
Abstract
Background
Non-small-cell lung cancer (NSCLC), the most common lung cancer, leads to the largest number of cancer-related deaths worldwide. There are many studies to identify the differentially expressed genes (DEGs) between NSCLC and normal control (NC) tissues by means of microarray technology. Because of the inconsistency of the microarray data sets, we performed an integrated analysis to identify DEGs and analyzed their biological function.
Methods and Results
We combined 15 microarray data sets and identified 1063 DEGs between NSCLC and NC tissues; in addition, we found that the DEGs were enriched in regulation of cell proliferation process and focal adhesion signaling pathway. The protein–protein interaction network analysis for the top 20 significantly DEGs revealed that CAV1, COL1A1, and ADRB2 were the significant hub proteins. Finally, we employed qRT-PCR to validate the meta-analysis approach by determining the expression of the top 10 most significantly DEGs and found that the expression of these genes were significantly different between tumor and NC tissues, in accordance with the results of meta-analysis.
Conclusion
qRT-PCR results indicated that the meta-analysis approach in our study was acceptable. Our data suggested that some of the DEGs, including MMP12, COL11A1, THBS2, FAP, and CAV1, may participate in the pathology of NSCLC and could be applied as potential markers or therapeutic targets for NSCLC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lung cancer leads to the largest number of cancer-related deaths worldwide, more than 85 % of which are lung adenocarcinoma, also known as non-small-cell lung cancer (NSCLC) [1, 2]. The predicted 5-year survival rate of NSCLC patients is 15.9 %, and little improvement has been reached during the past few decades [3]. Due to the poor clinical outcome, substantial researches focus on uncovering the molecular mechanism of NSCLC, providing insights into potential therapeutic targets.
Microarray analysis is widely used in the field of cancer genetics research, which can measure gene expression on a genome-wide scale simultaneously [4]. The technology of the microarray helps to better understand the mechanisms of various diseases [5]. Previous studies have used this technique to find the differentially expressed genes (DEGs) between NSCLC and normal tissues. However, the results of these studies are inconsistent, probably due to sample sources, microarray platforms, and analysis techniques. In order to avoid these problems, a meta-analysis method is developed to detect DEGs by integrating multiple microarray studies [6]. This method has been performed in various types of tumors including hepatocellular carcinoma [7], nasopharyngeal carcinoma [8], colorectal cancer [9], and osteosarcoma [10] to detect key genes, i.e., oncogenes or tumor suppressor genes involved in the development of cancers.
In this study, we employed meta-analysis method to identify DEGs between NSCLC and normal control (NC) tissues, and then we performed functional annotation of these genes to discover the biological processes and signaling pathways associated with NSCLC. Finally, we utilized qRT-PCR to validate the meta-analysis approach.
Materials and Methods
Strategy for Identification of NSCLC Gene Expression Datasets
We searched PubMed database and gene expression omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) database to identify NSCLC expression profiling studies by microarray. The following keywords and their combinations were used: lung adenocarcinoma and homo sapiens. The original studies that compared gene expression profiling between NSCLC and NC biopsy tissues or cultured cells were included in this study. Non-human studies, reviews, and meta-analysis articles were excluded.
After the background correction and normalization of raw data, we used significance analysis of microarray (SAM) to normalize the data and identified the DEGs by t test. The false discovery rate (FDR) <0.01 was selected as the criterion for significant differences.
Functional Classification of DEGs
We performed gene ontology (GO) enrichment analysis of the DEGs to investigate their biological functions in NSCLC using the online software GENECODIS (http://genecodis.cnb.csic.es) [11]. We also performed the pathway enrichment analysis by utilizing the Kyoto encyclopedia of genes and genomes (KEGG) database.
PPI Network Construction
The protein–protein interactions (PPIs) play central role in the regulation of biological processes and reveal the function of proteins at molecular level. The construction of PPI Network in a genome-wide scale is important for the interpretation of its functions. Biological general repository for interaction datasets (BioGRID) (http://thebiogrid.org/) was used to construct PPI network, and the top 10 up- and down-regulated DEGs were visualized in the network in Cytoscape [12].
