
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that is neuropathologically characterized by degeneration of dopaminergic neurons of the substantia nigra (SN) and formation of Lewy bodies and Lewy neurites composed of aggregated α-synuclein. Proteolysis of α-synuclein by matrix metalloproteinases was shown to facilitate its aggregation and to affect cell viability. One of the proteolysed fragments, Gln79-α-synuclein, possesses a glutamine residue at its N-terminus. We argue that glutaminyl cyclase (QC) may catalyze the pyroglutamate (pGlu)79-α-synuclein formation and, thereby, contribute to enhanced aggregation and compromised degradation of α-synuclein in human synucleinopathies. Here, the kinetic characteristics of Gln79-α-synuclein conversion into the pGlu-form by QC are shown using enzymatic assays and mass spectrometry. Thioflavin T assays and electron microscopy demonstrated a decreased potential of pGlu79-α-synuclein to form fibrils. However, size exclusion chromatography and cell viability assays revealed an increased propensity of pGlu79-α-synuclein to form oligomeric aggregates with high neurotoxicity. In brains of wild-type mice, QC and α-synuclein were co-expressed by dopaminergic SN neurons. Using a specific antibody against the pGlu-modified neo-epitope of α-synuclein, pGlu79-α-synuclein aggregates were detected in association with QC in brains of two transgenic mouse lines with human α-synuclein overexpression. In human brain samples of PD and dementia with Lewy body subjects, pGlu79-α-synuclein was shown to be present in SN neurons, in a number of Lewy bodies and in dystrophic neurites. Importantly, there was a spatial co-occurrence of pGlu79-α-synuclein with the enzyme QC in the human SN complex and a defined association of QC with neuropathological structures. We conclude that QC catalyzes the formation of oligomer-prone pGlu79-α-synuclein in human synucleinopathies, which may—in analogy to pGlu-Aβ peptides in Alzheimer’s disease—act as a seed for pathogenic protein aggregation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most frequent progressive neurodegenerative disorder after Alzheimer’s disease (AD) [7, 12]. The brains of PD patients are neuropathologically characterized by the degeneration of dopaminergic neurons of the substantia nigra (SN) pars compacta, which results in dopamine depletion of the striatum [52, 68]. This dopaminergic hypoactivity affects functions of the complex basal ganglia network, leading to clinical symptoms, such as hypokinesis and tremor [15, 34]. Another typical feature of PD is the appearance of Lewy bodies and Lewy neurites that are mainly composed of aggregated α-synuclein [28, 82]. Under physiological conditions, α-synuclein is believed to be a natively unfolded protein of 140 amino acids, but it may also exist as α-helically folded multimers [8, 25]. It is predominantly localized to presynaptic nerve terminals and has been shown to act as a molecular chaperone in the formation of SNARE complexes being involved in the regulation of dopamine release [35]. In the course of PD, however, α-synuclein conformation is altered to form Lewy inclusions and various aggregation conformers, ranging from small oligomers to amyloid fibrils, with distinct structural and biochemical features [5, 59, 100]. Recent studies provided evidence that aggregated α-synuclein may propagate its structural alterations and loss or gain of function via prion-like spreading [47, 54, 60, 106]. Similarly, α-synuclein aggregates are also present in brains of patients suffering from multiple systems atrophy and from dementia with Lewy bodies (DLB), where the pathology also affects cortical association areas [40, 75, 101].
Structurally, α-synuclein is composed of 3 domains: an amphipathic N-terminal region (aa 1–60), a central hydrophobic domain involved in protein aggregation (non-Abeta component (NAC) region; aa 61–95) and a highly acidic, proline-rich C-terminus (aa 96–140) [33]. The full-length α-synuclein can be post-translationally modified by phosphorylation, ubiquitination, nitration, glycation, SUMOylation and truncation [4, 37, 105, 109]. C-terminal truncations of α-synuclein by defined protease activities, such as m-calpain and 20S proteasome, have been linked to increased aggregation, fibril formation and neurotoxicity [55, 63, 74, 99]. In addition, N-terminally truncated α-synuclein fragments are generated by matrix metalloproteinases (MMPs) -1, -3 and -9 [61, 102]. Most importantly, limited proteolysis of α-synuclein by MMP-1 and MMP-3, but not by MMP-9, was shown to generate fragments that increase de novo aggregation of α-synuclein in vitro [61]. Since α-synuclein is cleaved by MMP-3 preferentially within the NAC domain, the resultant fragments do not form fibrils but rather oligomers that compromise cell viability [102]. One of the MMP-3-generated α-synuclein fragments, Gln79-α-synuclein, possesses a glutamine residue at its N-terminus (Fig. 1a).

QC-catalyzed pGlu79-α-synuclein formation. a Schematic representation of N-terminal α-synuclein truncation by MMP-3 resulting in the formation of N-terminal glutamine (Gln) residue at position 79 of α-synuclein. b Schematic illustration of pGlu formation from N-terminal Gln under liberation of ammonia catalyzed by QC. c Kinetic characteristics of QC-catalyzed pGlu79-α-synuclein formation revealed by a continuous coupled spectrophotometric test. Values were obtained from 3 to 4 independent determinations and are displayed as mean ± SD. d Mass spectrometric analysis of pGlu formation at the N-terminus of the synthetic α-synuclein79–90 fragment. The maternal α-synuclein79–90 fragment was detected at the predicted mass of 1130.6 Da at all time points without any spontaneous degradation or modification. When the enzyme QC was added, a new peak (blue) was detected at a molecular weight of 17 g/mol below the maternal α-synuclein79–90 fragment, consistent with the liberation of ammonia during enzyme-catalyzed pGlu-formation. The conversion of α-synuclein to pGlu-α-synuclein79-90 was completely prevented by addition of the QC inhibitor PBD150
Peptides with an N-terminal glutamate or glutamine residue may serve as substrates for glutaminyl cyclase (QC), giving rise to pyroglutamate (pGlu)-modified peptides [92, 97]. This pGlu modification confers stability against proteolytical degradation and increases the biological activity of neuropeptides and peptide hormones, such as orexin A, gastrin, gonadotropin- and thyrotropin-releasing hormones and neurotensin in hypothalamus and pituitary [14, 18, 29, 81].
Under pathological conditions in AD, however, QC catalyzes the pGlu modification of N-truncated Aβ peptides that are highly pathogenic and act as seeds for Aβ oligomer and plaque formation [3, 23, 76, 93]. QC expression is developmentally regulated [43] and highly abundant in brain structures affected by amyloid pathology in AD, such as nucleus basalis Meynert, locus coeruleus and Edinger–Westphal nucleus [73], hippocampus [42] and neocortex [72]. Pharmacological inhibition of QC activity [93] and genetic ablation of QC in experimental animal models [3, 50] reduced pGlu-Aβ generation and total Aβ load and ameliorated learning and memory deficits. It is tempting to speculate that QC—if expressed by dopaminergic SN neurons—may catalyze the pGlu79-α-synuclein formation and, thereby, contribute to enhanced aggregation and compromised degradation of α-synuclein in human synucleinopathies (Fig. 1b).
Therefore, we here analyzed the enzymatic formation of pGlu79-α-synuclein by QC in vitro, its aggregation characteristics and neurotoxic profile, its co-localization with QC and increased formation in the SN of human PD and DLB brains as well as in animal models for synucleinopathies. Together, we demonstrate the existence of a novel pathogenic post-translational α-synuclein modification. Since this modification is QC-catalyzed and QC inhibitors are already in clinical trials for AD treatment, there might be novel therapeutic options for interfering with α-synuclein aggregation in PD as well.
Materials and methods
QC-catalyzed formation of pGlu79-α-synuclein
Peptide synthesis
The synthetic α-synuclein79–90 peptide was synthesized according to standard Fmoc solid phase protocols on a Tetras peptide synthesizer (Advanced ChemTech, Louisville, USA) at 60 µmol scale as C-terminal amide on Rink amide resin (Iris Biotech; Marktredwitz, Germany) using standard Fmoc/tBu-protected amino acids (Iris Biotech). Coupling was done using O-(benzotriazol-1-yl)-N,N,N',N′-tetramethyluronium tetrafluoroborate (TBTU) and N-methylmorpholine (NMM). Fmoc-deprotection was carried out using 20% piperidine in DMF. Final cleavage and deprotection of the peptides was performed using TFA:EDT:H2O:TIS (50:2:2:1 v/v). After precipitation with cold diethylether, the peptides were purified by preparative reversed phase (RP)–HPLC (Phenomenex Luna C18(2) column) and eluted with an increasing water:acetonitrile gradient starting with 5% containing 0.04% TFA. The identity and purity were assessed by analytical RP–HPLC and ESI MS.
Enzymatic activity assay
The kinetics of QC-catalyzed pGlu-α-synuclein79–90 formation was measured by a continuous coupled spectrophotometric test in 50 mM Tris/HCl buffer, pH 8.0 using glutamate dehydrogenase as auxiliary enzyme as described in detail by Schilling et al. [90, 91]. Kinetic parameters were calculated by non-linear regression as indicated before [95].
Mass spectrometry
The pGlu formation at the N-terminus of the synthetic α-synuclein79–90 peptide was monitored by mass spectrometry. 22.6 µg of this peptide were incubated in a total volume of 200 µl 50 mM Tris/HCl buffer, pH 8.0, (100 µM) for 10, 30 and 60 min in the absence or presence of the enzyme QC (0.7 µg/ml; 20 nM) with and without the QC inhibitor PBD150 (100 µM). The analytes were ionized by a nitrogen laser pulse (337 nm) and accelerated under 20 kV with a time-delayed extraction before entering the time-of-flight mass spectrometer (Voyager De Pro, Sciex). The maternal synthetic α-synuclein79–90 peptide was detected at the mass of 1130.6 Da, whereas after pGlu79 modification and liberation of ammonia the molecular weight was reduced to 1113.8 Da.
Aggregation of recombinant α-synuclein and pGlu79-α-synuclein
Expression and purification of α-synuclein proteins
The human full-length α-synuclein and Gln79-α-synuclein proteins were recombinantly expressed following procedures described recently [57]. Purification included Ni2+-chelating chromatography on a Streamline Chelating resin (Streamline Chelating, GE Healthcare Life Sciences, Uppsala, Sweden). Fractions containing the expression construct were subjected to a second purification step via a glutathione sepharose resin (Glutathione Sepharose 4FF, GE Healthcare Life Sciences). The removal of glutathione was achieved by overnight dialysis against buffer containing 100 mM NaCl, 30 mM Tris/HCl pH 7.6, 0.1 mM DTT and a membrane with 6–8 kDa cutoff. Separation of the GST- and His-tag from the α-synuclein sequence by a TEV protease cleavage left an native N-terminus [53] followed by cyclization of Gln79-α-synuclein to pGlu79-α-synuclein with QC overnight at room temperature. The fractions obtained were analyzed and subjected to reversed phase chromatography (Source 15 RPC, GE Healthcare Life Sciences), followed by lyophilization and anion exchange chromatography (MonoQ 5/50GL, GE Healthcare Life Sciences). The final buffer used for the experiments was 20 mM Tris/HCl, pH 7.0, containing 100 mM NaCl. The purity of the samples was assessed by SDS PAGE and mass spectrometry. Protein concentrations were determined using UV absorption at 280 nm.
Thioflavin T assay
The thioflavin T (ThT) assay was carried out as described previously [94] on a FluoStar Optima (BMG Labtech, Ortenberg, Germany) plate reader using a 96-well plate (λex = 440 nm and λem = 490 nm). For monitoring the fibrillation process of the recombinant full-length or pGlu79-α-synuclein, 20 µM ThT (Sigma-Aldrich) were added to the aggregation buffer (20 mM Tris/HCl, 100 mM NaCl, pH 7.0). Signals were recorded at 37 °C under continuous shaking (300 rpm) with a time interval of 15 min for 110 h. Analyses of the obtained aggregation curves were conducted according to [46]. For each peptide, measurements were performed in six cavities of one plate. Obtained data were analyzed with one-way ANOVA and post-hoc Tukey test.
Transmission electron microscopy
Potential fibril formation from full-length α-synuclein and pGlu79-α-synuclein was initiated in aggregation buffer (20 mM Tris/HCl, 100 mM NaCl, pH 7.0) at 37 °C under continuous shaking (300 rpm) for 110 h. Samples (5 µl) were placed on a formvar carbon-coated copper grid (Plano, Wetzlar, Germany) for 10 min and washed three times with distilled water. Staining was obtained with 2% (v/v) phosphotungstic acid (Sigma-Aldrich) for 5 min. Grids were imaged with a TEM/STEM FEI-Tecnai G2 F20 (FEI Company, Hillsboro, USA) in STEM-mode at 200 kV. The electron micrographs were detected using a high-angle annular dark-field detector, and finally processed by contrast-inversion.
Size exclusion chromatography and dot blot analysis
To analyze the formation of oligomers by size exclusion chromatography (SEC), 50–120 µg of the recombinant full-length or pGlu79-α-synuclein, either untreated (monomers) or agitated for aggregation as described in ThT assay, was centrifuged at 10,000×g for 60 min to remove large aggregated particles. Centrifuged α-synuclein samples of each variant were next diluted with SEC running buffer (50 mM Tris/HCl pH 7.2 buffer with 200 mM NaCl) to a total volume of 300 µl, filtered by a Whatman PVDF filter device (pore size 0.2 µm) and subsequently loaded onto a Yarra SEC 3000 column (Phenomenex, Aschaffenburg, Germany). SEC was performed using an isocratic elution with the SEC running buffer at a flow rate of 0.3 ml/min on an ÄKTA pure 25 M system (Cytiva, Freiburg, Germany). A total of 30 fractions of each analysis were collected with 0.5 ml per fraction. The eluted peaks were monitored at 215 and 280 nm. The elution time and quantity of the monomers and oligomers were determined using Unicorn software (Cytiva). For immunodetection of the fractions containing α-synuclein, the collected fractions were applied onto a nitrocellulose membrane using a Minifold Dot-Blot System (Schleicher & Schuell) and probed with Syn1 (BD Transduction Laboratories™), a monoclonal mouse antibody against pan α-synuclein (1:1000), or the polyclonal rabbit anti-pGlu79-α-synuclein antibody (described below, 1:700). For immunodetection, horseradish peroxidase conjugated anti-mouse or anti-rabbit antibody (Dianova) and chemiluminescent substrates (SuperSignal West Chemiluminescent Substrate kits, Thermo Fisher Scientific) were used.
Cell culture and toxicity assay
The toxic effect of full-length and pGlu79-α-synuclein on SH-SY5Y neuroblastoma cells was assessed using a WST-1 assay (ThermoFisher, Darmstadt, Germany). SH-SY5Y cells were grown in DMEM medium supplemented with 10% FBS at 37 °C, 10% CO2. To induce differentiation towards a neuronal phenotype, 1.83 × 104 cells/well were seeded in a transparent 96-well plate and the medium was changed towards DMEM supplemented with 5% FBS and 10 µM all-trans retinoic acid (ThermoFisher, Darmstadt, Germany) for 3 days. The medium was further exchanged to Neurobasal-A medium without phenol red, supplemented with 1% (v/v) Glutamax, 1% (v/v) N-2 supplement (ThermoFisher, Darmstadt, Germany) and human BDNF (ThermoFisher, Darmstadt, Germany) at a concentration of 50 ng/ml (v/v) for additional 4 days. On day 7 of differentiation, the assay was carried out according to the manufacturer’s protocol. In brief, cells were exposed to the different peptide species and cultured at 37 °C in a humidified atmosphere containing 10% CO2 for 72 h. Afterwards, 10% WST-1 was added to the cell medium and incubated for 30 min. The absorbance was determined at 440 nm using a plate reader (Tecan Sunrise, Switzerland). The values were normalized to the PBS control and directly correlated to the number of viable cells.
Mouse brain tissue
The expression of endogenous α-synuclein was analyzed in brains of C57Bl/6 wild-type mice (N = 4) obtained from the Animal Care Facility of the Medical Faculty, Leipzig University. α-synuclein knock-out (KO) mice (N = 2; Charles River; JAX strain 003692) were used to demonstrate the specificity of α-synuclein antibodies employed in this study. Two transgenic mouse lines were investigated for the formation of α-synuclein aggregates: (1) mice overexpressing human wild-type α-synuclein under the Thy-1 promoter (termed ASO; N = 4) [20, 83] and (2) mice overexpressing human wild-type α-synuclein from a bacterial artificial chromosome (termed BAC-SNCA; N = 4) [71, 108]. All mouse lines were on C57Bl/6 background. Animals were housed at 12 h day/12 h night cycles with food and water ad libitum in cages that contained nest building material. All experimental protocols were approved by Landesdirektion Sachsen, license number T28/16 and the local ethical board of the District Government of Lower Franconia, Bavaria, Germany (approval # 55.2-DMS 2532-2-218). All methods were carried out in accordance with the relevant guidelines and regulations.
Tissue preparation
Mice were sacrificed by CO2 inhalation and perfused transcardially with 0.9% saline followed by perfusion with 4% paraformaldehyde in phosphate buffer (0.1 M, pH 7.4). The brains were removed from the skull and post-fixed by immersion in the same fixative overnight at 4 °C. After cryoprotection in 30% sucrose in 0.1 M phosphate buffer for 3 days, 30 µm thick coronal sections were cut on a sliding microtome and collected in phosphate buffer supplemented with 0.025% sodium azide for storage.
