. Lapis 6:13–42
Wenk E (1955) Eine Strukturkarte der Tessineralpen. Schweiz Mineral Petrogr Mitt 35:311–319
Yardley WD, Lloyd GE (1995) Why metasomatic fronts are really metasomatic sides. Geology 23:53–56
Zen E (1961) The zeolite facies: an interpretation. Am J Sci 259:401–409
Acknowledgments
We are grateful to Giovanni and Alfredo Polti for permission to do field work in the active quarry. Special thanks go to the technicians and staff of the Institute of Geosciences, Mineralogy—Geochemistry, University of Freiburg and particularly H. Müller-Sigmund for her useful advise during EMP analyses and her patience with us at the electron microprobe. A. Leemann from the Swiss Federal Laboratories for Materials Testing and Research for impregnation of rock samples. We thank J. Ferry, L. Machiels and an anonymous reviewer for their very detailed and constructive comments that have greatly improved our paper and J. Hoefs for his editorial efforts and the editorial handling of the paper. A special thanks deserved to the Friedrich Rinne foundation for the financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Hoefs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material 1:
(DOC 203 kb)
Electronic supplementary material 2:
X-ray images (TS 12.1) showing a relict plagioclase grain surrounded by albite, showing increased porosity around plagioclase. (a) Ca element map. Plagioclase shows Ca enrichment relative to the core. (b) K element map. (c) Na element map. Same colour codes are used as in figure 6 (EPS 7989 kb)
Electronic supplementary material 3:
(DOC 124 kb)
Electronic supplementary material 4:
(DOC 93 kb)
Electronic supplementary material 5:
(DOC 67 kb)
Electronic supplementary material 6:
X-ray images showing element distribution in a prehnite aggregate indicating a Fe ⇔ Al substitution during growth. (a) Fe element map showing an iron-enrichment in the core. (b) Al element map showing an Al-depletion in the core. The same colour code are used as in figure 6 (EPS 4755 kb)
Electronic supplementary material 7:
(DOC 59 kb)
Electronic supplementary material 8:
(DOC 89 kb)
Electronic supplementary material 9:
(a) Extra-framework cation (Ca+Sr+Mg-Na-K) distribution of zeolites. (b) R2+ - R+ - Si compositional diagram of zeolites. Si/Al ratio increases in chronologic order. Dashed area marks the chemical composition of zeolites found in granites and gneisses in the Swiss Alps (Weisenberger and Bucher 2010) (EPS 428 kb)
Rights and permissions
About this article
Cite this article
Weisenberger, T., Bucher, K. Mass transfer and porosity evolution during low temperature water–rock interaction in gneisses of the simano nappe: Arvigo, Val Calanca, Swiss Alps. Contrib Mineral Petrol 162, 61–81 (2011). https://doi.org/10.1007/s00410-010-0583-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-010-0583-2