The clinical specimens were provided by First Affiliated The Collection of Clinical Specimens
Hospital of PLA General Hospital, with the permission of the patients. Before the study, the protocols and documents were approved by the Medical Ethics Committee of the hospital. The written informed consent forms were obtained from the patients or legal guardians of the patients. The utilization of the samples was in strict accordance with the National Regulation of Clinical Sampling in China. The tumor tissues were immediately frozen and were stored in liquid nitrogen until RNA extraction.
RNA Preparation and qRT-PCR
Total RNA was extracted from each sample using RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Two micrograms total RNA was reverse transcribed into single-stranded cDNA using superscript II reverse transcriptase (Invitrogen/Life Technologies, Carlsbad, CA). We utilized PrimerPlex 2.61 (PREMIER Biosoft, Palo Alto, CA) to design primers (see Supplementary Table 1 for primers used). cDNA was amplified using Power SYBR Green PCR Master Mix (Applied Biosystems/Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Quantitative PCR was performed with ABI 7500 real-time PCR system (Applied Biosystems, Carlsbad CA). The results were analyzed using Ct method using data assist software version 3.0 (Applied Biosystems/Life Technologies). The data were normalized to ACTIN gene expression.
Results
Differential Gene Expression Analysis by Meta-analysis
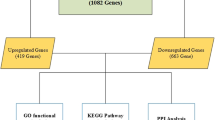
We identified 15 expression profiling studies eligible for the meta-analysis, including a total of 637 cases of NSCLC and 298 cases of NC. The details of the selected studies are summarized in Table 1. We found 1063 DEGs with FDR <0.01, among which 464 genes were up-regulated and 599 genes were down-regulated in NSCLC tissues. The top 20 significantly DEGs are listed in Table 2. The full list of the DEGs can be found in Supplementary Table 2.
Functional Annotation
We conducted a GO categories enrichment analysis to investigate the biological roles of identified DEGs. We separately examined the three groups of GO categories, including biological process, cellular component, and molecular function, by web-based software GENECODIS. Genes of P < 0.01 were selected and were tested against the background set of all genes with GO annotations. We found that the enriched GO terms for biological process was regulation of cell proliferation (GO: 0042127), and while for cellular component was plasma membrane part (GO: 0044459), for molecular function was growth factor binding (GO: 0019838). The full list of GO terms is given in Table 3.
We also performed the KEGG pathway enrichment analysis to further explore the biological significance of the DEGs. Hypergeometric test with P value <0.05 was used as the criterion for pathway detection. From the KEGG pathway analysis, we found that 37 genes were enriched in focal adhesion signaling pathway, indicating that they may relate with NSCLC metastasis (Fig. 1).

The enriched KEGG pathway of DEGs (focal adhesion pathway). The red icons mean DEGs identified in this study
PPI Network Construction
Based on PPI data in BioGRID, PPI networks of the top 20 significantly DEGs were constructed by Cytoscape software. The PPI network consisted of nodes and edges, which represents proteins and interactions. There were 410 edges and 404 nodes in the PPI network, among which ADRB2, CAV1, and COL1A1 were connected with more proteins (Fig. 2).

PPI networks of the top ten most significantly up- or down-regulated DEGs. Nodes and edges represent proteins and interactions between proteins. The up-regulated genes in NSCLC were marked with red color and the down-regulated genes in NSCLC were marked with deep blue color
qRT-PCR Validation
Five pairs of NSCLC and NC tissues were used to validate the results of meta-analysis. We selected the top ten up- or down-regulated genes for validation. MMP12, COL11A1, THBS2, ADAM12, and FAP were selected as the up-regulated genes in NSCLC, while FABP4, CDH5, CAV1, TCF21, and ADRB2 were selected as the down-regulated genes in NSCLC.
The qRT-PCR results showed that the expression patterns of selected genes in NSCLC and NC tissues were similar to those in the meta-analysis. The expression profiling of up- and down-regulated genes in each sample is shown in Fig. 3. The average fold changes of the up-regulated genes MMP12, COL11A1, THBS2, ADAM12, and FAP were 6.6-, 17.76-, 4.46-, 4.69-, and 2.77-folds, respectively. MMP12 and COL11A1 were dramatically up-regulated in three of five samples, while the other three genes were mild up-regulated in most of the NSCLC samples. The average fold changes of the down-regulated genes FABP4, CDH5, CAV1, TCF21, and ADRB2 were 24.9-, 3.5-, 18.28-, 8.3-, and 3.4-folds. FABP4 and CAV1 were dramatically down-regulated in four of five patient samples.

qRT-PCR validation of the top ten most significantly up- or down-regulated DEGs in five pairs of NSCLC and NC tissues. ACTIN was used as an internal reference gene for normalization. The graph showed the relative expression levels of ADAM12, ADRB2, CAV1, CDH5, COL11A1, FABP4, FAP, MMP12, TCF21, and THBS2 between each pair of NSCLC and NC tissues. Bar graph represents mean ± SEM. Z means normal control tissues; C means non-small-cell lung cancer tissues
Discussion
Lung cancer is still the leading cause of cancer-related mortality all over the word [13]. Although there is a huge development in molecular techniques and lung cancer biology, many of the genetic alterations related to lung carcinogenesis still remain unknown. Microarray analysis can discover the expression alteration of a large number of genes simultaneously within tumors, which may help discover new signaling pathways or molecular mechanisms associated with tumorigenesis. In this study, we combined 15 microarray data sets to detect DEGs. We also performed GO term, KEGG, and PPI analysis of the DEGs and detected some important molecules and signaling pathways which may extend our understanding of the pathology of NSCLC and further guide the development of new therapeutic targets.
In this study, we found 1063 DEGs between NSCLC and NC tissues. GO analysis showed that 266 DEGs were enriched in cell proliferation regulation and 96 DEGs were enriched in DNA binding and growth factor binding. The results indicated that cell proliferation was dysregulated in NSCLC tissues. The fundamental abnormality of cancer cells was the continual unregulated cell proliferation [14], and numerous studies had proved the important role of EGFR in NSCLC, indicating that the GO analysis in this study was reasonable.
KEGG pathway analysis revealed that the DEGs between NSCLC and normal tissues are enriched in focal adhesion signaling pathways. Focal adhesions are the contact sites between the cytoskeleton and extracellular matrix through transmembrane proteins, integrins [15]. Cells received signals from extracellular microenvironment through focal adhesions to maintain proper cell survival, proliferation, differentiation, and motility through integrin-related signaling pathways [16–18]. The loss of the tight regulation of focal adhesions can lead to cancer progression and metastasis [19–21]. PPI network analysis for the top 20 significantly DEGs indicated that the significant hub proteins were CAV1 and COL1A1, which are the important components of focal adhesion [21]. Our data indicated that focal adhesion components and related signaling pathways may play important roles in pathology of NSCLC and may shed light on discovery of new therapeutic targets of NSCLC.
In order to validate our meta-analysis data, we performed qRT-PCR to detect the expression of the top ten significant DEGs in NSCLC and NC tissues. We found that the expression patterns of the selected ten DEGs, including MMP12, COL11A1, THBS2, ADAM12, FAP, FABP4, CDH5, CAV1, TCF21, and ADRB2, were consistent with our meta-analysis and previous reports.
qRT-PCR results show that the mRNA levels of MMP12, COL11A1, THBS2, ADAM12, and FAP are significantly higher in NSCLC tissues than those in NC tissues. MMP12 is a 22 kDa metal-dependent proteinase which can degrade elastin, type IV collagen, fibronectin, laminin, gelatin, vitronectin, entactin, heparin, and chondroitin [22]. Many of the MMP12 substrates, such as collagen, laminin, and fibronectin, are important extracellular matrix molecules which can regulate cell shape, migration, and survival through focal adhesion [23]. Previous studies also reveal that MMP12 is correlated with early cancer-related deaths in NSCLC, especially for the patients who exposed to tobacco cigarette smoke [24].
Both COL11A1 and THBS2 participate in focal adhesion signaling pathways. COL11A1 is linked to ovarian cancer recurrence and poor survival. The invasion ability and oncogenic potential of ovarian cancer cells are suppressed by COL11A1 knockdown [25]. COL11A1 and THBS2 are overexpressed in lung cancer and can be recognized as a marker of lung cancer [26–28]. FAP is a serine protease selectively and highly expressed on the surface of cancer-associated fibroblasts, and it is important in the progression and prognosis in diverse malignancies [29]. The expression level of FAP is closely associated with tumor recurrence and poor clinical outcome in rectal and pancreatic cancer [30, 31]. However, the functions of FAP in NSCLC are poorly understood. One study reports that FAP is highly expressed in lung cancer stroma, and its high expression is a predictor of poor survival of NSCLC patients [32].
The expression of CAV1 and ADRB2 is lower in NSCLC tissues than that in NC tissues, and they are also the significant hub proteins of the PPI network in our study. Furthermore, CAV1 is involved in the focal adhesion signaling pathway. CAV1 is a major structural component of caveolae which is a specialized plasma membrane invagination [33]. The function of CAV1 in tumorigenesis is depending on tumor type and tumor stage. The expression of CAV1 is down-regulated in tumor cells and tissues isolated from breast, cervix, lung, and ovary [34–38], indicating that CAV1 may act as a tumor suppressor. Currently, the in vivo function of ADRB2 in NSCLC is largely unknown.
In summary, we used a meta-analysis approach to integrate 15 microarray data sets of NSCLC and identified DEGs and their biological function. We used qRT-PCR to validate the meta-analysis approach by detecting the expression of top ten significantly DEGs. Our study suggested that some DEGs, including MMP12, COL11A1, THBS2, FAP, and CAV1, might participate in the pathology of NSCLC and they might be potential therapeutic targets.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63(1):11–30
Ettinger DS, Akerley W, Borghaei H, Chang AC, Cheney RT, Chirieac LR et al (2013) Non-small cell lung cancer, version 2.2013. J Natl Compr Cancer Netw 11(6):645–653 quiz 53
DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M et al (1996) Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet 14(4):457–460
Lakhani SR, Ashworth A (2001) Microarray and histopathological analysis of tumours: the future and the past? Nat Rev Cancer 1(2):151–157
Ramasamy A, Mondry A, Holmes CC, Altman DG (2008) Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med 5(9):e184
Xu W, Huang H, Yu L, Cao L (2015) Meta-analysis of gene expression profiles indicates genes in spliceosome pathway are up-regulated in hepatocellular carcinoma (HCC). Med Oncol 32(4):425
Tulalamba W, Larbcharoensub N, Sirachainan E, Tantiwetrueangdet A, Janvilisri T (2015). Transcriptome meta-analysis reveals dysregulated pathways in nasopharyngeal carcinoma. Tumour biol. doi:10.1007/s13277-015-3268-7
Letellier E, Schmitz M, Baig K, Beaume N, Schwartz C, Frasquilho S et al (2014) Identification of SOCS2 and SOCS6 as biomarkers in human colorectal cancer. Br J Cancer 111(4):726–735
Yang Z, Chen Y, Fu Y, Yang Y, Zhang Y, Li D (2014) Meta-analysis of differentially expressed genes in osteosarcoma based on gene expression data. BMC Med Genet 15:80
Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A (2012) GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res 40:W478–W483 (Web Server issue)
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
World Health Organization (2011) Cancer: fact sheet (N297). http://www.who.int/mediacentre/factsheets/fs297/en/
Cooper GM, Hausman RE (2000) The cell. Sinauer Associates, Sunderland, pp 725–730
Dubash AD, Menold MM, Samson T, Boulter E, Garcia-Mata R, Doughman R et al (2009) Chapter 1. Focal adhesions: new angles on an old structure. Int Rev Cell Mol Biol 277:1–65
Legate KR, Fassler R (2009) Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci 122(Pt 2):187–198
Legate KR, Wickstrom SA, Fassler R (2009) Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev 23(4):397–418
Vehlow A, Cordes N (2013) Invasion as target for therapy of glioblastoma multiforme. Biochim Biophys Acta 1836(2):236–244
Bissell MJ, Hines WC (2011) Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17(3):320–329
Bissell MJ, Kenny PA, Radisky DC (2005) Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb Sym 70:343–356
Nelson CM, Bissell MJ (2006) Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol 22:287–309
Gronski TJ Jr, Martin RL, Kobayashi DK, Walsh BC, Holman MC, Huber M et al (1997) Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastase. J Biol Chem 272(18):12189–12194
Seong J, Wang N, Wang Y (2013) Mechanotransduction at focal adhesions: from physiology to cancer development. J Cell Mol Med 17(5):597–604
Hofmann HS, Hansen G, Richter G, Taege C, Simm A, Silber RE et al (2005) Matrix metalloproteinase-12 expression correlates with local recurrence and metastatic disease in non-small cell lung cancer patients. Clin Cancer Res 11(3):1086–1092
Wu YH, Chang TH, Huang YF, Huang HD, Chou CY (2014) COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene 33(26):3432–3440
Wang KK, Liu N, Radulovich N, Wigle DA, Johnston MR, Shepherd FA et al (2002) Novel candidate tumor marker genes for lung adenocarcinoma. Oncogene 21(49):7598–7604
Chong IW, Chang MY, Chang HC, Yu YP, Sheu CC, Tsai JR et al (2006) Great potential of a panel of multiple hMTH1, SPD, ITGA11 and COL11A1 markers for diagnosis of patients with non-small cell lung cancer. Oncol Rep 16(5):981–988
Metodieva SN, Nikolova DN, Cherneva RV, Dimova II, Petrov DB, Toncheva DI (2011) Expression analysis of angiogenesis-related genes in Bulgarian patients with early-stage non-small cell lung cancer. Tumori 97(1):86–94
Liu R, Li H, Liu L, Yu J, Ren X (2012) Fibroblast activation protein: a potential therapeutic target in cancer. Cancer Biol Ther 13(3):123–129
Saigusa S, Toiyama Y, Tanaka K, Yokoe T, Okugawa Y, Fujikawa H et al (2011) Cancer-associated fibroblasts correlate with poor prognosis in rectal cancer after chemoradiotherapy. Int J Oncol 38(3):655–663
Cohen SJ, Alpaugh RK, Palazzo I, Meropol NJ, Rogatko A, Xu Z et al (2008) Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 37(2):154–158
Liao Y, Ni Y, He R, Liu W, Du J (2013) Clinical implications of fibroblast activation protein-alpha in non-small cell lung cancer after curative resection: a new predictor for prognosis. J Cancer Res Clin Oncol 139(9):1523–1528
Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68(4):673–682
Sloan EK, Stanley KL, Anderson RL (2004) Caveolin-1 inhibits breast cancer growth and metastasis. Oncogene 23(47):7893–7897
Williams TM, Medina F, Badano I, Hazan RB, Hutchinson J, Muller WJ et al (2004) Caveolin-1 gene disruption promotes mammary tumorigenesis and dramatically enhances lung metastasis in vivo. Role of Cav-1 in cell invasiveness and matrix metalloproteinase (MMP-2/9) secretion. J Biol Chem 279(49):51630–51646
Racine C, Belanger M, Hirabayashi H, Boucher M, Chakir J, Couet J (1999) Reduction of caveolin 1 gene expression in lung carcinoma cell lines. Biochem Biophys Res Commun 255(3):580–586
Sunaga N, Miyajima K, Suzuki M, Sato M, White MA, Ramirez RD et al (2004) Different roles for caveolin-1 in the development of non-small cell lung cancer versus small cell lung cancer. Cancer Res 64(12):4277–4285
Wiechen K, Diatchenko L, Agoulnik A, Scharff KM, Schober H, Arlt K et al (2001) Caveolin-1 is down-regulated in human ovarian carcinoma and acts as a candidate tumor suppressor gene. Am J Pathol 159(5):1635–1643
Acknowledgments
This research was supported by a grant from Special project for the transformation of major scientific and technological achievements of Hebei province (No. 15277732D).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tian, ZQ., Li, ZH., Wen, SW. et al. Identification of Commonly Dysregulated Genes in Non-small-cell Lung Cancer by Integrated Analysis of Microarray Data and qRT-PCR Validation. Lung 193, 583–592 (2015). https://doi.org/10.1007/s00408-015-9726-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-015-9726-6