Human brain tissue
Case recruitment and characterization of human brain tissue
Case recruitment and autopsy were performed in accordance with guidelines effective at the Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program [10]. The required consent was obtained for all cases. Cases were staged for synuclein pathology using the Unified Staging System [2, 9]. The definite diagnosis of PD was based on clinical findings of 2 of 3 cardinal signs (rigidity, bradykinesia and rest tremor) as well as depigmentation with Lewy bodies in the SN. DLB was defined as dementia occurring either at presentation or within 1 year of the onset of parkinsonism, with a brain distribution of α-synuclein pathology meeting DLB Consortium criteria for “intermediate” or “high” likelihood [70]. The Unified Staging System and McKeith criteria received good inter-rater reliability scores in a multi-centre comprehensive analysis defining consensus criteria for the evaluation of Lewy body pathology in post mortem brains [6]. Three out of 10 DLB cases were additionally diagnosed with AD, due to intermediate or high AD neuropathological changes according to [21].
Tissue preparation
Transverse midbrain sections (40 µm thick) comprising SN at the level of the red nucleus, exit of the oculomotor nerve and superior colliculus from 10 controls, 10 idiopathic PD cases and 10 DLB cases (Table 1) were used for QC and pGlu79-α-synuclein immunohistochemistry. Anatomical regions were identified on Nissl and anti-HuC/D-stained sections using a human brain atlas [66].
Antibody generation
Polyclonal anti-pGlu79-α-synuclein-specific antibodies were produced from rabbits immunized with synthetic pGlu-α-synuclein79–90 peptide conjugated to a carrier according to the manufacturer’s standard protocol (Davids Biotechnology, Germany). Rabbits were immunized five times (days 1, 14, 28, 42 and 56) with the optimal amount of antigen followed by a final bleed at day 63. After day 35, a test serum was taken and the ELISA titer was determined. The antiserum was affinity purified and characterized for specificity (Suppl. Figure 1, online resource).
Antibody specificity
The specificity of the rabbit antiserum against pGlu79-α-synuclein was verified by dot blot analysis against recombinant human full-length α-synuclein, β-synuclein, γ-synuclein and the target pGlu79-α-synuclein fragment, spotted at descending amounts onto nitrocellulose membranes (Suppl. Figure 1, online resource). After chemiluminescent detection, membranes were stripped and re-probed with the Syn1 antibody (BD Transduction; 1:2000). In addition, immunohistochemistry was performed on wild-type, α-synuclein overexpressing and α-synuclein KO mouse brain sections, demonstrating the specificity of the pGlu79-α-synuclein antiserum for this application (Suppl. Figure 1, online resource). The specificity of the goat antiserum directed against QC has been recently demonstrated comparing immunohistochemical labelling in wild type and QC KO mouse brain sections [41].
Immunohistochemistry
Single labelling of pGlu79-α-synuclein and QC in mouse brain sections
To detect pGlu79-α-synuclein and QC in wild type, ASO and BAC-SNCA mice, single labelling immunohistochemistry was performed on free-floating coronal brain sections. Brain sections were washed in 0.1 M phosphate buffer (pH 7.4) for 5 min and endogenous peroxidases were inactivated by treating brain slices with 60% methanol containing 1% H2O2 for 60 min followed by three washing steps with Tris buffered saline (TBS, 0.1 M, pH 7.4) for 5 min each. After masking unspecific binding sites with blocking solution (5% normal donkey serum in TBS containing 0.3% Triton X-100) for 60 min, sections were incubated with the primary rabbit anti-pGlu79-α-synuclein (1:200) or goat anti-QC (1:200) antibodies for 40 h at 4 °C. Brain sections were then washed three times in TBS for 5 min each before being incubated with biotinylated secondary donkey anti-goat or donkey anti-rabbit antibodies (Dianova; 1:1000) in TBS containing 2% bovine serum albumin (BSA) for 60 min. After three washing steps in TBS for 5 min each, slices were incubated with ExtrAvidin peroxidase (Sigma; 1:2000) in TBS/2% BSA followed by washing steps and pre-incubation in Tris buffer (0.05 M, pH 7.6) for 5 min. Finally, visualization of peroxidase binding was performed by incubation with 4 mg 3,3′-diaminobenzidine (DAB) and 2.5 µl 30% H2O2 per 5 ml Tris buffer. After washing, sections were mounted onto glass slides and cover slipped.
Triple immunofluorescent labellings in mouse brain
To reveal the expression of QC by tyrosine hydroxylase (TH)-positive dopaminergic SN neurons and its possible co-localization with full-length α-synuclein and with pGlu79-α-synuclein in mouse SN, the goat anti-QC antibody was applied in cocktails with primary guinea pig antibodies against TH (Synaptic Systems; #213104), mouse anti-α-synuclein (Syn1; BD Transduction Laboratories) or rabbit anti-pGlu79-α-synuclein as specified in Table 1. Brain sections were incubated with cocktails of primary antibodies for 40 h at 4 °C. Sections were then washed three times with TBS followed by incubation with cocktails of Cy2-, Cy3- or Cy5-conjugated donkey anti-mouse, -rabbit, -guinea pig or -goat, respectively, antisera (1:200 each; Dianova) in TBS containing 2% BSA for 60 min at room temperature. After washing, sections were mounted onto glass slides and cover slipped. Switching the fluorescent labels of the secondary antibodies generated similar results as when following the procedure outlined above (not shown).
Detection of pGlu79-α-synuclein and QC in human SN
To reveal presence of pGlu79-α-synuclein and QC in the SN of post mortem human control, DLB and PD tissue, single labelling immunohistochemistry was performed on free-floating transverse midbrain sections. Sections were washed in phosphate buffered saline (PBS, pH 7.4) for 5 min and endogenous peroxidases were inactivated by treating brain slices with 60% methanol containing 1% H2O2 for 30 min followed by rinses with PBS containing 0.02% Tween 20 (PBS-T) for 5 min each. Unspecific staining was then blocked in PBS-T containing 2% BSA, 0.3% milk powder and 0.5% normal donkey serum before incubating brain sections in the same solution containing the primary antibodies rabbit anti-pGlu79-α-synuclein (1:200) or goat anti-QC (1:200) in a humid chamber for 40 h at 4 °C. Subsequently, sections were washed in PBS-T (three times for 5 min) and were then incubated with secondary biotinylated donkey anti-rabbit or donkey anti-goat antibodies (Dianova; 1:1,000) in a mixture of blocking solution and PBS-T (1:2) for 60 min at room temperature. Following washing steps, the ABC method was applied which comprised incubation with complexed streptavidin and biotinylated horseradish peroxidase (Sigma; 1:2000) in PBS-T. Binding of peroxidase was visualized by incubation with 2 mg DAB, 20 mg nickel ammonium sulfate and 2.5 µl 30% H2O2 per 5 ml Tris buffer (0.05 M; pH 8.0) for 3–4 min. DAB-Ni staining resulted in black visualization of pGlu79-α-synuclein and QC which allowed for the co-localization with brown, neuromelanin-positive (NM+) neurons in the SN.
For all single and triple immunohistochemical labellings in brain sections described above, control experiments in the absence of primary antibodies were carried out. In each case, this resulted in unstained brain sections (not shown).
Microscopy
Light microscopy
Mouse and human brain tissue sections immunohistochemically stained with DAB or DAB-Ni for pGlu79-α-synuclein and QC expression were examined with an Axio-Scan.Z1 slide scanner connected with a Colibri.7 light source and a Hitachi HV-F202SCL camera (Carl Zeiss, Göttingen, Germany). High resolution images of midbrain sections containing the SN were taken using a 20× objective lens with 0.5 numerical aperture (Zeiss). Images were digitized by means of ZEN 2.6 software and analyzed using the ZEN imaging tool.
Confocal laser scanning microscopy
Laser scanning microscopy (LSM 880 Airyscan, Zeiss, Oberkochen, Germany) using an Axioplan2 microscope was performed to reveal co-localization of QC with its substrate pGlu79-α-synuclein, with full-length α-synuclein and with TH, respectively. For Cy2-labelled antigens (green fluorescence), an argon laser with 488 nm excitation was used and emission from Cy2 was recorded at 510 nm applying a low-range band pass (505–550 nm). For Cy3-labelled antigens (red fluorescence), a helium–neon laser with 543 nm excitation was applied and emission from Cy3 at 570 nm was detected applying high-range band pass (560–615 nm) and Cy5-labelled antigens (blue fluorescence) were detected using excitation at 650 nm and emission at 670 nm. Images of areas of interest were taken using a 20× objective lens with 0.75 numerical aperture (Zeiss). Photoshop CS2 (Adobe Systems, CA) was used to process the images obtained by light and confocal laser scanning microscopy. Care was taken to apply the same brightness, sharpness, color saturation and contrast adjustments in the various pictures.
Quantification of QC staining in SN of human midbrain
Analysis of QC expression in the SN pars compacta (SNc) from control subjects (CO), as well as from DLB and PD patients (N = 10, each; Table 2) was performed using the ZEN 2.6 imaging software. Transverse sections (40 µm) of the ventral midbrain approx. at the level of the center of Ncl. ruber were evaluated with respect to intra- and extracellular QC immunoreactivity. For each individual case, the area of the SNc was delineated at lower magnification according to the distribution of the nigral matrix and nigrosomes, respectively, enclosing pigmented NM-containing neurons in the SNc [24]. Hereby, even at high zoom levels, orientation within the SN was assured to restrict examination of QC immunoreactive structures to the pars compacta subregion.
Evaluation of intracellular QC staining
Zooming from overview to high-resolution magnification in a given specimen, neuronal QC immunoreactivity (DAB-Ni; black) was assessed in each individual neuron throughout the medioventral to dorsolateral extent of the SNc by an investigator blinded to the origin of the case. The presence of intracellular QC, either bound to NM or independently distributed in the cytoplasm, was evaluated with respect to the presence or absence of NM (brown) and vice versa. Then, each neuron was assigned to one of the following three categories: (1) QC and NM positive (QC+/NM+), (2) QC positive and NM negative (QC+/NM−), or (3) QC negative and NM positive (QC−/NM+).
Evaluation of pathological QC-positive structures
In addition to apparently intact neuronal cell bodies, QC immunoreactive, potentially pathogenic intra- and extracellular structures were identified and counted in the SNc. These were (1) degenerating neurons larger than 10 µm in diameter with very strong QC-immunoreactivity, which displayed a clearly aberrant form and/or were fractionated; (2) axonal varicosities; (3) axonal bulbs and Lewy neurites; (4) smaller (< 5 µm); and (5) larger (> 5 µm) Lewy body-like structures. QC-immunoreactive morphological features were termed axonal or dendritic varicosities when at least three punctate labellings smaller than 3 µm in diameter appeared along a neuronal process being arranged in a typical “beads on a string” manner, whereas slightly larger structures within axonal shafts (3–5 µm in diameter) were referred to as axonal bulbs.
Statistical analysis
The total number of identified neurons, as well as numbers of QC neurons and pathological structures in each category were compared between groups. Statistical analyses of the acquired data were performed by an unpaired t test. Differences between groups were considered statistically significant for p values < 0.05.
Results
The enzymatic characterization of Gln79-α-synuclein conversion to the pGlu variant and the mass spectrometric analyses shown in Fig. 1c, d were carried out using a 12 amino acid peptide starting with Gln79 and termed α-synuclein79–90 and pGlu-α-synuclein79–90, respectively. The further analyses of aggregation properties and toxicity profiles were performed with recombinant pGlu79–140-α-synuclein, termed pGlu79-α-synuclein.
QC-catalyzed generation of pGlu79-α-synuclein
To address the question whether Gln79-α-synuclein represents a QC substrate, the N-terminal part (79–90) of this α-synuclein fragment was synthesised and spontaneous as well as QC-catalyzed pGlu modification was followed by enzyme kinetic analysis (Fig. 1c). The subsequent calculation of kinetic parameters revealed a Vmax of 0.5999 mM/h, Km of 0.1333 mM and Kcat of 14.9/s. This is supportive for Gln79-α-synuclein being a QC substrate and compares well with other enzyme-catalyzed reactions, such as the conversion of the known physiological QC substrates gastrin, gonadotropin-releasing hormone and neurotensin [91].
Mass spectrometric analysis revealed that incubation of the α-synuclein79–90 fragment alone for up to 60 min did not lead to spontaneous pGlu modification of the N-terminus (Fig. 1d). By contrast, addition of QC to the incubation solution resulted in rapid pGlu-α-synuclein79–90 formation, which was already detectable after 10 min of incubation (Fig. 1d). By 1 h of incubation, the α-synuclein79–90 fragment was completely converted into pGlu-α-synuclein79–90. Addition of the QC inhibitor PBD150 prevented the pGlu modification (Fig. 1d).
Aggregation characteristics and toxicity of pGlu79-α-synuclein
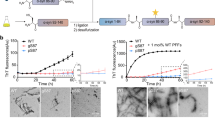
The aggregation characteristics of recombinant pGlu79-α-synuclein were compared to those of full-length wild-type α-synuclein by continuous agitation of the α-synuclein variants and simultaneous monitoring of fibril formation by ThT assay. The full-length α-synuclein displayed a typical sigmoidal fibril formation behaviour (Fig. 2a). In contrast, no fibril formation from pGlu79-α-synuclein was detected by ThT assay (Fig. 2a). Transmission electron microscopy substantiated fibril formation from full-length α-synuclein and the presence of aggregates lacking fibrillary structures from pGlu79-α-synuclein (Fig. 2b). The labelled material analysed by electron microscopy represents most likely small oligomers, as shown for the maternal MMP-3-cleaved α-synuclein fragments [61, 102]. After 72 h of agitation, to confirm the formation of oligomers from pGlu79-α-synuclein, full-length and pGlu79-α-synuclein were centrifuged to remove the insoluble fibrils, and the supernatants were analysed by SEC (Fig. 2c). The chromatograms of agitated α-synuclein variants (Fig. 2c) were compared to the respective chromatograms of their monomers (Suppl. Figure 2, online resource). Indeed, SEC analysis of the soluble fraction of agitated pGlu79-α-synuclein revealed a remarkably increased formation of oligomers. The levels of oligomers formed from agitated pGlu79-α-synuclein were three times as high as those from full-length α-synuclein (Fig. 2c). Together, data from ThT assays, electron microscopy and SEC demonstrated that pGlu79-α-synuclein is prone to form oligomers, while full-length α-synuclein preferred forming ThT-positive amyloid fibrils (Fig. 2d).

Aggregation characteristics of pGlu79-α-synuclein. a ThT assay to follow the characteristics of fibril formation from recombinant full-length α-synuclein and from pGlu79-α-synuclein. Note the typical, sigmoid-shaped curve of fibril formation from full-length α-synuclein (black trace) during the 110 h agitation period. In contrast, no fibril formation was observed for pGlu79-α-synuclein (blue trace). b Electron microscopic analysis of aggregates formed from full-length α-synuclein and from pGlu79-α-synuclein. Note the absence of fibril formation from pGlu79-α-synuclein but the presence of oligomers. c SEC and dot blot analysis of oligomers from full length or pGlu79-α-synuclein after 72 h agitation for protein aggregation. Agitated full-length and pGlu79-α-synuclein were centrifuged and the supernatants were analyzed by SEC. Their unagitated monomeric counterparts were also analyzed for determining the elution times of the monomers (Suppl. Figure 2, online resource). Peak elution times of full-length and pGlu79-α-synuclein monomers are 28.23 min and 30.74 min, respectively (see also Suppl. Figure 2, online resource). The representative chromatogram of agitated pGlu79-α-synuclein (right) demonstrates a remarkable increase in oligomers, characterized by a peak with an elution time of 29.93 min (indicated by O and red line), in addition to the peak for monomers at 31.86 min (M and black line). By contrast, the representative chromatogram of agitated full-length α-synuclein (left) is characterized by the presence of small peaks for oligomers with elution times between 5 and 25 min, however, to a much lesser extent (highlighted in the red inset), when compared to the peak for monomers at 29.40 min (black line). Dot blot analysis of SEC fractions confirmed the specificity of the peaks. For immunodetection, SEC fractions were either analyzed by the Syn1 antibody or the anti-pGlu79-α-synuclein antibody for detection of full-length α-synuclein and of pGlu79-α-synuclein, respectively. Quantification of three independent aggregation and SEC analyses shows significantly higher oligomer levels in agitated pGlu79-α-synuclein than in full-length α-synuclein. Statistical significance at **p < 0.01 defined by t test. d ThT assay and SEC, for analyzing fibril and oligomer formation, respectively, reveals that pGlu79-α-synuclein is more prone to form oligomers, however, is unable to form ThT-positive amyloid fibrils. e Analysis of cellular toxicity of monomers and aggregates of full-length α-synuclein (5 µM) and pGlu79-α-synuclein (5 µM). Cell viability was assessed by WST-1 assay in differentiated SH-SY5Y cells after 72 h of treatment with the peptides (mean ± SD, n = 3, *p < 0.05; **p < 0.01 defined by one-way ANOVA followed by Tukey post-hoc analysis)
To study the toxicity of full-length α-synuclein and pGlu79-α-synuclein, a WST-1 assay was performed using SH-SY5Y neuroblastoma cells. Monomers of both α-synuclein species and aggregates thereof produced by 72 h agitation were tested at concentrations of 5 µM and normalized to the vehicle control PBS. Compared to the monomeric peptides, a significant cytotoxic potential of aggregates of full-length α-synuclein and pGlu79-α-synuclein was observed (full-length α-synuclein monomers: 82% viability, full-length α-synuclein aggregates: 54% viability, pGlu79-α-synuclein monomers: 73% viability, pGlu79-α-synuclein aggregates: 50% viability, Fig. 2e).
QC expression by SN dopaminergic neurons
The co-expression of QC and full-length α-synuclein by dopaminergic SN neurons is a prerequisite for pGlu79-α-synuclein formation in this brain region. Therefore, we first analyzed the co-expression of these proteins together with TH, the marker enzyme of dopaminergic neurons, in the SN of wild-type mouse brain sections by triple immunofluorescent labellings. As shown in Fig. 3a, QC is abundantly expressed by TH-positive neurons that also display α-synuclein immunoreactivity.

QC and pGlu79-α-synuclein in wild type and transgenic mouse brain. a QC expression by mouse TH-positive SN neurons and co-localization with α-synuclein. Triple immunofluorescent labellings demonstrate the expression of QC (green) by dopaminergic SN neurons (blue) and co-expression of maternal full-length α-synuclein (red) in mouse brain. b In brain sections of ASO mice (top), immunoreactivity for pGlu79-α-synuclein is displayed in fine, disperse aggregates in stratum lacunosum (slm) and pyramidal cell layer (Py) of the CA2 and CA3 hippocampal subregions, respectively, while larger aggregates and Lewy body-like structures are depicted in lateral hypothalamus (LH) and SN. Aggregates of pGlu79-α-synuclein in BAC-SNCA mice (bottom) are shown in hippocampus, either rod-shaped in CA2-slm or finely scattered in CA1-, CA3-Py and subiculum (Sub). Triple immunofluorescent labelling revealed a frequent co-localization of these aggregates with QC, as exemplarily shown for LH in ASO and for CA2 in BAC-SNCA mouse brain
pGlu79-α-synuclein aggregates in transgenic mouse models
Next, we wanted to test whether pGlu79-α-synuclein aggregates contribute to histopathology in brains of transgenic ASO and BAC-SNCA mice overexpressing human wild-type α-synuclein. Single pGlu79-α-synuclein DAB labellings revealed the presence of such aggregates in hippocampal and subcortical structures (Fig. 3b). In hippocampus, pGlu79-α-synuclein deposits were detected in all subregions. However, they were particularly prominent in CA2 stratum lacunosum moleculare, where densely dispersed labellings emerged in ASO mouse brain and where rod-shaped, neuritic structures appeared in BAC-SNCA mice. In addition, Lewy body-like aggregates were detected in lateral hypothalamus and SN in ASO mouse brain. Brain regions affected by these deposits differed between experimental animal models, which might be due to different transgene expression patterns. In both transgenic animal models, there was a frequent co-localization of QC and pGlu79-α-synuclein in these aggregates as exemplarily shown for lateral hypothalamus in ASO and for CA2 in BAC-SNCA mouse brain (Fig. 3b).
pGlu79-α-synuclein aggregates in human substantia nigra
The main goal of our study was to reveal the presence and potential aggregation of pGlu79-α-synuclein in human clinical conditions of PD and DLB. Therefore, well-characterized high quality human brain tissue of short post mortem delay (1.5–4.2 h) was analyzed by immunohistochemistry. The dopaminergic SN neurons in human brain tissue can be easily identified by the intracellular presence of brown NM. However, this excludes the possibility of simultaneous immunohistochemical detection of intracellular antigens by brown DAB labelling in these neurons. We, therefore, visualized pGlu79-α-synuclein using DAB-Ni as histochemical substrate, resulting in black labelling.
In human control subjects, only a small proportion of the numerous NM-positive neurons contained pGlu79-α-synuclein (Fig. 4). In PD and DLB cases, the number of NM-positive SN neurons was drastically reduced, consistent with the known degeneration of this cell group in the course of both clinical conditions (for quantification see Fig. 5). The remaining NM-containing neurons frequently displayed pGlu79-α-synuclein immunoreactivity and morphological signs of degeneration, such as shrinkage and irregular shape. In addition, PD-typical features, such as Lewy bodies and Lewy neuritis, were pGlu79-α-synuclein immunoreactive (Fig. 4). We conclude that a fraction of deposited α-synuclein in Lewy bodies and Lewy neurites consists of or contains pGlu79-α-synuclein.

pGlu79-α-synuclein in human SN. a Typical examples of pGlu79-α-synuclein immunoreactivity in human SN of control subjects as well as PD and DLB patients. In control subjects, neuromelanin-containing SN neurons only sparsely contain pGlu79-α-synuclein (black DAB-Ni labelling). In PD and in DLB, the density of neuromelanin-containing neurons is markedly reduced, consistent with the degeneration of dopaminergic SN neurons in these clinical conditions. In addition, a high proportion of neuromelanin-positive neurons contains pGlu79-α-synuclein in PD and DLB cases. b pGlu79-α-synuclein immunoreactivity is also present in pathological structures, such as Lewy bodies (arrows) and Lewy neurites (asterisk) in PD and DLB cases

QC in human SN: Relation to neuromelanin. a Typical examples of immunohistochemical QC labellings (black) in SN of control, PD and DLB cases. Note the reduced numbers of neuromelanin-containing neurons (brown) in PD and DLB as compared to control and the differential association of QC (black) with brown, neuromelanin-containing neurons in the high magnification images (a’). b Quantification of all neuromelanin-containing (NM+) neurons (light bars) and NM+ neurons expressing QC (NM+/QC+; dark bars) illustrating the substantial loss of NM neurons in DLB and PD and the high proportion of NM+ neurons expressing QC in DLB and PD clinical conditions. c Proportions of NM+ neurons expressing QC (NM+/QC+) versus not expressing QC (NM+/QC−). Note the higher proportion of NM+ neurons expressing QC in DLB and PD clinical conditions. d Proportions of QC-expressing neurons associated with neuromelanin (QC+/NM+) versus not associated with neuromelanin (QC+/NM−). e Pie charts illustrating the drastically low proportion of NM+ only neurons in PD (6.1%) and in DLB (15.1%) compared to controls (45.7%), whereas the proportions of QC+ only and of NM+/QC+ neurons are increased in these groups. Mean ± SEM, n = 10, Statistical significance at *p < 0.05; **p < 0.005; ***p < 0.001 defined by t test
QC expression by human SN neurons: relation to neuromelanin
In analogy to the pGlu79-α-synuclein labelling, the expression of the enzyme QC catalyzing the pGlu modification was evaluated in human SN of control, PD and DLB cases (Fig. 5). Typically, in all conditions analyzed there were NM-containing neurons without QC immunoreactivity (NM+/QC−), NM-positive neurons expressing QC (NM+/QC+) and NM-negative neurons solely immunoreactive for QC (NM−/QC+) (Fig. 5a; highlighted in Fig. 5a’). As described above, the total number of NM-positive neurons (NM+) was much lower in DLB (587 ± 107 per brain section) and in PD (425 ± 77 per brain section) as compared to controls (1731 ± 270 per brain section) (Fig. 5b). In contrast, the numbers of QC+ neurons were only reduced from 964 ± 155 in controls to 580 ± 110 in DLB and to 512 ± 80 in PD (Fig. 5b).
When calculating the proportions of NM+/QC+ and of NM+/QC− neurons in the various groups, controls displayed a 52% NM+/QC+ to 48% NM+/QC− ratio, that was significantly different from DLB (82.5% to 17.5%) and from PD (92% to 8%) (Fig. 5c). Thus, in both clinical conditions a greater proportion of NM+ neurons displayed QC immunoreactivity.
From the perspective of all QC immunoreactive neurons, 91% were associated with NM in control SN, whereas this proportion was reduced to 84% in DLB and to 74% in PD (Fig. 5d). This was accompanied by a concomitant increase in the proportion of QC+/NM− neurons from 9% in controls to 16% in DLB and 26% in PD (Fig. 5d). Out of these, a subgroup of morphologically degenerating and often shrunken or fractionated neurons stood out, which displayed excessive QC immunoreactivity (see below).
The summary pie charts (Fig. 5e) illustrate the drastic reduction of the proportion of NM+/QC− neurons in DLB (15%) and in PD (6%) compared to controls (46%), whereas the proportions of QC+/NM+ and of QC+/NM− neurons are increased.
QC in human SN: presence in neuropathological structures
QC was not only found to be associated with NM-positive neurons in the SN, but was additionally detected in typical neuropathological structures of synucleinopathies. These structures include degenerating neurons (Fig. 6a), axonal or dendritic varicosities and Lewy neurites/axonal bulbs (Fig. 6b), as well as Lewy body-like aggregates, smaller and larger than 5 µm in diameter (Fig. 6c, d). The corresponding quantifications demonstrate a much higher abundance of QC-immunoreactive degenerating neurons in DLB (4.9%) and in PD (12.1%) than in control conditions (0.9%) (Fig. 6a). While the numbers of QC-immunoreactive varicosities did not differ between clinical groups, counts of QC-immunoreactive Lewy neurites/axonal bulbs in DLB (164 ± 33) and PD (149 ± 48) were significantly higher compared to controls (23 ± 6) (Fig. 6b). Interestingly, both, the numbers of QC-immunoreactive Lewy body-like aggregates of smaller and of larger size, were significantly higher in SN of PD, but not of DLB, subjects compared to controls (Fig. 6c, d). This is consistent with the more cortical pathology of DLB as compared to the SN pathology in PD [51].

QC in human SN: presence in neuropathological structures. QC (labelled in black) was found to be associated with degenerating neurons (a), with varicosities (arrows) and Lewy neurites/axonal bulbs (asterisks) (b), as well as with small (c) and large (d) Lewy body-like structures. The quantification of these structures revealed a high association of QC with Lewy body-like aggregates in PD, but not in DLB (c, d) no significant differences in the number of QC-positive varicosities between groups, but a similarly strong increase in the number of QC immunoreactive Lewy neurites and axonal bulbs in DLB and PD compared to controls (CO) (b), and a more abundant association of QC with degenerating neurons in PD than in DLB (a). Brown color arises from neuromelanin. Mean ± SEM, n = 10, Statistical significance at *p < 0.05; **p < 0.01; ***p < 0.001 defined by t-test
However, it should be noted that these QC-immunoreactive structures were also present in control subjects; although to a much lower extent. These aggregates, therefore, may represent early pathological events in clinically silent subjects.
Discussion
Formation of pGlu79-α-synuclein by QC
Post-translational modifications of α-synuclein have been extensively studied with respect to the regulation of its physiological functions and their contribution to pathological processes in clinical conditions, such as PD and DLB [17, 37]. The most prominent disease-related modification is α-synuclein phosphorylation at serine129 [4, 32, 86], which could become an early peripheral diagnostic marker [1, 26, 30, 67, 104].
In the present study, we focused on proteolytically cleaved fragments of α-synuclein. MMP-3 cleavage of α-synuclein generates a number of defined fragments with distinct biophysical and cell biological properties [80, 102], for review see [13, 98]. One of the identified MMP-3-generated α-synuclein fragments, Gln79-α-synuclein, possesses a glutamine residue at its N-terminus and could, therefore, serve as QC substrate. In cell free assays, we demonstrate QC-catalyzed conversion of this fragment into the pGlu-modified form that follows kinetic characteristics of enzyme-catalyzed reactions (see Fig. 1). When we compared the aggregation behavior of pGlu79-α-synuclein to that of maternal, full-length α-synuclein applying ThT assays, we noticed a lack of fibril formation from pGlu79-α-synuclein. This is consistent with the loss of a significant part of the NAC domain and observations by others, analyzing the aggregation behavior of different α-synuclein fragments [33, 61, 102]. Cryo-EM structures of full-length α-synuclein reveal that various residues (46–95) are associated with fibril formation and stabilization by the generation of hydrophobic cores of a single protofilament and steric zippers of two protofilaments [39, 62]. Since most of these residues critical for fibril stabilization are absent in pGlu79-α-synuclein, fibrillation is most likely disabled and aggregation is limited to the oligomeric state. In addition, it is speculated that fibril elongation by hydrophobic interaction might be related to residues V74–V82, because β-synuclein lacking these residues is incapable of forming fibrils [36]. However, the lack of fibril formation does not exclude the generation of oligomeric α-synuclein assemblies after MMP-3 cleavage as established in two independent studies employing different analytical methods [61, 102]. Indeed, uncleaved α-synuclein oligomers have been shown to be a neurotoxic species in vivo [84, 107] and, intriguingly, α-synuclein oligomers generated following processing by MMP-3 have also been shown to exert a more toxic effect on cultured cells than oligomers composed of full-length α-synuclein [61, 102]. Here, using ThT assay and size exclusion chromatography, we demonstrate distinct aggregation behaviors of full-length and pGlu79-α-synuclein. Under continuous agitation, a classic in vitro condition for fibrillization, a large fraction of full-length α-synuclein is converted into amyloid fibrils, whereas pGlu79-α-synuclein is prone to form soluble oligomers. The lack of formation of ThT positive amyloid fibrils of the latter suggests an off-pathway in the aggregation of this cleaved and modified α-synuclein species. These observations are also reflected in the cytotoxicity assay. Although pGlu79-α-synuclein does not tend to form fibrils, its toxicity on neuroblastoma cells is comparable to aggregates of full-length α-synuclein. This is in line with evidence that MMP-3 knock-out mice display attenuated neuronal death in the MPTP mouse PD model in vivo [56]. Moreover, prion-like seeding of misfolded α-synuclein is propagated from oligomer-like species, but not from insoluble aggregates [11, 27, 87] and α-synuclein oligomers were reported to stabilize pre-existing defects in supported bilayers and propagate membrane damage [19]. In living cells, the stabilization of α-synuclein oligomers resulted in increased cytotoxicity, which could be rescued by Hsp70 via suppression of oligomer formation [77]. Thus, oligomer formation from α-synuclein fragments lacking parts of the NAC domain is likely to lead to pathogenic protein assemblies.
Detection of pGlu79-α-synuclein in transgenic animal models
If QC-catalyzed modification of α-synuclein does occur in vivo, the maternal α-synuclein and QC should be co-localized. QC is not ubiquitously distributed throughout the brain but is rather restricted to defined regions, such as hypothalamus and pituitary, where physiological substrates reside [14, 43]. In addition, QC is abundant in brain nuclei, such as nucleus basalis Meynert, locus coeruleus and Edinger–Westphal nucleus, which are affected by amyloid pathology in AD [45, 73]. Here, we demonstrate co-expression of α-synuclein and QC by TH-positive dopaminergic SN neurons in brain of wild-type mice.
In addition, two transgenic mouse lines with overexpression of human α-synuclein—ASO and BAC-SNCA—were analyzed. Aged BAC-SNCA mice have a 2.7-fold amount of endogenous α-synuclein and show highest transgene expression levels in cortex, striatum and hippocampus [108]. They display a high abundance of monomeric α-synuclein but also higher order SDS-resistant α-synuclein [71]. In ASO mice, Thy-1-driven transgene expression results in 1.5–3.4-fold overexpression of α-synuclein in many brain regions including olfactory bulb, SN, thalamus, cortex, hippocampus and striatum [20]. Already in 5-month-old transgenic mice, proteinase K-resistant α-synuclein aggregates are present in SN and oligomeric assemblies were detected by Western blot analysis [20]. The transgene is inserted in the X chromosome, which leads to diminished motor deficits in females, most likely due to random inactivation of the X chromosome carrying the mutation [20]. Therefore, only male ASO mice were used in the present study.
When detecting pGlu79-α-synuclein in both α-synuclein overexpressing mouse models, we found increased immunoreactivity in subcellular sites of physiological α-synuclein presence, such as synapses, confirming the specificity of the antibody. Focusing on histopathological features, we identified a co-localization of QC and pGlu79-α-synuclein in Lewy body-like structures predominantly in ASO mice and in disperse aggregates in brain tissue of both mouse models. These aggregates were found in brain structures that are known for the formation of α-synuclein deposits in these animal models but also in different hippocampal subregions, such as subiculum, CA1, CA3 and pronounced in the stratum lacunosum sublayer of CA2. Distinct hippocampal CA2 synuclein pathology has been associated with cholinergic degeneration in PD with cognitive decline [64]. Thus, there is co-localization of QC with α-synuclein in SN of mouse brain and a spatial association of both proteins in protein aggregates of transgenic animal models of synucleinopathies with relevance to the clinical condition in human patients.
Localization of QC and pGlu79-α-synuclein in human brains affected by synucleinopathies
Animal models for protein aggregation disorders do not always reflect the exact scenario present in human disease. Therefore, it is mandatory to validate observations derived from experimental animals in human brain tissue. Here, using well-characterized human brain tissue sections from control subjects as well as PD and DLB patients, we demonstrate robust deposition of pGlu79-α-synuclein in SN in a disease-specific manner. The detection of pGlu79-α-synuclein in Lewy bodies and Lewy neurites is reminiscent of the classical α-synuclein aggregates representing typical pathological hallmarks in Lewy body diseases, such as PD and DLB [16, 88]. The synucleinopathy typically affects NM-containing dopaminergic neurons of the SN complex [48, 69, 96]. The precise biochemical composition of Lewy bodies has not yet been decoded. However, in addition to the main component full-length α-synuclein, the presence of truncated fragments [110] and of pSer129-α-synuclein [4] has been reported. Here, we show for the first time pGlu79-α-synuclein immunoreactivity in neuromelanin positive SN neurons and accumulation in Lewy bodies and Lewy neurites. The post-translational α-synuclein modification present in this fragment is catalyzed by QC, which we demonstrated to be associated with neuropathological structures in SN of PD and DLB subjects in addition to its expression in neuronal somata. It was not in the scope of this study to define the exact nature of the varieties of QC- and pGlu79-α-synuclein-immunoreactive axonal and parenchymal neuronal structures with pathological appearance. Descriptions of the morphology and nature of small pathogenic alterations and even the subcellular localization of varicosities, axonal or dendritic, in human PD and DLB brain tissue differ considerably in the literature [78, 103], for review see [38].
Interestingly, the proportion of NM-containing SN neurons that express QC is increased in PD and DLB (see Fig. 5e), indicating a contribution of QC to both disorders. This finding can be interpreted in two different ways. On the one hand one could reason that neuronal degeneration mainly affects the subset of NM+ neurons which is devoid of QC, assuming that its presence in surviving neurons is neuroprotective. On the other hand QC expression may be newly induced in a neuronal subfraction of SNc in the course of the disease, rendering the respective neurons more vulnerable to the pathological process.
In PD brains, compared to the DLB or the control group, we observed a statistically significant increase in the actual number of neurons in the SNc that solely express QC without containing neuromelanin. This can only be explained by de novo synthesis of QC in a substantial fraction of this neuronal category (QC+/NM−). Furthermore, the percentage of degenerating neurons with excessively high QC immunoreactivity is augmented five and twelve times, respectively, in DLB and PD cases in comparison to control subjects (Fig. 6a). Taken together, it appears most likely that QC expression by SNc neurons is a factor which contributes to neuropathology rather than to neuroprotection. However, while the incidence of QC-positive structures in Lewy neurites and varicosities is comparable in PD and DLB, the association of QC with Lewy body-like structures in SN was more pronounced in PD than in DLB (Fig. 6), which is consistent with a more cortical pathology in DLB [51]. It would be interesting to reveal whether the same type of association between QC and pGlu79-α-synuclein and neuropathological structures is also present in other types of synucleinopathies, such as multiple system atrophy or Alzheimer’s disease with amygdala-restricted Lewy bodies that were not analyzed here.
We propose that QC-catalyzed pGlu-modification of pathogenic proteins might be a more general mechanism in human clinical conditions than previously thought. In AD, it is well established that the pGlu modification of Aβ peptides initially described by the Saido and Roher groups [58, 85] compromises Aβ degradation and increases its toxicity [76] as well as aggregation propensity in human cortex, hippocampus and subcortical brain nuclei affected by amyloid pathology [42, 72, 73]. There are currently two independent therapeutic strategies being tested in preclinical studies and in clinical settings: (1) inhibition of QC to prevent pGlu-Aβ formation and (2) targeting existing amyloid assemblies by pGlu-Aβ-specific antibodies.
Pharmacological inhibition of QC activity and genetic ablation of QC in transgenic animal models reduced pGlu-Aβ generation and total Aβ load and ameliorated learning and memory deficits [3, 44, 50, 93]. In addition, clinical trials indicated safety, tolerability and efficacy of the QC inhibitor PQ912 in human subjects [65, 89]. For information on ongoing clinical trials, see https://www.alzforum.org/therapeutics/varoglutamstat.
Targeting pGlu-Aβ using a well-characterized monoclonal antibody [79] reduced amyloid plaques and improved cognition in APP transgenic mice [22, 31, 49]. Using this specific passive immunization approach, an attenuated cognitive decline was reported in people with early AD in the Phase 2 TRAILBLAZER-ALZ study (https://www.alzforum.org/news/research-news/phase-2-donanemab-curbs-cognitive-decline-early-alzheimers).
Thus, there are well-characterized tools with defined cell biological, pharmacological and safety properties already available that can be tested in cellular and animal models of synucleinopathies in a straightforward way, to interfere with pGlu79-α-synuclein generation and to test its contribution to motor, cognitive and histological disturbances.
Conclusions
We demonstrate the QC-catalyzed formation of pGlu79-α-synuclein, its oligomerization and neurotoxic profiles and its accumulation in brain in two transgenic mouse models of synucleinopathy. In human brain, the presence of QC and pGlu79-α-synuclein is largely increased in neurons of the SN and associated with pathological structures in PD and DLB subjects. Given the resistance of pGlu-modified proteins against proteolytical degradation in general and the high oligomer formation velocity of pGlu79-α-synuclein in particular, these molecules might be interesting novel targets for pharmacologically interfering with human synucleinopathies.
Availability of data and materials
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- BSA:
-
Bovine serum albumin
- DAB:
-
3,3′-Diaminobenzidine
- DLB:
-
Dementia with Lewy bodies
- DMF:
-
Dimethyl formamide
- EDT:
-
1,2-Ethanedithiol
- ESI:
-
Electrospray ionization
- MMP:
-
Matrix metalloproteinase
- NAC:
-
Non-Abeta component
- NM:
-
Neuromelanin
- PBS:
-
Phosphate-buffered saline
- PD:
-
Parkinson’s disease
- QC:
-
Glutaminyl cyclase
- SN:
-
Substantia nigra
- SNc:
-
Substantia nigra pars compacta
- TBS:
-
Tris-buffered saline
- TFA:
-
Trifluoroacetic acid
- TH:
-
Tyrosine hydroxylase
- ThT:
-
Thioflavin T
- TIS:
-
Triisopropylsilane
References
Abd Elhadi S, Grigoletto J, Poli M, Arosio P, Arkadir D, Sharon R (2019) alpha-Synuclein in blood cells differentiates Parkinson’s disease from healthy controls. Ann Clin Transl Neurol 6:2426–2436
Adler CH, Beach TG, Zhang N, Shill HA, Driver-Dunckley E, Caviness JN et al (2019) Unified staging system for Lewy body disorders: Clinicopathologic correlations and comparison to Braak staging. J Neuropathol Exp Neurol 78:891–899
Alexandru A, Jagla W, Graubner S, Becker A, Bäuscher C, Kohlmann S et al (2011) Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation. J Neurosci 31:12790–12801
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ et al (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281:29739–29752
Araki K, Yagi N, Aoyama K, Choong CJ, Hayakawa H, Fujimura H et al (2019) Parkinson’s disease is a type of amyloidosis featuring accumulation of amyloid fibrils of α-synuclein. Proc Natl Acad Sci USA 116:17963–17969
Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G et al (2021) Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta Neuropathol 141:159–172
Bartels AL, Leenders KL (2009) Parkinson’s disease: the syndrome, the pathogenesis and pathophysiology. Cortex 45:915–921
Bartels T, Choi JG, Selkoe DJ (2011) α-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477:107–110
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J et al (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634
Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG et al (2015) Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathology 35:354–389
Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2017) Alpha-synuclein oligomers: a new hope. Acta Neuropathol 134:819–838
Bloem BR, Okun MS, Klein C (2021) Parkinson’s disease. Lancet. https://doi.org/10.1016/S0140-6736(21)00218-X
Bluhm A, Schrempel S, von Hörsten S, Schulze A, Roßner S (2021) Proteolytic cleavage of α-synuclein in health and disease. Int J Mol Sci 22:5450
Böckers TM, Kreutz MR, Pohl T (1995) Glutaminyl-cyclase expression in the bovine/porcine hypothalamus and pituitary. J Neuroendocrinol 7:445–453
Braak H, Del Tredici K (2008) Cortico-basal ganglia-cortical circuitry in Parkinson’s disease reconsidered. Exp Neurol 212:226–229
Brás IC, Dominguez-Meijide A, Gerhardt E, Koss D, Lázaro DF, Santos PI et al (2020) Synucleinopathies: where we are and where we need to go. J Neurochem 153:433–454
Burré J, Sharma M, Südhof TC (2018) Cell biology and pathophysiology of α-synuclein. Cold Spring Harb Perspect Med 8(3):a024091
Busby WH, Quackenbush GE, Humm J, Youngblood WW, Kizer JS (1987) An enzyme(s) that converts glutaminyl-peptides into pyroglutamyl-peptides. J Biol Chem 262:8532–8536
Chaudhary H, Iyer A, Subramaniam V, Claessens MM (2016) alpha-synuclein oligomers stabilize pre-existing defects in supported bilayers and propagate membrane damage in a fractal-like pattern. Langmuir 32:11827–11836
Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR (2012) A progressive mouse model of Parkinson’s disease: The Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 9:297–314
Consensus recommendations for the postmortem diagnosis of Alzheimer's disease (1997) The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 18:S1–2
Crehan H, Liu B, Kleinschmidt M, Rahfeld JU, Le KX, Caldarone BJ et al (2020) Effector function of anti-pyroglutamate-3 Aβ antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimers Res Ther 12:12
Cynis H, Scheel E, Saido TC, Schilling S, Demuth HU (2008) Amyloidogenic processing of amyloid precursor protein: evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-beta. Biochemistry 47:7405–7413
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain: I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D28K immunohistochemistry. Brain 122:1421–1436
Dettmer U, Selkoe D, Bartels T (2015) New insights into cellular alpha-synuclein homeostasis in health and disease. Curr Opin Neurobiol 36:15–22
Doppler K (2021) Detection of dermal alpha-synuclein deposits as a biomarker for Parkinson's disease. J Parkinson's Dis (in press)
Du XY, Xie XX, Liu RT (2020) The role of α-synuclein oligomers in Parkinson’s disease. Int J Mol Sci 21:8645
Fares MB, Jagannath S, Lashuel HA (2021) Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci 22:111–131
Fischer WH, Spiess J (1987) Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc Natl Acad Sci USA 84:3628–3632
Foulds PG, Mitchell JD, Parker A, Turner R, Green G, Diggle P et al (2011) Phosphorylated alpha-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson’s disease. FASEB J 25:4127–4137
Frost JL, Liu B, Rahfeld JU, Kleinschmidt M, O’Nuallain B, Le KX et al (2015) An anti-pyroglutamate-3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol Aging 36:3187–3199
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS et al (2002) α-Synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biol 4:160–164
Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM et al (2014) Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat Commun 5:3827
Galvan A, Wichmann T (2008) Pathophysiology of parkinsonism. Clin Neurophysiol 119:1459–1474
Garcia-Reitböck P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E et al (2010) SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 133:2032–2044
Giasson BI, Murray IV, Trojanowski JQ, Lee VM (2001) A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem 276:2380–2386
González N, Arcos-López T, König A, Quintanar L, Menacho Márquez M, Outeiro TF et al (2019) Effects of alpha-synuclein post-translational modifications on metal binding. J Neurochem 150:507–521
Gu C (2021) Rapid and reversible development of axonal varicosities: A new form of neural plasticity. Front Mol Neurosci 14:1–13
Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R et al (2018) Cryo-EM structure of alpha-synuclein fibrils. Elife 7:e36402
Halliday GM, Holton JL, Revesz T, Dickson DW (2011) Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 122:187–204
Hartlage-Rübsamen M, Bluhm A, Piechotta A, Linnert M, Rahfeld JU, Demuth HU et al (2018) Immunohistochemical evidence from APP-Transgenic mice for glutaminyl cyclase as drug target to diminish pE-Abeta formation. Molecules 23:924
Hartlage-Rübsamen M, Morawski M, Waniek A, Jäger C, Zeitschel U, Koch B et al (2011) Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Aβ deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol 121:705–719
Hartlage-Rübsamen M, Staffa K, Waniek A, Wermann M, Hoffmann T, Cynis H et al (2009) Developmental expression and subcellular localization of glutaminyl cyclase in mouse brain. Int J Devl Neurosci 27:825–835
Hoffmann T, Meyer A, Heiser U, Kurat S, Böhme L, Kleinschmidt M et al (2017) Glutaminyl cyclase inhibitor PQ912 improves cognition in mouse models of Alzheimer’s Disease-Studies on relation to effective target occupancy. J Pharmacol Exp Ther 362:119–130
Höfling C, Indrischek H, Höpcke T, Waniek A, Cynis H, Koch B et al (2014) Mouse strain and brain region-specific expression of the glutaminyl cyclases QC and isoQC. Int J Dev Neurosci 36:64–73
Hortschansky P, Schroeckh V, Christopeit T, Zandomeneghi G, Fändrich M (2005) The aggregation kinetics of Alzheimer’s beta-amyloid peptide is controlled by stochastic nucleation. Protein Sci 14:1753–1759
Iljina M, Garcia GA, Horrocks MH, Tosatto L, Choi ML, Ganzinger KA et al (2016) Kinetic model of the aggregation of α-synuclein provides insights into prion-like spreading. Proc Natl Acad Sci USA 113:E1206–E1215
Isaias IU, Trujillo P, Summers P, Marotta G, Mainardi L, Pezzoli G et al (2016) Neuromelanin imaging and dopaminergic loss in Parkinson’s disease. Front Aging Neurosci 8:196
Janssens J, Hermans B, Vandermeeren M, Barale-Thomas E, Borgers M, Willems R et al (2021) Passive immunotherapy with a novel antibody against 3pE-modified Aβ demonstrates potential for enhanced efficacy and favorable safety in combination with BACE inhibitor treatment in plaque-depositing mice. Neurobiol Dis 154:105365
Jawhar S, Wirths O, Schilling S, Graubner S, Demuth HU, Bayer TA (2011) Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J Biol Chem 286:4454–4460
Jellinger KA (2018) Dementia with Lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J Neural Transm 125:615–650
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386:896–912
Kapust RB, Tözsér J, Copeland TD, Waugh DS (2002) The P1’ specificity of tobacco etch virus protease. Biochem Biophys Res Commun 294:949–955
Killinger BA, Kordower JH (2019) Spreading of alpha-synuclein – relevant or epiphenomenon? J Neurochem 150:605–611
Kim HJ, Lee D, Lee CH, Chung KC, Kim J, Paik SR (2006) Calpain-resistant fragment(s) of alpha-synuclein regulates the synuclein-cleaving activity of 20S proteasome. Arch Biochem Biophys 455:40–47
Kim YS, Choi DH, Block ML, Lorenzl S, Yang L, Kim YJ et al (2007) A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J 21:179–187
Köppen J, Schulze A, Machner L, Wermann M, Eichentopf R, Guthardt M et al (2020) Amyloid-beta peptides trigger aggregation of alpha-synuclein in vitro. Molecules 25:580
Kuo YM, Emmerling MR, Woods AS, Cotter RJ, Roher AE (1997) Isolation, chemical characterization, and quantitation of A beta 3-pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits. Biochem Biophys Res Commun 237:188–191
Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38–48
Leak RK, Frosch MP, Beach TG, Halliday GM (2019) Alpha-synuclein: prion or prion-like? Acta Neuropathol 138:509–514
Levin J, Giese A, Boetzel K, Israel L, Högen T, Nübling G et al (2009) Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp Neurol 215:201–208
Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G et al (2018) Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun 9:3609
Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L et al (2005) Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci USA 102:2162–2167
Liu AKL, Chau TW, Lim EJ, Ahmed I, Chang RC, Kalaitzakis ME et al (2019) Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta Neuropathol Commun 7:61
Lues I, Weber F, Meyer A, Bühring U, Hoffmann T, Kühn-Wache K et al (2015) A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects. Alzheimers Dement (N Y) 1:182–195
Mai JK, Assheuer J, Paxinos G (2004) Atlas of the human brain. Academic Press, San Diego
Majbour NK, Vaikath NN, van Dijk KD, Ardah MT, Varghese S, Vesterager LB et al (2016) Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol Neurodegener 11:7
Marsden CD (1990) Parkinsons’s disease. Lancet 335:948–952
Martín-Bastida A, Lao-Kaim NP, Roussakis AA, Searle GE, Xing Y, Gunn RN et al (2019) Relationship between neuromelanin and dopamine terminals within the Parkinson’s nigrostriatal system. Brain 142:2023–2036
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D et al (2017) Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 89:88–100
Minakaki G, Canneva F, Chevessier F, Bode F, Menges S, Timotius IK et al (2019) Treadmill exercise intervention improves gait and postural control in alpha-synuclein mouse models without inducing cerebral autophagy. Behav Brain Res 363:199–215
Morawski M, Schilling S, Kreuzberger M, Waniek A, Jäger C, Koch B et al (2014) Glutaminyl cyclases in human cortex—correlation with (pGlu)-Abeta load and cognitive decline in Alzheimer’s disease. J Alzheimers Dis 39:385–400
Morawski M, Hartlage-Rübsamen M, Jäger C, Waniek A, Schilling S, Schwab C et al (2010) Distinct glutaminyl cyclase expression in Edinger-Westphal nucleus, locus coeruleus and nucleus basalis Meynert contributes to pGlu-Aβ pathology in Alzheimer’s disease. Acta Neuropathol 120:195–207
Murray IVJ, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H et al (2003) Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42:8530–8540
Nicastro N, Mak E, Surendranathan A, Rittman T, Rowe JB, O’Brien JT (2021) Altered structural connectivity networks in dementia with lewy bodies. Brain Imaging Behavior. https://doi.org/10.1007/s11682-020-00444-x
Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T et al (2012) Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485:651–655
Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F et al (2008) Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One 3:e1867
Patt S, Gertz HJ, Gerhard L, Cervós-Navarro J (1991) Pathological changes in dendrites of substantia nigra neurons in Parkinson’s disease: a Golgi study. Histol Histopathol 6:373–380
Piechotta A, Parthier C, Kleinschmidt M, Gnoth K, Pillot T, Lues I et al (2017) Structural and functional analyses of pyroglutamate-amyloid-β-specific antibodies as a basis for Alzheimer immunotherapy. J Biol Chem 292:12713–12724
Pieri L, Chafey P, Le Gall M, Clary G, Melki R, Redeker V (2016) Cellular response of human neuroblastoma cells to α-synuclein fbrils, the main constituent of Lewy bodies. Biochim Biophys Acta 1860:8–19
Pohl T, Zimmer M, Mugele K, Spiess J (1991) Primary structure and functional expression of a glutaminyl cyclase. Proc Natl Acad Sci USA 88:10059–10063
Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P (2004) Alpha-synuclein and Parkinson’s disease. FASEB J 18:617–626
Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I et al (2002) Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res 68:568–578
Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C et al (2014) Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137:1496–1513
Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S (1995) Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 14:457–466
Samuel F, Flavin WP, Iqbal S, Pacelli C, Sri Renganathan SD, Trudeau LE et al (2016) Effects of serine 129 phosphorylation on alpha-synuclein aggregation, membrane association, and internalization. J Biol Chem 291:4374–4385
Sano K, Atarashi R, Satoh K, Ishibashi D, Nakagaki T, Iwasaki Y et al (2018) