
Abstract
Porokeratosis is a chronic skin disorder characterized by the presence of patches with elevated, thick, keratotic borders, with histological cornoid lamella. Classic porokeratosis of Mibelli (PM) frequently appears in childhood with a risk of malignant transformation. Disseminated superficial actinic porokeratosis (DSAP) is the most common subtype of porokeratosis with genetic heterogeneities, and mevalonate kinase gene (MVK) mutations have been identified in minor portion of DSAP families of Chinese origin. To confirm the previous findings about MVK mutations in DSAP patients and test MVK’s role(s) in PM development, we performed genomic sequence analysis for 3 DSAP families and 1 PM family of Chinese origin. We identified a splicing mutation of MVK gene, designated as c.1039+1G>A, in the PM family. No MVK mutations were found in three DSAP families. Sequence analysis for complementary DNA templates from PM lesions of all patients revealed a mutation at splice donor site of intron 10, designated as c.1039+1G>A, leading to the splicing defect and termination codon 52 amino acids after exon 10. Although no MVK mutations in DSAP patients were found as reported previously, we identified MVK simultaneously responsible for PM development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porokeratosis is a chronic skin disorder characterized by the presence of patches with elevated, thick, keratotic borders histologically featuring cornoid lamella [23]. Classically, five clinical subtypes of porokeratosis are recognized [2]: porokeratosis of Mibelli (PM), disseminated superficial porokeratosis (DSP), disseminated superficial actinic porokeratosis (DSAP), porokeratosis palmaris et plantaris disseminate (PPPD) and linear porokeratosis (LP). Porokeratosis lesions, especially large and linear lesions, can be associated with squamous cell carcinoma [15, 17, 24, 31].
PM was originally described in 1893. PM can present as a single plaque or a small number of plaques of up to 20 cm in diameter. Lesions are usually located unilaterally on limbs, although other parts of the body may be affected (e.g., palms, soles, lips, genitals or mucous membranes). PM frequently appears in childhood—especially if it is hereditary—and occurs with a higher incidence in males. Linkage analysis mapped the disease gene of PM on chromosome 3p, but no mutations have been detected. The most common subtype of porokeratosis, DSAP, is an autosomal dominant keratinization disorder with genetic heterogeneity characterized by multiple superficial keratotic lesions surrounded by a slightly raised keratotic border [19]. Generally, the lesions begin to develop in adolescents and reach near-complete penetrance by the third or fourth decade of life [34].
MVK, located on 12q24, comprises ten coding exons and one non-coding exon spanning over 21 kb [32]. Previous studies indicated that MVK mutations are associated with Hyper-IgD and periodic fever syndrome [1, 4, 5–12, 14, 20, 21, 25–27, 29, 30], mevalonic aciduria [10], neonatal-onset chronic hepatitis and other significant liver disease [13, 16, 28], mevalonate kinase deficiency and dyserythropoietic anemia [22]. Recently, Zhang et al. [35] reported MVK mutations in 33 % familiar and 16 % sporadic patients with DSAP by exome sequencing. Previous linkage analysis identified five chromosomal regions (12q23.2–24.1, 12q24.1–q24.2, 15q25.1–26.1, 1p31.3–p31.1 and 16q24.1–24.3) harboring potential genetic risks for DSAP. The low frequency of MVK mutations in DSAP patients suggested that other underlying causative genes or environmental factors may affect the disease developments.
Materials and methods
Subjects
Three families with porokeratosis consisting of 24 affected and 48 unaffected individuals were identified through probands, respectively, from Guangdong province, Fujian province and the Inner Mongolia Autonomous Region in China (Fig. 1). All family members were carefully examined by at least two dermatologists. The diagnosis was confirmed by histological examination of skin biopsy specimens. The diagnosis of HIDS and Mevalonic aciduria was excluded based on the lack of recognizable symptoms and normal serum IgD levels. Genetic epidemiological surveys to all family members were performed and informational data are described in Table 1. After written informed consent was obtained, blood and skin biopsies were collected from available family individuals and 100 unrelated, unaffected controls. The study was carried out in accordance with the Declaration of Helsinki. This study was approved by the ethical committee of Southern Medical University Review Board.

Chinese DSAP Pedigrees. The left below arrow indicates the proband; affected and unaffected individuals are, respectively, represented by black and open symbols; circles and squares, respectively, indicated females and males. The a, b, c represent the degrees of family 1, 2 and 3, respectively
Skin samples
The lesional and non-lesional skin samples from patients in each family were obtained. The healthy skin samples were obtained from human foreskin or of non-DSAP/PM patients undergoing surgeries. Each skin sample was divided into two parts as follows: one part was directly frozen in liquid nitrogen, and another part was formalin-fixed and paraffin-embedded for section.
Polymerase chain reaction (PCR) and sequencing
The molecular genetic analysis of MVK gene was carried out at the central laboratory of Nanfang Hospital of the Southern Medical University in Guangzhou, China. Genomic DNAs were extracted from peripheral blood using the E.Z.N.A.™ Blood DNA Kit (Omega, Norcross, GA, Cat. No. D3392). All coding exons of the NF1 gene together with boundary exon–intron sequences were amplified using published primers [33]. PCR was carried out in a 25 uL total volume, containing 20 ng genomic DNA, 10 mM Tris–HCl (pH 8.3), 50 mM KCl, 3.0 mM MgCl2, 0.01 % gelatine, 0.2 mM dNTPs, 10 pmol of each primer, and 0.75 U HotStarsTaq (QIAgene, Germany). The PCR program was set as below: HotStarsTaq activation at 94° for 5 min, followed by 38 cycles, each having denaturation at 94 °C for 30 s, annealing at 54° for 30 s and extension at 72 °C for 45 s, except that in the first 12 cycles the annealing temperature decreased from 60° to 54° by 0.5 °C per cycle, and the final extension was 72 °C for 7 min. PCR products were directly sequenced on an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA). Direct sequencing of the PCR products was performed with a BigDye Direct Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and analyzed on the ABI 3130 genetic analyzer (Applied Biosystems, USA). The new variants were then analyzed in 200 normal chromosomes regarding the possibility of polymorphisms.
cDNA sequence analysis
Total RNA of lesional skin tissues from three patients in family 1 (II:7 and III:1 and III:3) and unaffected skins from healthy controls was isolated using TRIzol from Invitrogen. One microgram of cellular RNA was used to conduct reverse transcription with a Takara RT kit (6210A). Seven pairs of primers (shown in Table 1) were designed using the online Primer 3 (http://frodo.wi.mit.edu/primer3/input.htm). Polymerase chain reactions were carried out in a 25 μl volume as described above, with a touchdown program: HotStarsTaq activation at 94° for 5 min, followed by 35 cycles, each having denaturation at 94° for 30 s, annealing at 55° for 30 s and extension at 72° for 45 s, except that in the first 10 cycles the annealing temperature decreased from 58° to 55° by 0.5° per cycle, and the final extension was 72° for 7 min. PCR products were directly sequenced on the ABI 3130 genetic analyzer.
Results
Clinical findings
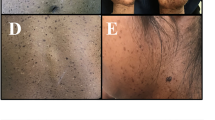
One PM family (family 1, Fig. 1a), consisting of 1 affected male and 3 affected females, and two DSAP families [family 2 (Fig. 1b) and family 3 (Fig. 1c)] were included in this study, including 12 affected males and 6 female patients, vertically inherited in an autosomal dominant model. The PM family (family 1) was from Fujian province in eastern China. The onset age of patients of PM family was, respectively, 12, 13, and 15 years old, firstly observed on buttocks or hands. One DSAP family (family 2) was from Guangdong province in southern China, and the other DSAP family (family 3) was Chinese Mongols from the Inner Mongolia Autonomous Region at western China, with initial skin lesions on the face, and onsets of age between 25 and 44. In comparison to family 2 (Fig. 2d) and family 3 (Fig. 2e), patients in family 1 were recognized with different geographical location and onset site, earlier onset age, more severe disease conditions, including bigger and widespread keratinized plaques, verrucous proliferation, and nail dystrophy (Fig. 2a–c).

Clinical and histopathological presentation. The cutaneous lesions of the proband (II:7) in PM family (family 1) were characterized by keratinized plaques (a), verrucous proliferation (b), and nail dystrophy (c). Representative clinical lesions of the other two families were shown by probands of family 2 (d) and family 3 (e), indicating milder disease conditions compared to that of the proband in family 1 who had MVK splicing mutation. Histopathologically (f), lesional examination for the proband in PM family (family 1) revealed cornoid lamella with absent granular layers, infiltration of inflammatory cells, and melanocyte clusters below the parakeratotic column (hematoxylin and eosin, original magnification ×100; scale bar 100 μm)
Hematoxylin and eosin staining of skin specimens from probands of family 1 showed histopathological features (Fig. 2f) described as below (1) atrophy of the epidermis, (2) parakeratotic columns (cornoid lamella) in the affected epidermis of the peripheral and central white tracks, and (3) a non-specific perivascular infiltrate of chronic inflammatory cells, (4) slightly dilated vessels and (5) melanin granules in the upper dermis close to cornoid lamella.
Mutation screening for MVK genomic sequence
Direct sequencing of all coding and exon–intron boundary sequences identified a substitution mutation at the splice donor site of intron 10, designated as c.1039+1G>A, in all patients in the PM family (Fig. 3a). Unaffected individuals in family 1 and unrelated controls did not show this change (Fig. 3b). However, no mutations of MVK gene were found in all members of three DSAP families.

MVK mutation analysis. A splicing mutation, c.1039+1G>A, was found in all patients of family 1 (a), but no MVK mutations in family 2 and family 3 (b). This splicing mutation resulted in the inclusion of the entire intron 10 in the complementary DNA (c), and less transcription of the mutated allele judged from sequencing diagram. The GAPDH cDNA was sequenced as internal control (d)
To further explore whether the splicing mutation affects MVK biological functions, complementary DNA (cDNA) sequence was analyzed. Simultaneously, analysis of GAPDH coding sequence and the Exon 8–Exon 9 validated the use of cDNA templates (Fig. 3c). Overlapping sequences between exon 10 and exon 11 were amplified by 7 pairs of primers. Direct sequencing revealed the inclusion of intron 10 in the cDNA of all patients in family 1 (Fig. 3d).
Discussion
The genetic basis and pathologic mechanisms of porokeratosis are still not well known. Linkage analysis has localized the genes to chromosome 12q23.2–24.1, 1p31.3–p31.1, 18p11.3, 16q24.1–24.3, 12q21.2–24.21, responsible for disseminated superficial actinic porokeratosis [3, 18, 19, 33, 34]. However, no causative genes had been identified until a recent study using the routine strategies, such as gene cloning or functional candidate screening. Recently, Zhang et al. first described the association of MVK mutations with 33 % familial and 16 % sporadic DSAP patients, including a splice site mutation (c.1039+2T>C) on MVK [3]. Zhang et al’s functional studies implicated that MVK might play important roles for keratinocyte differentiation by regulating the expression of keratin 1 in the spinous layer and involucrin in the granular layer. However, this discovery was not confirmed by other independent research groups, ethnics, and different geographical locations, since DSAP had broad phenotypic variants. Furthermore, it remained unclear what the differences are between MVK-mutated DSAP and MVK-unrelated DSAP. It is also unknown if other subtypes of porokeratosis, such as PM, share the same genetic abnormalities of MVK gene. To answer these questions, we collected two DSAP families, respectively, from southern China (Guangdong) and western China (Inner Mongolia Autonomous Region), and one PM family from eastern China (Fujian), with Chinese Han origin. We identified a splicing mutation (c.1039+1G>A) in intron 10 of MVK gene in the PM family, but failed to confirm MVK mutations as the cause of DSAP.
To assess the effect of the splice site mutation (c.1039+1G>A, located at position +1 of the splice donor site of intron 10 of MVK gene, we amplified the complementary DNA fragments between exons 10 and 11 by PCR from all three affected patients of this family. Sequence analysis of the cDNA fragments demonstrated that the entire intron 10 was included as part of the cDNA sequences between exons 10 and 11, resulting in the completely altered C-terminal domain of the MVK protein after residue 346, with functional domain(s) for keratinocyte differentiation.
DSAP was first described by Chernosky and Freeman in 1967. The cutaneous lesions, which occur primarily in sun-exposed areas of the skin, begin to develop in the teenagers of affected families, with penetrance nearly complete by the third and fourth decades of life. The low frequency of MVK mutations in DSAP patients suggested that other genetic (genes harbored in other linkage loci) or environmental (repeated exposure to NB-UVB) modifiers play roles on the development of DSAP. However, no genotype–phenotype correlations were analyzed by previous studies. To confirm the MVK’s mutations as the genetic basis responsible for DSAP development, it is essential to obtain more MVK genotype data in a spectrum of DSAP families from different countries and origins. In this study, three DSAP families, respectively, from western China and eastern China did not carry MVK mutations, suggesting that the impact of geographical locations was not critical. The PM patients (family 1) had a splicing mutation in MVK gene, having firstly developed DSAP lesions on hands and buttocks, with onset during the early first decade and more severe disease conditions. All patients of the three DSAP families started the development of typical lesions on face between the second and fourth decade, suggesting that MVK mutations induced early development of porokeratosis of Mibelli.
Taken together, it is worthy to note that MVK mutations are also responsible for PM, although MVK mutations were not confirmed in two DSAP families.
Web resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/.
SwissProt, UniProtKB/TREMBL, http://www.ebi.ac.uk/uniprot/.
Genome Browser and Blat, http://genome.ucsc.edu/.
dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/.
The Human Gene Mutation Database, http://www.hgmd.org/.
Abbreviations
- PM:
-
Porokeratosis of Mibelli
- DSAP:
-
Disseminated superficial actinic porokeratosis
- MVK :
-
Mevalonate kinase gene
- cDNA:
-
Complementary DNA
- PCR:
-
Polymerase chain reaction
- GADPH:
-
Glyceraldehyde-3-phosphate dehydrogenase
References
Ammouri W, Cuisset L, Rouaghe S, Rolland MO, Delpech M, Grateau G, Ravet N (2007) Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology (Oxford) 46:1597–1600
Bozdag KE, Bicakci H, Ermete M (2004) Giant porokeratosis. Int J Dermatol 43:518–520
Cao HM, Wang ZY, Zhang GW, Liu CF, Pan CM, Zhao SX, Song ZY, Song HD, Zhang L (2012) Identification of a locus (DSP2) for disseminated superficial porokeratosis at chromosome 12q21.2-24.21. Clin Exp Dermatol 37:672–676
Cuisset L, Drenth JP, Simon A, Vincent MF, van der Velde Visser S, van der Meer JW, Grateau G, Delpech M, International Hyper-IgD Study Group (2001) Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet 9:260–266
Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, Beckmann JS, van der Meer JW, Delpech M (1999) Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 22:178–181
Frenkel J, Houten SM, Waterham HR, Wanders RJ, Rijkers GT, Duran M, Kuijpers TW, van Luijk W, Poll-The BT, Kuis W (2001) Clinical and molecular variability in childhood periodic fever with hyperimmunoglobulinaemia D. Rheumatology (Oxford) 40:579–584
Hager EJ, Piganelli JD, Tse HM, Gibson KM (2012) Aberrant expression of costimulatory molecules in splenocytes of the mevalonate kinase-deficient mouse model of human hyper-IgD syndrome (HIDS). J Inherit Metab Dis 35:159–168
Houten SM, Frenkel J, Rijkers GT, Wanders RJ, Kuis W, Waterham HR (2002) Temperature dependence of mutant mevalonate kinase activity as a pathogenic factor in hyper-IgD and periodic fever syndrome. Hum Mol Genet 11:3115–3124
Hospach T, Lohse P, Heilbronner H, Dannecker GE, Lohse P (2005) Pseudodominant inheritance of the hyperimmunoglobulinemia D with periodic fever syndrome in a mother and her two monozygotic twins. Arthritis Rheum 52:3606–3610
Houten SM, Koster J, Romeijn GJ, Frenkel J, Di Rocco M, Caruso U, Landrieu P, Kelley RI, Kuis W, Poll-The BT et al (2001) Organization of the mevalonate kinase (MVK) gene and identification of novel mutations causing mevalonic aciduria and hyperimmunoglobulinaemia D and periodic fever syndrome. Eur J Hum Genet 9:253–259
Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, Frenkel J, Dorland L, de Barse MM, Huijbers WA et al (1999) Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet 22:175–177
Houten SM, van Woerden CS, Wijburg FA, Wanders RJ, Waterham HR (2003) Carrier frequency of the V377I (1129G>A) MVK mutation, associated with Hyper-IgD and periodic fever syndrome, in the Netherlands. Eur J Hum Genet 11:196–200
Kodell RL, Young JF, Delongchamp RR, Turturro A, Chen JJ, Gaylor DW, Howard PC, Zheng Q (2001) A mechanistic approach to modelling the risk of liver tumours in mice exposed to fumonisin B1 in the diet. Food Addit Contam 18:237–253
Lainka E, Neudorf U, Lohse P, Timmann C, Bielak M, Stojanov S, Huss K, von Kries R, Niehues T (2012) Incidence and clinical features of hyperimmunoglobulinemia D and periodic fever syndrome (HIDS) and spectrum of mevalonate kinase (MVK) mutations in German children. Rheumatol Int 32:3253–3260
Lee HR, Han TY, Son SJ, Lee JH (2011) Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol 23:536–538
Leyva-Vega M, Weiss PF, Ganesh J, Conlin L, Spinner NB, Matthews RP (2011) Significant liver disease in a patient with Y116H mutation in the MVK gene. Am J Med Genet A 155A:1461–1464
Li JH, Yang ZH, Li B, Chen HD (2011) Squamous cell carcinoma arising from giant porokeratosis. Dermatol Surg 37:855–857
Liu P, Zhang S, Yao Q, Liu X, Wang X, Huang C, Huang X, Wang P, Yuan M, Liu JY et al (2008) Identification of a genetic locus for autosomal dominant disseminated superficial actinic porokeratosis on chromosome 1p31.3-p31.1. Hum Genet 123:507–513
Luan J, Niu Z, Zhang J, Crosby ME, Zhang Z, Chu X, Wang Z, Huang W, Xiang L, Zheng Z (2011) A novel locus for disseminated superficial actinic porokeratosis maps to chromosome 16q24.1-24.3. Hum Genet 129:329–334
Mizuno T, Sakai H, Nishikomori R, Oshima K, Ohara O, Hata I, Shigematsu Y, Ishige T, Tamura K, Arakawa H (2012) Novel mutations of MVK gene in Japanese family members affected with hyperimmunoglobulinemia D and periodic fever syndrome. Rheumatol Int 32:3761–3764
Naruto T, Nakagishi Y, Mori M, Miyamae T, Imagawa T, Yokota S (2009) Hyper-IgD syndrome with novel mutation in a Japanese girl. Mod Rheumatol 19:96–99
Samkari A, Borzutzky A, Fermo E, Treaba DO, Dedeoglu F, Altura RA (2010) A novel missense mutation in MVK associated with MK deficiency and dyserythropoietic anemia. Pediatrics 125:e964–e968
Schena D, Papagrigoraki A, Frigo A, Girolomoni G (2010) Eruptive disseminated porokeratosis associated with internal malignancies: a case report. Cutis 85:156–159
Scola N, Skrygan M, Wieland U, Kreuter A, Gambichler T (2012) Altered gene expression in squamous cell carcinoma arising from congenital unilateral linear porokeratosis. Clin Exp Dermatol 37:781–785
Sornsakrin M, Wenner K, Ganschow R (2009) B cell cytopenia in two brothers with hyper-IgD and periodic fever syndrome. Eur J Pediatr 168:825–831
Stojanov S, Lohse P, Lohse P, Hoffmann F, Renner ED, Zellerer S, Kéry A, Shin YS, Haas D, Hoffmann GF et al (2004) Molecular analysis of the MVK and TNFRSF1A genes in patients with a clinical presentation typical of the hyperimmunoglobulinemia D with periodic fever syndrome: a low-penetrance TNFRSF1A variant in a heterozygous MVK carrier possibly influences the phenotype of hyperimmunoglobulinemia D with periodic fever syndrome or vice versa. Arthritis Rheum 50:1951–1958
Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL (2003) Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 48:2645–2651
Tahara M, Sakai H, Nishikomori R, Yasumi T, Heike T, Nagata I, Inui A, Fujisawa T, Shigematsu Y, Nishijima K et al (2011) Patient with neonatal-onset chronic hepatitis presenting with mevalonate kinase deficiency with a novel MVK gene mutation. Mod Rheumatol 21:641–645
Tas DA, Dinkci S, Erken E (2012) Different clinical presentation of the hyperimmunoglobulin D syndrome (HIDS) (four cases from Turkey). Clin Rheumatol 31:889–893
van der Hilst JC, Bodar EJ, Barron KS, Frenkel J, Drenth JP, van der Meer JW, Simon A, International HIDS Study Group (2008) Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 87:301–310
Vivas AC, Maderal AD, Kirsner RS (2012) Giant ulcerating squamous cell carcinoma arising from linear porokeratosis: a case study. Ostomy Wound Manage 58:18–20
Wang P, Wei Z, Yan B, Huang T, Gou K, Dai Y, Zheng M, Wang M, Cheng X, Wang X et al (2012) Establishment of a transgenic mouse model with liver-specific expression of secretory immunoglobulin D. Sci China Life Sci 55:219–227
Wei S, Zhang TD, Zhou Y, Zhang XB, Zhu HL, Li J, Huang ZM, Deng L, Zhang XJ (2010) Fine mapping of the disseminated superficial porokeratosis locus to a 2.7 Mb region at 18p11.3. Clin Exp Dermatol 35:664–667
Xia JH, Yang YF, Deng H, Tang BS, Tang DS, He YG, Xia K, Chen SX, Li YX, Pan Q et al (2000) Identification of a locus for disseminated superficial actinic porokeratosis at chromosome 12q23.2-24.1. J Invest Dermatol 114:1071–1074
Zhang SQ, Jiang T, Li M, Zhang X, Ren YQ, Wei SC, Sun LD, Cheng H, Li Y, Yin XY et al (2012) Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis. Nat Genet 44:1156–1160
Acknowledgment
This work was supported by a grant from Nanfang Hospital (JQ201201) to Yan-Hua Liang.
Conflict of interests
The authors have declared that no competing interests exist.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zeng, K., Zhang, QG., Li, L. et al. Splicing mutation in MVK is a cause of porokeratosis of Mibelli. Arch Dermatol Res 306, 749–755 (2014). https://doi.org/10.1007/s00403-014-1465-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00403-014-1465-7