
Abstract
Purpose
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder that affects women of reproductive age and is characterized by ovulatory dysfunction and/or androgen excess or polycystic ovaries. Women with PCOS present a number of systemic symptoms in addition to those related to the reproductive system. It has been associated with functional derangements in adipose tissue, metabolic syndrome, type 2 diabetes, and an increased risk of cardiovascular disease (CVD).
Methods
A detailed literature search on Pubmed was done for articles about PCOS, adipokines, insulin resistance, and metabolic syndrome. Original articles, reviews, and meta-analysis were included.
Results
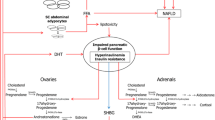
PCOS women are prone to visceral fat hypertrophy in the presence of androgen excess and the presence of these conditions is related to insulin resistance and worsens the PCO phenotype. Disturbed secretion of many adipocyte-derived substances (adipokines) is associated with chronic low-grade inflammation and contributes to insulin resistance. Abdominal obesity and insulin resistance stimulate ovarian and adrenal androgen production, and may further increase abdominal obesity and inflammation, thus creating a vicious cycle.
Conclusion
The high prevalence of metabolic disorders mainly related to insulin resistance and CVD risk factors in women with PCOS highlight the need for early lifestyle changes for reducing metabolic risks in these patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Hyperandrogenism represents the most common endocrine disorder in women of reproductive age with prevalence between 6 and 15% according to the used criteria. The most common hyperandrogenic disorder is the polycystic ovary syndrome (PCOS), with approximately an 80–85% prevalence among women with androgen excess [1]. Diagnosis of PCOS is currently based on Rotterdam diagnosis criteria [2], which broadened the previous NIH classification [3]. At least two of the following features among oligo-anovulation, hyperandrogenism and polycystic ovaries by ultrasound should be present. In 2006, the Androgen Excess Society criteria suggested that the presence of clinical and/or biochemical hyperandrogenism together with oligo-anovulation and polycystic ovaries by ultrasound is needed for the diagnosis of PCOS [4]. Ultrasound has emerged as a useful tool to help in the diagnosis of PCOS. Indeed, the increased proportion between the stroma and the ovary surface in the median section was a strong predictor of elevated androgen and testosterone levels [5]. According to the National Institutes of Health (NIH), four PCOS phenotypes have been identified. These specific clinical phenotypes include the following: (1) a classic phenotype, with polycystic ovaries in addition to hyperandrogenism and ovulatory dysfunction; (2) a phenotype with ovulation in addition to polycystic ovaries and hyperandrogenism; (3) a phenotype with hyperandrogenism, ovulatory dysfunction without polycystic ovaries; and (4) a phenotype without polycystic ovaries but with androgen and ovulatory dysfunction. These different clinical pictures have been recommended in all research studies and during clinical care. Although the clinical syndrome has been described more than 80 years ago, the underlying cause of the disorder continues to be uncertain. PCOS is considered a multifactorial disorder with various genetic, metabolic, endocrine and environmental abnormalities [6]. Until recently, the main concerns in women with PCOS were menstrual irregularity, hirsutism, and infertility. Weight gain and obesity are also common features of PCOS and often precede the onset of anovulatory cycles. Moreover, recent studies have clearly shown that insulin resistance (IR) can be an integral part of the syndrome being present in 65–80% of women with PCOS, and plays a significant role in its aetiology [7]. In addition, women with PCOS are at a higher risk for diabetes and heart disease than the general population. In light of its association with IR, PCOS is now considered as a general health disorder in women instead of just a cosmetic or fertility problem. Evidence also showed that a central distribution of body fat carries higher risk of cardiovascular disease (CVD) and diabetes than does a gynoid fat distribution [8]. This fat topography tends to change in middle-aged female, when there is a change to an android type of fat distribution, whereas women with PCOS tend to accumulate fat in a central distribution also during the fertile period. The changes are related to conditions of absolute or relative (i.e. menopause) androgen excess. These influences of hormones on body fat distribution appear to be related both to adipose tissue-specific expression of steroid receptors and to local tissue steroid hormone metabolism [9]. The considerable overlap between features of PCOS and metabolic syndrome (MS) seems to suggest that dysregulation of adipocyte functions might contribute to many of the metabolic complications observed in women with PCOS.
In this review, we will focus on the role of adipose tissue in the pathophysiological mechanisms involved in PCOS and its complications.
PCOS: clinical and biochemical profile
PCOS is a heterogeneous disorder, the principal features of which include androgen excess, ovary dysfunction and/or polycystic ovaries. PCOS has a complex multifactorial aetiology, in which a variety of predisposing genes interact with environmental factors to produce diseases [10]. Many attempts have being made to identify specific genes that underline the intrinsic cause of PCOS [11]. Genes involved in steroidogenesis, carbohydrate metabolism, sex hormone binding globulin (SHBG) gene, the androgen receptor, and the genetic loci associated with insulin sensitivity and susceptibility to obesity are the likely ones involved [11]. The pathogenesis of PCOS may be associated with CYP17 gene regulation, as CYP17 activity is fourfold greater in patients with PCOS [12]. Despite repeated attempts to identify the putative gene or genes involved in this disorder, no gene has clearly emerged as most important in PCOS, and many positive results have not been confirmed in subsequent studies. Therefore, it is likely that the variety of pathways involved in PCOS and the lack of a common pathogenetic mechanism suggest that genes coding for transcriptional factors or signalling pathway components that may affect the organs involved in PCOS are globally involved in the pathogenesis of the syndrome.
Patients with PCOS have a supraphysiological androgen production, and hyperandrogenism is the biochemical hallmark of PCOS [13]. Elevated circulating androgen levels are observed in 80–90% of women with PCOS [13, 14]. Hyperandrogenism is caused by derangements of androgen production and metabolism, and biochemically is usually assessed by assay of total and free testosterone, androstenedione, 17-hydroxy progesterone, dehydroepiandrosterone sulphate (DHEAS) and SHBG. Elevated levels of free testosterone usually reflect the fact that SHBG levels are generally decreased in women with PCOS due to the effects of testosterone and insulin to decrease hepatic production of SHBG [15]. Women with PCOS often have higher than normal serum levels of some or all these hormones. Elevated levels of free testosterone account for the vast majority of abnormal finding, whereas DHEAS and androstenedione are also used for diagnostic testing in the diagnosis of hyperandrogenism. Roughly, 80% of the total dehydroepiandrosterone (DHEA) is of adrenal origin, while androstenedione is secreted approximately in equal amounts by the adrenal and ovaries in normal premenopausal women. Although the ovaries are the main source of increased androgen in PCOS [16], adrenal androgen excess can be present in approximately 20–25% among women with PCOS [17]. Women with PCOS have increased secretion of adrenocortical precursor steroids basally and after adrenocorticotropic hormone (ACTH) stimulation [18]. Thus, hyperandrogenism has a multifactorial origin attributed mostly to the ovaries with a contribution from the adrenal. The ovarian hyperandrogenism of PCOS is demonstrated directly by the gonadotropin-releasing hormone (GnRH) test or the human chorionic gonadotropin test [11]. The PCOS ovary is typically hypersensitive to luteinizing hormone (LH) stimulation associated with an increased androgen responsiveness. This hypersensitive steroid response to LH stimulation was initially postulated to include insulin excess, which is known to sensitize to LH action [19]. However, support for an intrinsic theca cell defect that can account for excess androgen production has been obtained from in vivo and in vitro studies, by demonstrating that an overactive androgenic phenotype is constitutively present in theca cells [11, 20]. Alterations in P450c17 at transcriptional and post-transcriptional level have been implicated in the aetiology of PCOS. A relative inhibition of 17–20 lyase activity with respect to 17-hydroxylase, leading to an increased 17-hydroxy progesterone/androstenedione ratio has been demonstrated [21]. Low aromatase activity has also been demonstrated in women with PCOS, thus contributing to theca cell overproduction of androgens [21, 22]. Elevated levels of androgens may have a negative impact on follicular developments, causing atresia, premature luteinization and hindering ovulation by impairing selection of the dominant follicle [11].
LH excess is common in the disorder and a high percentage of patients with PCOS have an elevated LH/follicle-stimulating hormone (FSH) ratio presumably due to high levels of LH. LH is necessary for the expression of gonadal steroidogenic enzymes, but it seems unlikely that the LH increase might be the primary cause of ovarian androgen excess, due to the normal process of LH-induced desensitization of theca cells. LH-releasing hormone stimulation causes, indeed, excessive LH secretion [23] probably due to a higher frequency or amplitude of hypothalamic GnRH. Altered hypothalamic neurotransmission has been suggested in these patients (e.g. altered dopaminergic and opioid tone), although not confirmed by pharmacological manipulation of these neuronal systems [23].
IR, as defined as a decreased ability of insulin to mediate the metabolic action on glucose and lipid metabolism, is very common in PCOS [11, 24, 25]. In some studies, this has been shown to be related to lower expression of the insulin-sensitive glucose transporter-4 (GLUT-4). However, IR selectively affects tissue-specific metabolic action, whereas mitogenic and steroidogenic actions are usually preserved. Thus, this compensatory hyperinsulinism sensitizes ovarian theca cells to secret androgen in response to LH and seems to have a similar effect on the adrenal androgens secretion induced by ACTH: hyperinsulinism is thought to contribute to hyperandrogenism in these patients [26] since it stimulates ovarian steroidogenesis by a direct thecal production of androgens, increases secretion of androgen mediated by LH. Moreover, insulin inhibits hepatic SHBG synthesis leading to increased testosterone availability. Insulin may also act via the insulin-like growth factor-1 (IGF-I) receptor when insulin reaches high concentrations, as compensatory hyperinsulinemia [27]. IGF-I is a growth factor which is mainly synthetized by the liver, but is also produced by other tissues, including the ovaries, where it has paracrine/autocrine functions. IGF-I, when overexpressed, induces increased androgen production by the theca cells and synergically with LH and FSH modulates the expression of aromatase in granulosa cells [28].
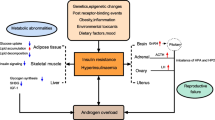
Many studies have demonstrated that obesity is a common feature of PCOS but the prevalence of obesity in affected women ranges from 50 to 80% [29,30,31]. These discrepancies might be due to differences in the diagnostic criteria for PCOS. However, when comparable diagnostic criteria were applied, the prevalence rates were still different in various studies [17], suggesting that environmental factors, such as lifestyle, contribute to the presence of obesity in PCOS [32, 33]. Moreover, PCOS is also a common clinical aspect in women with obesity. Regional differences in adipose tissue anatomy have been reported in PCOS that contribute to the clinical manifestations and complications of the syndrome. Many studies based on anthropometric measurements have suggested that women with PCOS have increased abdominal fat distribution, regardless of body mass index (BMI) [34], although global adiposity rather than regional fat distribution characterizes women with PCOS [35]. By whatever mechanism, it is clear that an alteration of lean-to-fat mass ratio plays an important role in the clinical expression of the metabolic features and complications of PCOS.
Biochemical presentation of PCOS changes with aging [36], but little is known to PCOS women after menopause. Postmenopausal women without PCOS had decreased estrogen production as well as a reduction of androstenedione circulating levels. Similar biochemical characteristics have been described in PCOS woman after the transition to menopause. Indeed, testosterone level in PCOS woman was similar to non-PCOS women in perimenopausal period, thus explaining the tendency to cycle regularly as PCOS women grow older [37]. On the other hand, another study showed that PCOS women after menopause had raised levels of testosterone level in comparison to control women without PCOS [38].
The adipose tissue
There is considerable evidence that adipose tissue, besides the biological repertoire necessary for storing and releasing energy, plays a wide-ranging role in metabolic regulation and physiological homeostasis, also including neuroendocrine function and immune function.
It is now well acknowledged that adipose tissue excess, particularly in the visceral compartment, is associated with IR, diabetes, hypertension, prothrombotic and proinflammatory states, and CVD [39].
So far, many adipocyte-derived signals have been identified (Table 1) and significant progress has been made in understanding their specific functions. Several molecules that were previously established as signals from non-adipose tissues, are now known to be secreted by adipose cells as well. Using adipokines as one of the major communication tools, adipocytes and stromal cells affect a large number of other tissues and organs, such as the liver, muscle, adrenal cortex, brain, the sympathetic nervous system, the reproductive system, and pancreatic β-cells and participate in appetite and energy balance, blood pressure, lipid metabolism as well as angiogenesis and haemostasis.
Several studies have clearly demonstrated that obese people with visceral adiposity are associated with more IR and increased morbidity than individuals with a similar degree of overall adiposity [40]. Moreover, individuals with glucose intolerance or diabetes have larger adipocytes than do those with normal glucose tolerance [41, 42].
Approximately 75% of PCOS patients are overweight and central obesity is observed in both normal and overweight PCOS women [43, 44], whereas PCOS has been reported to be a common finding in obese or overweight women [45].
IR is also a common feature of PCOS since it is present in 50–80% of these patients. Although IR is clearly associated with obesity, lean women with PCOS may have IR that is caused by genetic disorders. However, the available data suggest that patients with PCOS can have 5–8 times increased diabetes risk compared to age- and weight-matched controls [46]. Thus, adipose tissue excess directly contributes to the pathogenesis of obesity-related disorders.
The role of obesity in PCOS pathogenesis and metabolic complications
As noted in a preceding section, weight gain and central obesity are common features of PCOS, often precede the onset of anovulatory cycles, and are associated with a greater prevalence of hirsutism and menstrual irregularity [36]. Moreover, obese women should be screened for PCOS since the syndrome has been shown to be a common finding also in obese and overweight women [45, 47]. IR hyperinsulinemic is a major factor in the excessive adipogenesis and lipogenesis of PCOS, and obesity in turn seems to aggravate the hyperandrogenism by exaggerating IR (Fig. 1). These clinical aspects are in large part explained by the increased prevalence of visceral fat not only in obese and in overweight but also in many normal-weight patients with PCOS. Visceral fat contributes more to the IR in PCOS than does subcutaneous fat because of its enhanced lipolytic response to catecholamines, whereas the lipolytic effect was decreased in subcutaneous fat [48, 49]. The adverse metabolic impact of visceral adipose tissue (VAT) has been attributed to distinct biological properties of adipocytes in this compartment compared with other adipose tissue depots. VAT has been suggested to possess a large amount of β-adrenergic receptors with a higher lipolytic activity [50]. This increased catecholamine-induced lipolysis in VAT has been attributed to increased expression of β1 and β2 receptor [50] as well as enhancement of β3 receptor function [51]. Therefore, excess VAT would lead to greater free fatty acid delivery to the liver, whereby impairing insulin clearance and action [52]. The adipose tissue produces and releases a number of bioactive proteins collectively referred as adipokines [53]. Most of these substances are primarily secreted by adipose tissue-resident macrophages [54] that are increased in both subcutaneous and VAT. Proinflammatory cytokines arising from the mononuclear cells of adipose tissue are other mediators of the IR of PCOS. Excess abdominal adipose tissue triggers an inflammatory response, which is an additional important element in the pathogenesis of IR. Thus, the dysregulation of adipose tissue mass associated with obesity might be a cause of altered endocrine system function as well as its consequence.

Link of insulin resistance between PCOS and metabolic syndrome
Adipose tissue expresses a variety of enzymes for activation, interconversion and inactivation of steroid hormones. Cytochrome P450-dependent aromatase and 17β-hydroxysteroid dehydrogenases (17-βHSD) are highly expressed in adipose tissue stromal cells and preadipocytes. 17-βHSD converts weak androgens and estrogens to their more potent counterparts (androstenedione to testosterone and estrone to estradiol). The ratio of 17-βHSD to P450 aromatase is positively correlated with central obesity, indicating an increased local androgen production in visceral adiposity [55, 56]. Given the mass of adipose tissue, the contribution of adipose tissue to whole-body steroid metabolism is important, with adipose tissue contributing up to 100% of circulating estrogen in postmenopausal women and 50% of circulating testosterone in premenopausal women.
Adipose tissue is also involved in the regulation of glucocorticoid metabolism. This tissue-specific glucocorticoid metabolism is primarily determined by the enzyme 11β-hydroxysteroid dehydrogenases (11-βHSDs), which catalyses interconversion of active cortisol and inert cortisone. Two isoenzymes have been discovered, each with unique properties and powerful biological roles. 11-βHSD type 1 is highly expressed in adipose tissue, particularly in VAT, where it regenerates metabolically active cortisol from cortisone. 11-βHSD type 2 inactivates cortisol by catalysing interconversion of active cortisol to inert cortisone, thereby protecting key tissues [57]. Omental adipocytes have a twofold higher rate of insulin-stimulated glucose uptake compared with subcutaneous adipose tissue [58], whereas glucocorticoids down-regulate glucose uptake capacity and insulin-signalling protein in omental but not subcutaneous adipocytes. These data further support the interaction between endogenous (circulating and adipocyte generated) cortisol and visceral fat in the development of IR.
Several observations have associated 11-βHSD type 1 dysregulation with a variety of medical conditions including obesity, diabetes, hypertension, CVD as well as polycystic ovarian syndrome [59,60,61]. The importance of VAT glucocorticoid metabolism by 11-βHSD type 1 is demonstrated in experimental animals overexpressing 11-βHSD in all adipose tissues [62]. These mice develop visceral obesity and features of the MS including IR, dyslipidemia and hypertension. All had reduced levels of adiponectin and increased concentrations of leptin, tumour necrosis factor-α (TNFα), angiotensinogen and free fatty acid. In contrast, mice with deletion of 11-βHSD type 1 have a favourable metabolic phenotype. Taken together, these observations support a role of glucocorticoid metabolism by visceral fat in the pathogenesis of visceral obesity and the MS.
Thus, the influence of hormones on body fat distribution appears to be related both to adipose tissue-specific expression of steroid receptors and to local tissue steroid hormone metabolism [9]. The concomitant glucocorticoid receptor activation by local regeneration of cortisol and androgens [63] together with the decline in the growth hormone (GH)–IGF-I axis observed in visceral obesity represents additional mechanism for stimulation of visceral fat accumulation [64].
The involvement of growth hormone (GH) in the regulation of visceral fat is clearly documented in acromegaly where there is a reduction in VAT [65]. The main action of GH on adipose tissue is to prevent lipid accumulation and to stimulate lipid mobilization, which requires synergism with steroid hormones. These effects are mediated via inhibition of lipoprotein lipase activity [66] together with an enhancement of catecholamine-induced lipolysis via β adrenergic receptors [67]. In obesity, there is a decrease in circulating GH and a blunted response to provocative stimuli [68]. The degree of GH attenuation correlates with the amount of total and visceral fat [69,70,71]. Intra-abdominal fat, as measured by computed tomography, is inversely related to serum IGF-I concentrations in obesity suggesting that visceral fat may be inversely related to 24-h GH secretion in both sexes.
Adipokines in PCOS
Leptin
Leptin acts as an afferent satiety signal at a hypothalamic central level as a regulatory factor. Adipocytes secrete leptin in direct proportion to adipose tissue mass as well as nutritional status, and this secretion is greater from subcutaneous relative to VAT [72]. Many of leptin effects on energy homeostasis, particularly on energy intake and expenditure, are mediated via hypothalamic pathways [73]. An increase in leptin directly suppresses the orexigenic peptides neuropeptide Y (NPY) and agouti-related peptide in the arcuate nucleus. This putative action of leptin on NPY may further depend on an intermediate signal through melanocortin-4 receptor [74]. Leptin increases the levels of anorectic peptides, α-melanocyte stimulating hormone and cocaine- and amphetamine-regulated transcript, produced by neurons in the lateral arcuate nucleus. The net action of leptin is to inhibit appetite, stimulate thermogenesis and reduce body weight and fat. Although initially viewed as an anti-obesity hormone, leptin’s primary role is to guard against starvation rather than to prevent obesity [75]. Leptin levels rapidly decline with caloric restriction and weight loss. It should be noted that although leptin administration can produce profound effects on energy expenditure and food intake when compared with the leptin-deficient state, administration of leptin to normal or obese people produces unremarkable effects [76]. The mechanism for this leptin resistance is unknown but may depend on defects in leptin signalling or transport across the blood–brain barrier [77].
Most obese humans have an elevated circulating level of leptin due to their enlarged fat mass. However, they do not respond to this increased endogenous leptin level by reducing their food intake. The treatment of obese patients with exogenous leptin was generally ineffective. Only subjects receiving high doses of leptin (the average serum leptin concentration was 30–40 fold higher than baseline values) presented a significant reduction of body weight [78]. Thus, to overcome a leptin-resistant state in humans, it is likely that very high levels of this hormone must be achieved. The mechanism underlying leptin resistance is not yet known, although part of this mechanism may be due to limited entry of leptin into central nervous system, as leptin transport to brain is a saturable carrier-mediated process [79]. A decreased ability of leptin to activate hypothalamic receptor due to expression of inhibitory molecules in the regulation of leptin receptor signalling is another hypothesis [80]. Capobianco et al. showed no significant differences in C-reactive protein (CRP) and leptin concentrations through the menstrual cycle in both the examined cycles, but inter-group analysis revealed significant differences of CRP and leptin levels between the ovulatory and anovulatory cycles with higher values at periovulatory phase in the ovulatory cycles [81]. A number of studies have provided evidence that hyperleptinemia is involved in the pathogenesis of atherosclerotic vascular disease [82]. Many studies have shown elevated plasma leptin levels in patients with hypertension and a positive correlation between leptin and blood pressure [83]. It has been proposed that leptin could play a role in atherogenesis acting synergistically with other inflammatory mediators [84]. Moreover, leptin, in vitro, stimulates the proliferation and hypertrophy of vascular smooth muscle cells and is able to induce CRP expression in human coronary artery endothelial cells [85, 86]. A recent report suggests that increased concentrations of both leptin and CRP confer the highest risk for CVD, although the complex relationship between leptin and CRP remains to be fully elucidated [87]. Therefore, in obese subjects, leptin may no longer be able to regulate caloric intake and energy balance. Moreover, high serum leptin concentration can have adverse metabolic effects and represent an independent risk factor for coronary heart disease [88]. Although leptin therapy has limited success in common obesity, it has impressive effects in patients with genetic leptin deficiency or lipodystrophy [89, 90].
Serum leptin levels and subcutaneous leptin messenger RNA were found to be higher in overweight/obese PCOS women compared to normal-weight controls [91,92,93]. However, most published studies on circulating leptin levels in PCOS and control, stratified or not by BMI, have clearly demonstrated that circulating leptin correlated with the amount of body fat, being significantly increased in overweight and obese women, independently of the presence of PCOS [94]. The data suggest that leptin secretion is regulated by the amount of body fat both in normal and PCOS women. Elevated levels of leptin have also been suggested to be associated with lack of follicular maturation.
Adiponectin
The discovery of adiponectin occurred at about the same time as the discovery of leptin [95,96,97,98] but did not receive major attention until its marked protective role in the pathogenesis of obesity-related complications was acknowledged. Adiponectin is almost exclusively expressed in white mature adipocytes [99].
Although adiponectin is exclusively synthesized and secreted by adipocytes, adiponectin levels are decreased in obesity, in condition of IR and diabetes, in CVD and in dyslipidemia [100, 101]. Obesity also decreases expression level of adiponectin receptors and/or reduces postreceptor signalling, which may contribute to IR [99, 102]. Weight loss, on the other hand, results in increased adiponectin expression with accompanying improvement in insulin sensitivity [103]. The improvement in coronary circulation observed in obese women following weight loss was associated with adiponectin level [104]. Genetic studies have demonstrated a clear association with polymorphism resulting in hypoadiponectinemia with IR and CVD [105, 106]. Low level of adiponectin has been linked to inflammatory atherosclerosis in humans, suggesting that normal adiponectin levels are required to maintain a non-inflammatory phenotype of the vascular wall [107]. An association between hypertension and adiponectin levels has been reported [108,109,110,111]. Although the mechanism by which adiponectin levels are lowered in patients with hypertension remains to be clarified, hypoadiponectinemia has recently been proposed as a novel predictor of hypertension [112]. On the contrary, high plasma adiponectin levels are associated with lower risk of myocardial infarction and with lower risk of acute coronary syndrome [113, 114]. Recent findings have also suggested that adiponectin, possibly through AMPK activation, may limit the progression of myocardial hypertrophy [115]. Adiponectin is able to modulate the production of other adipokines [116] and is involved in the regulation of inflammation and atherosclerosis. In fact, monocyte adhesion to endothelial cells, macrophage transformation to foam cells and endothelial cell activation are down-regulated by adiponectin [117, 118]. Many factors with a significant impact on adiponectin regulation have inhibitory effects. These include catecholamines, glucocorticoids, interleukin 1, interleukin 6, TNFα, GH and androgens [53, 119, 120]. Insulin decreases adiponectin levels in humans [53], whereas thiazolidinediones increase the expression of adiponectin probably through interaction with PPAR-gamma response element and thereby may account for the insulin-sensitizing effect of such ligands [121]. Finally, adiponectin can also down-regulate its own production and the expression of its receptors [122].
The potential role of adiponectin in the ovulatory dysfunction and metabolic abnormalities in PCOS has been investigated in several studies but the available data are still not conclusive [91, 92, 94, 123, 124], although decreased levels of the adipokine in PCOS patients compared to weight-matched controls have been found in the majority of the studies [125]. Plasma adiponectin levels were found to be correlated with insulin sensitivity in PCOS [126, 127]. Other studies reported that serum adiponectin levels were reduced in obese women with PCOS and that BMI, testosterone concentrations and IR were the major determinants of reduced levels of adiponectin [128]. In addition to its role on insulin sensitivity, adiponectin may affect the reproductive system since it decreases LH secretion from the pituitary while stimulating estradiol secretion in human granulosa. In conclusion, adiponectin could be considered as a marker of IR in PCOS since adiponectin circulates in inverse proportion to the degree of IR. Reduced adiponectin levels appear to play a role in promoting IR, increasing triglycerides and small and dense low-density lipoprotein particles in these patients [129]. The role of hyperandrogenism on adiponectin secretion is not clear and opposite results have been published. However, total testosterone levels were not associated with adiponectin in a meta-analyses study [124]. On the contrary, increased insulin sensitivity was associated with increased adiponectin levels despite unchanged testosterone, and changes in testosterone levels did not modify adiponectin levels [123]. Adiponectin may also have effects on ovarian function since decreased local adiponectin expression stimulated hyperandrogenism, whereas adiponectin stimulation was associated with decreased ovarian androgen production [130]. Additional studies have demonstrated that insulin sensitizers such as metformin or pioglitazone [131], when PCOS patients were treated, both increased adiponectin levels and insulin-stimulated glucose disposal.
Therefore, adiponectin could be considered as a marker of IR also in PCOS, thus contributing to increase the cardiovascular risk in these patients.
Resistin
This adipokine, which is involved in IR, lipid metabolism and diabetes has been found to be higher in obese and non-obese Chinese PCOS women, suggesting that resistin may be implicated in the regulation of IR in this PCOS ethnic population [132]. Supporting these data, resistin levels were independently associated with insulin resistance and BMI in 66 patients with PCOS [133]. However, in another study from the same geographic area did not confirmed these data [134]. Thus, the exact role of resistin in women with PCOS is not clear, but could explain the increased IR found in PCOS.
Visfatin
Visfatin is produced by visceral adipose tissue in humans [135]. Studies have shown higher visfatin levels in IR [136]. PCOS women, as compared with control of comparable BMI, showed that this adipokine might be a marker of insulin sensibility. However, other studies did not confirm these data [137] since similar visfatin levels have been found in cross-sectional studies involving PCOS women and controls. On the contrary, a correlation of visfatin levels with proinflammatory markers has been described by other authors [137] suggesting a role of visfatin in endothelial dysfunction in these patients. A recent study showed that the increased visfatin level in women with PCOS may induce the expression of pro-angiogenic factors, thus favouring the development of endothelial dysfunction [138].
TNF-α
TNF-α was originally described to induce the necrosis of tumours after acute bacterial infection [139]. However, this cytokine is also involved in several immunological functions as well as in viral replication, septic shock and fever. TNF-α expression is elevated in adipose tissue and is associated with whole-body insulin resistance [140, 141]. Adipocytes also express both types of TNF-α receptors as membrane bound and soluble forms [142]. The effects of TNF-α on adipocytes include increased lipolysis, decreased adiponectin secretion, decreased glucose transporter type 4 expression, impaired insulin signalling by activation of serine kinases, and indirectly by increasing serum free fatty acids levels [143]. TNF-α is also involved in oxidative stress and mitochondrial dysfunction and endoplasmic reticulum stress [144, 145]. TNF-α also induces insulin resistance by suppressing the production of adiponectin from adipocytes [116].
Some studies found that women with PCOS had increased TNF-α levels in comparison to healthy controls [146, 147], but the results were not univocal [148, 149]. A recent meta-analysis showed that elevated TNF-α levels are directly related to the IR and androgen excess of PCOS, but not to BMI. This finding might suggest that high TNF-α level could promote IR and hyperandrogenism of PCOS [150].
Low-grade chronic inflammation in PCOS
Low-grade chronic inflammation in PCOS women is indicated by the presence of minor but significant elevated CRP levels and inflammatory cytokines. These proteins are linked to the development of metabolic dysfunction of PCOS, such as diabetes, cardiovascular risk factors. Chemokines have also been found to be high in visceral fat than in subcutaneous fat [151] and such findings might explain increased chemokines secretion in PCOS.
Insulin resistance
IR is present in 50–80% of PCOS women, but even PCOS women may have IR that is secondary to genetic disorders. More than 2% of PCOS have a risk of developing diabetes each year. Moreover, a significant increase in the prevalence of type 2 diabetes mellitus (T2D) as well as of impaired glucose tolerance has been found in small-sized studies as well as in large-sized studies in PCOS vs. control [152, 153]. BMI was found to have an important role in the development of diabetes. Weight gain was also associated with worsening of glucose tolerance and a 1% increase in BMI was found to lead to 2% increase in diabetes risk [154]. A family history of diabetes is less common in diabetic women with PCOS than in those without [155, 156]. These findings would suggest that T2D with and without PCOS is under different genetic controls. Furthermore, genetic studies have failed to find an association between genes known to influence the heritability of T2D with the presence of PCOS [157, 158]. The age of onset of diabetes has been reported to be earlier in women with PCOS compared with those without [156, 159, 160]. This would suggest that IR is present at an earlier age; a longer duration of T2D is significantly associated with a more severe phenotype with higher prevalence of micro- and macro-vascular complications in PCOS. IR is also a central pathophysiologic element of non-alcoholic fatty liver disease (NAFLD). Although the prevalence of NAFLD remains poorly defined due to variation of the characteristics of the studied populations, it is estimated to affect 20–30% of adults in the general population in developed countries [161, 162]. Women with PCOS have been shown to have a high risk of NAFLD [163, 164]. Moreover, obese women with PCOS have elevated aminotransferase levels that correlate with testosterone concentrations [165]. Obesity and IR are the main factors related to NAFLD in PCOS. Androgen excess, which is related to IR, may be an additional contributing factor to the development of NAFLD in the syndrome. Estimates of NAFLD prevalence range as high as 75% in PCOS patients with obesity and T2D [166]. Therefore, women with PCOS should be identified early and all available procedure aimed at weight loss together with pharmacological interventions should be used in order to improve metabolism and CVD risk factors.
The metabolic syndrome
The MS is a complex of interrelated risk factors for CVD and diabetes. Clinical definition of the MS has been developed in the last two decades with the purpose of identifying those individuals at increased risk of these diseases in order to put in place preventive measures that can reduce this risk [167,168,169]. These risk factors include impaired fasting plasma glucose, raised blood pressure, elevated triglyceride levels, low high-density lipoprotein cholesterol levels (HDL-C) and abdominal obesity, which is highly correlated with IR.
Following the World Health Organization (WHO) criteria of the syndrome [170], the other major criteria came from the National Cholesterol Education Program Adult Treatment Panel III (ATP III) [171] and the International Diabetes Federation (IDF), which proposed a new definition based on clinical criteria and designed for global application in clinical practice. Among criteria, greater emphasis was placed on visceral obesity as the core feature of the syndrome. Thus, visceral obesity measured by waist circumference was suggested to be an essential requirement for the diagnosis [172], although there is now agreement that abdominal obesity (IR is usually found in patients with visceral obesity) should not be a prerequisite for diagnosis of MS but it is one of five criteria, so that the presence of any three of five risk factors constitutes a diagnosis of MS. Criteria for the diagnosis of MS are as follows: elevated waist circumference, elevated triglycerides, reduced HDL-C elevated blood pressure and elevated fasting glucose. Once CVD or diabetes develops, the MS is often present, and the number of components of the MS contributes to disease progression and risk. MS has a complex pathogenesis and hormones might play a role in its development [173,174,175].
Among PCOS, an abnormal degree of IR is reported in about two-thirds [11]. Obesity prevalence is also similar, with considerable variability among populations [176]. The risk of MS in women with PCOS is double that of women of similar age and BMI without PCOS [176]. Furthermore, the risk of MS is particularly high in younger women with PCOS compared with women from the general population [177]. Moreover, obese and also thin women with PCOS often present an atherogenic lipid profile [178], together with other biochemical cardiovascular risk factors [179, 180]. The association between MS and PCOS is not causative and both syndromes often coexist. Women with PCOS have a higher prevalence of MS, whereas women with MS often present endocrine trait of PCOS. However, the high androgen levels of PCOS, by aggravating the visceral adiposity and perpetuating IR, are the main endocrine modulator of MS.
Conclusion
PCOS is not only a reproductive disorder but also a systemic metabolic condition that is associated with increased risk of diabetes across a woman’s life span. The manifestations of PCOS are diverse but in most cases, the severity of clinical symptoms is related to abnormal visceral obesity. This condition causes increased low-grade chronic inflammation which can be measured by increased levels of adipokines, chemokine and interleukins predisposing these women to an increased risk of CVD. PCOS women have IR that, by genetic disorders, is clearly associated with visceral obesity and by the adverse metabolic consequence of dysregulation of adipose tissue mass. In particular, adiponectin is down-regulated in visceral obesity, and low levels of adiponectin are associated with increased risk for IR and T2D. The available data also suggest that patients with PCOS can have 5–8 times increased diabetes risk compared to age- and weight-matched controls [46]. The IR in PCOS affects selectively metabolic actions while mitogenic and steroidogenic actions are usually preserved. Insulin may also act via the IGF-I receptor when insulin reaches high concentrations, and induces increased androgen production by the theca cells and synergically with LH. The available data clearly suggest that there are important connections between hyperandrogenism, hyperinsulinemia and VAT hypertrophy and dysregulation in PCOS. In conclusion, PCOS is associated to an increased CVD risk. This association is related to several factors, but IR should be considered an important variable in the pathogenesis of metabolic complications. Adipocyte dysfunction has a main role in determining IR and androgen excess that is characteristic of the syndrome. However, it is not clear whether the increased CVD risk described in PCOS women translates into increased frequency of actual event. Indeed, a recent study longitudinally analysed over 21,000 women with PCOS showing that there was no increased risk of cardiovascular and cerebrovascular events [154].
Therefore, patients with PCOS should be fully evaluated to determine baseline metabolic parameters and all available procedure aimed at weight loss together with pharmacological interventions should be used in order to improve metabolism and CVD risk factors.
References
Yildiz BO (2006) Diagnosis of hyperandrogenism: clinical criteria. Best Pract Res Clin Endocrinol Metab 20:167–176
Rotterdam EA-SPcwg (2004) Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 19:41–47
Zawadski JK (1992) Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif AGJ, Haseltine FP, Merriam GR (eds) polycystic ovary syndrome. Blackwell Scientific Publications, Boston, pp 377–384
Azziz R (2006) Controversy in clinical endocrinology: diagnosis of polycystic ovarian syndrome: the Rotterdam criteria are premature. J Clin Endocrinol Metab 91:781–785
Fulghesu AM, Angioni S, Frau E, Belosi C, Apa R, Mioni R et al (2007) Ultrasound in polycystic ovary syndrome—the measuring of ovarian stroma and relationship with circulating androgens: results of a multicentric study. Hum Reprod 22:2501–2508
Kalinin VL, Suslova IN, Suslov AV (1984) Mutagenic effects of gamma-rays on plasmid DNA in Escherichia coli. Radiobiologiia 24:599–602
DeUgarte CM, Bartolucci AA, Azziz R (2005) Prevalence of insulin resistance in the polycystic ovary syndrome using the homeostasis model assessment. Fertil Steril 83:1454–1460
Van Pelt RE, Jankowski CM, Gozansky WS, Schwartz RS, Kohrt WM (2005) Lower-body adiposity and metabolic protection in postmenopausal women. J Clin Endocrinol Metab 90:4573–4578
Wajchenberg BL (2000) Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev 21:697–738
Goodarzi MO, Azziz R (2006) Diagnosis, epidemiology, and genetics of the polycystic ovary syndrome. Best Pract Res Clin Endocrinol Metab 20:193–205
Rosenfield RL, Ehrmann DA (2016) The pathogenesis of polycystic ovary syndrome (PCOS): the hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocr Rev 37:467–520
Wickenheisser JK, Quinn PG, Nelson VL, Legro RS, Strauss JF 3rd, McAllister JM (2000) Differential activity of the cytochrome P450 17alpha-hydroxylase and steroidogenic acute regulatory protein gene promoters in normal and polycystic ovary syndrome theca cells. J Clin Endocrinol Metab 85:2304–2311
Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W et al (2009) The androgen excess and PCOS society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril 91:456–488
Hull MG (1987) Epidemiology of infertility and polycystic ovarian disease: endocrinological and demographic studies. Gynecol Endocrinol 1:235–245
Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS et al (1991) A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab 72:83–89
Ehrmann DA, Barnes RB, Rosenfield RL (1995) Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev 16:322–353
Diamanti-Kandarakis E, Dunaif A (2012) Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev 33:981–1030
Yildiz BO, Azziz R (2007) The adrenal and polycystic ovary syndrome. Rev Endocr Metab Disord 8:331–342
Rosenfield RL, Barnes RB, Ehrmann DA (1994) Studies of the nature of 17-hydroxyprogesterone hyperresonsiveness to gonadotropin-releasing hormone agonist challenge in functional ovarian hyperandrogenism. J Clin Endocrinol Metab 79:1686–1692
Nelson VL, Qin KN, Rosenfield RL, Wood JR, Penning TM, Legro RS et al (2001) The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab 86:5925–5933
Diamanti-Kandarakis E (2008) Polycystic ovarian syndrome: pathophysiology, molecular aspects and clinical implications. Expert Rev Mol Med 10:e3
De Leo V, Musacchio MC, Cappelli V, Massaro MG, Morgante G, Petraglia F (2016) Genetic, hormonal and metabolic aspects of PCOS: an update. Reprod Biol Endocrinol 14:38
Kalro BN, Loucks TL, Berga SL (2001) Neuromodulation in polycystic ovary syndrome. Obstet Gynecol Clin N Am 28:35–62
Dunaif A, Wu X, Lee A, Diamanti-Kandarakis E (2001) Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS). Am J Physiol Endocrinol Metab 281:E392–E399
Ciaraldi TP, el-Roeiy A, Madar Z, Reichart D, Olefsky JM, Yen SS (1992) Cellular mechanisms of insulin resistance in polycystic ovarian syndrome. J Clin Endocrinol Metab 75:577–583
Bremer AA, Miller WL (2008) The serine phosphorylation hypothesis of polycystic ovary syndrome: a unifying mechanism for hyperandrogenemia and insulin resistance. Fertil Steril 89:1039–1048
Ciaraldi TP, Aroda V, Mudaliar S, Chang RJ, Henry RR (2009) Polycystic ovary syndrome is associated with tissue-specific differences in insulin resistance. J Clin Endocrinol Metab 94:157–163
De Leo V, la Marca A, Petraglia F (2003) Insulin-lowering agents in the management of polycystic ovary syndrome. Endocr Rev 24:633–667
Legro RS (1999) Polycystic ovary syndrome. Phenotype to genotype. Endocrinol Metab Clin N Am 28:379–396
Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J (1999) Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care 22:141–146
Kiddy DS, Hamilton-Fairley D, Bush A, Short F, Anyaoku V, Reed MJ et al (1992) Improvement in endocrine and ovarian function during dietary treatment of obese women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 36:105–111
Carmina E (2003) Genetic and environmental aspect of polycystic ovary syndrome. J Endocrinol Investig 26:1151–1159
Carmina E, Azziz R (2006) Diagnosis, phenotype, and prevalence of polycystic ovary syndrome. Fertil Steril 86(Suppl 1):S7–S8
Gambineri A, Pelusi C, Vicennati V, Pagotto U, Pasquali R (2002) Obesity and the polycystic ovary syndrome. Int J Obes Relat Metab Disord 26:883–896
Barber TM, Golding SJ, Alvey C, Wass JA, Karpe F, Franks S et al (2008) Global adiposity rather than abnormal regional fat distribution characterizes women with polycystic ovary syndrome. J Clin Endocrinol Metab 93:999–1004
Pasquali R, Gambineri A (2006) Polycystic ovary syndrome: a multifaceted disease from adolescence to adult age. Ann N Y Acad Sci 1092:158–174
Winters SJ, Talbott E, Guzick DS, Zborowski J, McHugh KP (2000) Serum testosterone levels decrease in middle age in women with the polycystic ovary syndrome. Fertil Steril 73:724–729
Birdsall MA, Farquhar CM (1996) Polycystic ovaries in pre and post-menopausal women. Clin Endocrinol (Oxf) 44:269–276
Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C, American Heart A et al (2004) Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 109:433–438
Despres JP, Lemieux I (2006) Abdominal obesity and metabolic syndrome. Nature 444:881–887
Weyer C, Hanson RL, Tataranni PA, Bogardus C, Pratley RE (2000) A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 49:2094–2101
Leonhardt W, Hanefeld M, Schneider H, Haller H (1972) Human adipocyte volumes: maximum size, and correlation to weight index in maturity onset-diabetes. Diabetologia 8:287–291
Kirchengast S, Huber J (2001) Body composition characteristics and body fat distribution in lean women with polycystic ovary syndrome. Hum Reprod 16:1255–1260
Horejsi R, Moller R, Rackl S, Giuliani A, Freytag U, Crailsheim K et al (2004) Android subcutaneous adipose tissue topography in lean and obese women suffering from PCOS: comparison with type 2 diabetic women. Am J Phys Anthropol 124:275–281
Cupisti S, Kajaia N, Dittrich R, Duezenli H, Beckmann MW, Mueller A (2008) Body mass index and ovarian function are associated with endocrine and metabolic abnormalities in women with hyperandrogenic syndrome. Eur J Endocrinol 158:711–719
Legro RS, Kunselman AR, Dodson WC, Dunaif A (1999) Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab 84:165–169
Glintborg D, Henriksen JE, Andersen M, Hagen C, Hangaard J, Rasmussen PE et al (2004) Prevalence of endocrine diseases and abnormal glucose tolerance tests in 340 Caucasian premenopausal women with hirsutism as the referral diagnosis. Fertil Steril 82:1570–1579
Rosen ED, Spiegelman BM (2014) What we talk about when we talk about fat. Cell 156:20–44
Ek I, Arner P, Ryden M, Holm C, Thorne A, Hoffstedt J et al (2002) A unique defect in the regulation of visceral fat cell lipolysis in the polycystic ovary syndrome as an early link to insulin resistance. Diabetes 51:484–492
Arner P, Hellstrom L, Wahrenberg H, Bronnegard M (1990) Beta-adrenoceptor expression in human fat cells from different regions. J Clin Investig 86:1595–1600
Lonnqvist F, Krief S, Strosberg AD, Nyberg S, Emorine LJ, Arner P (1993) Evidence for a functional beta 3-adrenoceptor in man. Br J Pharmacol 110:929–936
Boden G (1998) Free fatty acids (FFA), a link between obesity and insulin resistance. Front Biosci 3:d169–d175
Fasshauer M, Paschke R (2003) Regulation of adipocytokines and insulin resistance. Diabetologia 46:1594–1603
Fain JN (2006) Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm 74:443–477
Belanger C, Luu-The V, Dupont P, Tchernof A (2002) Adipose tissue intracrinology: potential importance of local androgen/estrogen metabolism in the regulation of adiposity. Horm Metab Res 34:737–745
Meseguer A, Puche C, Cabero A (2002) Sex steroid biosynthesis in white adipose tissue. Horm Metab Res 34:731–736
Seckl JR, Walker BR (2004) 11beta-hydroxysteroid dehydrogenase type 1 as a modulator of glucocorticoid action: from metabolism to memory. Trends Endocrinol Metab 15:418–424
Lundgren M, Buren J, Ruge T, Myrnas T, Eriksson JW (2004) Glucocorticoids down-regulate glucose uptake capacity and insulin-signaling proteins in omental but not subcutaneous human adipocytes. J Clin Endocrinol Metab 89:2989–2997
Michael AE, Evagelatou M, Norgate DP, Clarke RJ, Antoniw JW, Stedman BA et al (1997) Isoforms of 11beta-hydroxysteroid dehydrogenase in human granulosa-lutein cells. Mol Cell Endocrinol 132:43–52
Wake DJ, Rask E, Livingstone DE, Soderberg S, Olsson T, Walker BR (2003) Local and systemic impact of transcriptional up-regulation of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue in human obesity. J Clin Endocrinol Metab 88:3983–3988
Valsamakis G, Anwar A, Tomlinson JW, Shackleton CH, McTernan PG, Chetty R et al (2004) 11beta-hydroxysteroid dehydrogenase type 1 activity in lean and obese males with type 2 diabetes mellitus. J Clin Endocrinol Metab 89:4755–4761
Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR et al (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294:2166–2170
Rask E, Walker BR, Soderberg S, Livingstone DE, Eliasson M, Johnson O et al (2002) Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11beta-hydroxysteroid dehydrogenase type 1 activity. J Clin Endocrinol Metab 87:3330–3336
Fanciulli G, Delitala A, Delitala G (2009) Growth hormone, menopause and ageing: no definite evidence for ‘rejuvenation’ with growth hormone. Hum Reprod Update 15:341–358
Brummer RJ, Lonn L, Kvist H, Grangard U, Bengtsson BA, Sjostrom L (1993) Adipose tissue and muscle volume determination by computed tomography in acromegaly, before and 1 year after adenomectomy. Eur J Clin Investig 23:199–205
Bjorntorp P (1997) Obesity. Lancet 350:423–426
Beauville M, Harant I, Crampes F, Riviere D, Tauber MT, Tauber JP et al (1992) Effect of long-term rhGH administration in GH-deficient adults on fat cell epinephrine response. Am J Physiol 263:E467–E472
Glass AR (1989) Endocrine aspects of obesity. Med Clin N Am 73:139–160
Rudman D (1985) Growth hormone, body composition, and aging. J Am Geriatr Soc 33:800–807
Vahl N, Moller N, Lauritzen T, Christiansen JS, Jorgensen JO (1997) Metabolic effects and pharmacokinetics of a growth hormone pulse in healthy adults: relation to age, sex, and body composition. J Clin Endocrinol Metab 82:3612–3618
Clasey JL, Weltman A, Patrie J, Weltman JY, Pezzoli S, Bouchard C et al (2001) Abdominal visceral fat and fasting insulin are important predictors of 24-hour GH release independent of age, gender, and other physiological factors. J Clin Endocrinol Metab 86:3845–3852
Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW (2004) Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 145:2273–2282
Friedman JM, Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 395:763–770
Zemel MB (1998) Agouti/melanocortin interactions with leptin pathways in obesity. Nutr Rev 56:271–274
Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL (2002) Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J Clin Endocrinol Metab 87:2391–2394
Bjorbaek C, Kahn BB (2004) Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res 59:305–331
Flier JS (2004) Obesity wars: molecular progress confronts an expanding epidemic. Cell 116:337–350
Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T et al (1999) Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA 282:1568–1575
Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH et al (1996) Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet 348:159–161
Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS (1999) The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem 274:30059–30065
Capobianco G, de Muro P, Cherchi GM, Formato M, Lepedda AJ, Cigliano A et al (2010) Plasma levels of C-reactive protein, leptin and glycosaminoglycans during spontaneous menstrual cycle: differences between ovulatory and anovulatory cycles. Arch Gynecol Obstet 282:207–213
Koerner A, Kratzsch J, Kiess W (2005) Adipocytokines: leptin—the classical, resistin—the controversical, adiponectin—the promising, and more to come. Best Pract Res Clin Endocrinol Metab 19:525–546
Beltowski J (2006) Role of leptin in blood pressure regulation and arterial hypertension. J Hypertens 24:789–801
Karaduman M, Oktenli C, Musabak U, Sengul A, Yesilova Z, Cingoz F et al (2006) Leptin, soluble interleukin-6 receptor, C-reactive protein and soluble vascular cell adhesion molecule-1 levels in human coronary atherosclerotic plaque. Clin Exp Immunol 143:452–457
Beltowski J (2006) Leptin and atherosclerosis. Atherosclerosis 189:47–60
Singh P, Hoffmann M, Wolk R, Shamsuzzaman AS, Somers VK (2007) Leptin induces C-reactive protein expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol 27:e302–e307
Romero-Corral A, Sierra-Johnson J, Lopez-Jimenez F, Thomas RJ, Singh P, Hoffmann M et al (2008) Relationships between leptin and C-reactive protein with cardiovascular disease in the adult general population. Nat Clin Pract Cardiovasc Med 5:418–425
Wallace AM, McMahon AD, Packard CJ, Kelly A, Shepherd J, Gaw A et al (2001) Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation 104:3052–3056
Licinio J, Caglayan S, Ozata M, Yildiz BO, de Miranda PB, O’Kirwan F et al (2004) Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc Natl Acad Sci USA 101:4531–4536
Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P et al (2002) Leptin-replacement therapy for lipodystrophy. N Engl J Med 346:570–578
Lecke SB, Morsch DM, Spritzer PM (2011) Leptin and adiponectin in the female life course. Braz J Med Biol Res 44:381–387
Lecke SB, Morsch DM, Spritzer PM (2013) Association between adipose tissue expression and serum levels of leptin and adiponectin in women with polycystic ovary syndrome. Genet Mol Res 12:4292–4296
Spritzer PM, Comim FV, Capp E, D’Avila A (2005) Influence of leptin, androgens and insulin sensitivity on increased GH response to clonidine in lean patients with polycystic ovary syndrome. Horm Metab Res 37:94–98
Tan BK, Chen J, Hu J, Amar O, Mattu HS, Adya R et al (2013) Metformin increases the novel adipokine cartonectin/CTRP3 in women with polycystic ovary syndrome. J Clin Endocrinol Metab 98:E1891–E1900
Nakano Y, Tobe T, Choi-Miura NH, Mazda T, Tomita M (1996) Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J Biochem 120:803–812
Hu E, Liang P, Spiegelman BM (1996) AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem 271:10697–10703
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J et al (1999) Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79–83
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF (1995) A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749
Kadowaki T, Yamauchi T (2005) Adiponectin and adiponectin receptors. Endocr Rev 26:439–451
Matsuzawa Y, Funahashi T, Kihara S, Shimomura I (2004) Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol 24:29–33
Trujillo ME, Scherer PE (2006) Adipose tissue-derived factors: impact on health and disease. Endocr Rev 27:762–778
Chen MB, McAinch AJ, Macaulay SL, Castelli LA, O’Brien PE, Dixon JB et al (2005) Impaired activation of AMP-kinase and fatty acid oxidation by globular adiponectin in cultured human skeletal muscle of obese type 2 diabetics. J Clin Endocrinol Metab 90:3665–3672
Bottner A, Kratzsch J, Muller G, Kapellen TM, Bluher S, Keller E et al (2004) Gender differences of adiponectin levels develop during the progression of puberty and are related to serum androgen levels. J Clin Endocrinol Metab 89:4053–4061
Coppola A, Marfella R, Coppola L, Tagliamonte E, Fontana D, Liguori E et al (2009) Effect of weight loss on coronary circulation and adiponectin levels in obese women. Int J Cardiol 134:414–416
Stumvoll M, Tschritter O, Fritsche A, Staiger H, Renn W, Weisser M et al (2002) Association of the T-G polymorphism in adiponectin (exon 2) with obesity and insulin sensitivity: interaction with family history of type 2 diabetes. Diabetes 51:37–41
Vasseur F, Lepretre F, Lacquemant C, Froguel P (2003) The genetics of adiponectin. Curr Diab Rep 3:151–158
Lago F, Dieguez C, Gomez-Reino J, Gualillo O (2007) The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev 18:313–325
Kazumi T, Kawaguchi A, Sakai K, Hirano T, Yoshino G (2002) Young men with high-normal blood pressure have lower serum adiponectin, smaller LDL size, and higher elevated heart rate than those with optimal blood pressure. Diabetes Care 25:971–976
Furuhashi M, Ura N, Higashiura K, Miyazaki Y, Murakami H, Hyakukoku M et al (2005) Low adiponectin level in young normotensive men with a family history of essential hypertension. Hypertens Res 28:141–146
Lee HS, Lee M, Joung H (2007) Adiponectin represents an independent risk factor for hypertension in middle aged Korean women. Asia Pac J Clin Nutr 16:10–15
Chow WS, Cheung BM, Tso AW, Xu A, Wat NM, Fong CH et al (2007) Hypoadiponectinemia as a predictor for the development of hypertension: a 5-year prospective study. Hypertension 49:1455–1461
Imatoh T, Miyazaki M, Momose Y, Tanihara S, Une H (2008) Adiponectin levels associated with the development of hypertension: a prospective study. Hypertens Res 31:229–233
Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB (2004) Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 291:1730–1737
Wolk R, Berger P, Lennon RJ, Brilakis ES, Davison DE, Somers VK (2007) Association between plasma adiponectin levels and unstable coronary syndromes. Eur Heart J 28:292–298
Ouchi N, Shibata R, Walsh K (2006) Cardioprotection by adiponectin. Trends Cardiovasc Med 16:141–146
Degawa-Yamauchi M, Moss KA, Bovenkerk JE, Shankar SS, Morrison CL, Lelliott CJ et al (2005) Regulation of adiponectin expression in human adipocytes: effects of adiposity, glucocorticoids, and tumor necrosis factor alpha. Obes Res 13:662–669
Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H et al (2000) Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 102:1296–1301
Sell H, Dietze-Schroeder D, Eckardt K, Eckel J (2006) Cytokine secretion by human adipocytes is differentially regulated by adiponectin, AICAR, and troglitazone. Biochem Biophys Res Commun 343:700–706
Nilsson L, Binart N, Bohlooly YM, Bramnert M, Egecioglu E, Kindblom J et al (2005) Prolactin and growth hormone regulate adiponectin secretion and receptor expression in adipose tissue. Biochem Biophys Res Commun 331:1120–1126
Xu A, Chan KW, Hoo RL, Wang Y, Tan KC, Zhang J et al (2005) Testosterone selectively reduces the high molecular weight form of adiponectin by inhibiting its secretion from adipocytes. J Biol Chem 280:18073–18080
Combs TP, Wagner JA, Berger J, Doebber T, Wang WJ, Zhang BB et al (2002) Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology 143:998–1007
Bauche IB, Ait El Mkadem S, Rezsohazy R, Funahashi T, Maeda N, Miranda LM et al (2006) Adiponectin downregulates its own production and the expression of its AdipoR2 receptor in transgenic mice. Biochem Biophys Res Commun 345:1414–1424
Glintborg D, Stoving RK, Hagen C, Hermann AP, Frystyk J, Veldhuis JD et al (2005) Pioglitazone treatment increases spontaneous growth hormone (GH) secretion and stimulated GH levels in polycystic ovary syndrome. J Clin Endocrinol Metab 90:5605–5612
Toulis KA, Goulis DG, Farmakiotis D, Georgopoulos NA, Katsikis I, Tarlatzis BC et al (2009) Adiponectin levels in women with polycystic ovary syndrome: a systematic review and a meta-analysis. Hum Reprod Update 15:297–307
Li S, Huang X, Zhong H, Peng Q, Chen S, Xie Y et al (2014) Low circulating adiponectin levels in women with polycystic ovary syndrome: an updated meta-analysis. Tumour Biol 35:3961–3973
Spranger J, Mohlig M, Wegewitz U, Ristow M, Pfeiffer AF, Schill T et al (2004) Adiponectin is independently associated with insulin sensitivity in women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 61:738–746
Koleva DI, Orbetzova MM, Nikolova JG, Tyutyundzhiev SB (2016) Adipokines and soluble cell adhesion molecules in insulin resistant and non-insulin resistant women with polycystic ovary syndrome. Arch Physiol Biochem 122:223–227
Pangaribuan B, Yusuf I, Mansyur M, Wijaya A (2011) Serum adiponectin and resistin in relation to insulin resistance and markers of hyperandrogenism in lean and obese women with polycystic ovary syndrome. Ther Adv Endocrinol Metab 2:235–245
Tan BK, Chen J, Digby JE, Keay SD, Kennedy CR, Randeva HS (2006) Upregulation of adiponectin receptor 1 and 2 mRNA and protein in adipose tissue and adipocytes in insulin-resistant women with polycystic ovary syndrome. Diabetologia 49:2723–2728
Comim FV, Hardy K, Franks S (2013) Adiponectin and its receptors in the ovary: further evidence for a link between obesity and hyperandrogenism in polycystic ovary syndrome. PLoS One 8:e80416
Ibanez L, Valls C, Marcos MV, Ong K, Dunger DB, De Zegher F (2004) Insulin sensitization for girls with precocious pubarche and with risk for polycystic ovary syndrome: effects of prepubertal initiation and postpubertal discontinuation of metformin treatment. J Clin Endocrinol Metab 89:4331–4337
Wang Y, Xie X, Zhu W (2010) Serum adiponectin and resistin levels in patients with polycystic ovarian syndrome and their clinical implications. J Huazhong Univ Sci Technol Med Sci 30:638–642
Yilmaz M, Bukan N, Demirci H, Ozturk C, Kan E, Ayvaz G et al (2009) Serum resistin and adiponectin levels in women with polycystic ovary syndrome. Gynecol Endocrinol 25:246–252
Arikan S, Bahceci M, Tuzcu A, Kale E, Gokalp D (2010) Serum resistin and adiponectin levels in young non-obese women with polycystic ovary syndrome. Gynecol Endocrinol 26:161–166
Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K et al (2005) Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307:426–430
Plati E, Kouskouni E, Malamitsi-Puchner A, Boutsikou M, Kaparos G, Baka S (2010) Visfatin and leptin levels in women with polycystic ovaries undergoing ovarian stimulation. Fertil Steril 94:1451–1456
Lajunen TK, Purhonen AK, Haapea M, Ruokonen A, Puukka K, Hartikainen AL et al (2012) Full-length visfatin levels are associated with inflammation in women with polycystic ovary syndrome. Eur J Clin Investig 42:321–328
Dambala K, Vavilis D, Bili E, Goulis DG, Tarlatzis BC (2017) Serum visfatin, vascular endothelial growth factor and matrix metalloproteinase-9 in women with polycystic ovary syndrome. Gynecol Endocrinol 16:1–5. doi:10.1080/09513590.2017.1296425
Coppack SW (2001) Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc 60:349–356
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259:87–91
Katsuki A, Sumida Y, Murashima S, Murata K, Takarada Y, Ito K et al (1998) Serum levels of tumor necrosis factor-alpha are increased in obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 83:859–862
Ruan H, Lodish HF (2003) Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev 14:447–455
Juge-Aubry CE, Somm E, Pernin A, Alizadeh N, Giusti V, Dayer JM et al (2005) Adipose tissue is a regulated source of interleukin-10. Cytokine 29:270–274
Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M et al (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106:2067–2072
Dahlman I, Forsgren M, Sjogren A, Nordstrom EA, Kaaman M, Naslund E et al (2006) Downregulation of electron transport chain genes in visceral adipose tissue in type 2 diabetes independent of obesity and possibly involving tumor necrosis factor-alpha. Diabetes 55:1792–1799
Sayin NC, Gucer F, Balkanli-Kaplan P, Yuce MA, Ciftci S, Kucuk M et al (2003) Elevated serum TNF-alpha levels in normal-weight women with polycystic ovaries or the polycystic ovary syndrome. J Reprod Med 48:165–170
Araya AV, Aguirre A, Romero C, Miranda C, Molina MC, Ferreira A (2002) Evaluation of tumor necrosis factor alpha production in ex vivo short term cultured whole blood from women with polycystic ovary syndrome. Eur Cytokine Netw 13:419–424
Olszanecka-Glinianowicz M, Banas M, Zahorska-Markiewicz B, Janowska J, Kocelak P, Madej P et al (2007) Is the polycystic ovary syndrome associated with chronic inflammation per se? Eur J Obstet Gynecol Reprod Biol 133:197–202
Gonzalez F, Thusu K, Abdel-Rahman E, Prabhala A, Tomani M, Dandona P (1999) Elevated serum levels of tumor necrosis factor alpha in normal-weight women with polycystic ovary syndrome. Metabolism 48:437–441
Gao L, Gu Y, Yin X (2016) High serum tumor necrosis factor-alpha levels in women with polycystic ovary syndrome: a meta-analysis. PLoS One 11:e0164021
Glintborg D, Andersen M, Richelsen B, Bruun JM (2009) Plasma monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1alpha are increased in patients with polycystic ovary syndrome (PCOS) and associated with adiposity, but unaffected by pioglitazone treatment. Clin Endocrinol (Oxf) 71:652–658
Legro RS, Gnatuk CL, Kunselman AR, Dunaif A (2005) Changes in glucose tolerance over time in women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab 90:3236–3242
Norman RJ, Masters L, Milner CR, Wang JX, Davies MJ (2001) Relative risk of conversion from normoglycaemia to impaired glucose tolerance or non-insulin dependent diabetes mellitus in polycystic ovarian syndrome. Hum Reprod 16:1995–1998
Morgan CL, Jenkins-Jones S, Currie CJ, Rees DA (2012) Evaluation of adverse outcome in young women with polycystic ovary syndrome versus matched, reference controls: a retrospective, observational study. J Clin Endocrinol Metab 97:3251–3260
Mirzaei F, Kazemi N (2008) Prevalence of polycystic ovary syndrome in women with type 2 diabetes in Kerman, Iran. Metab Syndr Relat Disord 6:215–217
Sim SY, Chin SL, Tan JL, Brown SJ, Cussons AJ, Stuckey BG (2016) Polycystic ovary syndrome in type 2 diabetes: does it predict a more severe phenotype? Fertil Steril 106:1258–1263
Mendoza N (2011) Common genetic aspects between polycystic ovary syndrome and diabetes mellitus. Curr Diabetes Rev 7:377–391
Saxena R, Welt CK (2013) Polycystic ovary syndrome is not associated with genetic variants that mark risk of type 2 diabetes. Acta Diabetol 50:451–457
Peppard HR, Marfori J, Iuorno MJ, Nestler JE (2001) Prevalence of polycystic ovary syndrome among premenopausal women with type 2 diabetes. Diabetes Care 24:1050–1052
Kelestimur F, Unluhizarci K, Baybuga H, Atmaca H, Bayram F, Sahin Y (2006) Prevalence of polycystic ovarian changes and polycystic ovary syndrome in premenopausal women with treated type 2 diabetes mellitus. Fertil Steril 86:405–410
Clark JM, Diehl AM (2003) Nonalcoholic fatty liver disease: an underrecognized cause of cryptogenic cirrhosis. JAMA 289:3000–3004
Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S (2005) Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 42:44–52
Brzozowska MM, Ostapowicz G, Weltman MD (2009) An association between non-alcoholic fatty liver disease and polycystic ovarian syndrome. J Gastroenterol Hepatol 24:243–247
Setji TL, Holland ND, Sanders LL, Pereira KC, Diehl AM, Brown AJ (2006) Nonalcoholic steatohepatitis and nonalcoholic fatty liver disease in young women with polycystic ovary syndrome. J Clin Endocrinol Metab 91:1741–1747
Economou F, Xyrafis X, Livadas S, Androulakis II, Argyrakopoulou G, Christakou CD et al (2009) In overweight/obese but not in normal-weight women, polycystic ovary syndrome is associated with elevated liver enzymes compared to controls. Hormones (Athens) 8:199–206
Rector RS, Thyfault JP, Wei Y, Ibdah JA (2008) Non-alcoholic fatty liver disease and the metabolic syndrome: an update. World J Gastroenterol 14:185–192
Expert Panel on Detection E, Treatment of High Blood Cholesterol in A (2001) Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol In adults (adult treatment panel III). JAMA 285:2486–2497
Alberti KG (2007) Screening and diagnosis of prediabetes: where are we headed? Diabetes Obes Metab 9(Suppl 1):12–16
Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA et al (2009) Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120:1640–1645
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15:539–553
National Cholesterol Education Program Expert Panel on Detection E, Treatment of High Blood Cholesterol in A (2002) Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) final report. Circulation 106:3143–3421
Alberti KG, Zimmet P, Shaw J, Group IDFETFC (2005) The metabolic syndrome—a new worldwide definition. Lancet 366:1059–1062
Delitala AP, Fanciulli G, Pes GM, Maioli M, Delitala G (2017) Thyroid hormones, metabolic syndrome and its components. Endocr Metab Immune Disord Drug Targets. doi:10.2174/1871530317666170320105221
Niafar M, Pourafkari L, Porhomayon J, Nader N (2016) A systematic review of GLP-1 agonists on the metabolic syndrome in women with polycystic ovaries. Arch Gynecol Obstet 293:509–515
Fernandez-Miro M, Chillaron JJ, Pedro-Botet J (2016) Testosterone deficiency, metabolic syndrome and diabetes mellitus. Med Clin (Barc) 146:69–73
Ehrmann DA, Liljenquist DR, Kasza K, Azziz R, Legro RS, Ghazzi MN et al (2006) Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab 91:48–53
Coviello AD, Legro RS, Dunaif A (2006) Adolescent girls with polycystic ovary syndrome have an increased risk of the metabolic syndrome associated with increasing androgen levels independent of obesity and insulin resistance. J Clin Endocrinol Metab 91:492–497
Diamanti-Kandarakis E, Papavassiliou AG, Kandarakis SA, Chrousos GP (2007) Pathophysiology and types of dyslipidemia in PCOS. Trends Endocrinol Metab 18:280–285
Diamanti-Kandarakis E, Alexandraki K, Bergiele A, Kandarakis H, Mastorakos G, Aessopos A (2004) Presence of metabolic risk factors in non-obese PCOS sisters: evidence of heritability of insulin resistance. J Endocrinol Invest 27:931–936
Diamanti-Kandarakis E, Paterakis T, Kandarakis HA (2006) Indices of low-grade inflammation in polycystic ovary syndrome. Ann N Y Acad Sci 1092:175–186
Acknowledgements
Ph. Doctor School in Biomedical Sciences, Address in Gender Medicine, Men, Woman and Child, Sassari University, Italy, supported the study.
Author information
Authors and Affiliations
Contributions
APD: wrote the manuscript. GC: reviewed and edited the manuscript. GD: reviewed and edited the manuscript. PLC: reviewed and edited the manuscript. SD: reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Funding
Nothing to declare.
Conflict of interest
Alessandro P. Delitala declares that he has no conflict of interest. Giampiero Capobianco declares that he has no conflict of interest. Giuseppe Delitala declares that he has no conflict of interest. Pier Luigi Cherchi declares that he has no conflict of interest. Salvatore Dessole declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Delitala, A.P., Capobianco, G., Delitala, G. et al. Polycystic ovary syndrome, adipose tissue and metabolic syndrome. Arch Gynecol Obstet 296, 405–419 (2017). https://doi.org/10.1007/s00404-017-4429-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00404-017-4429-2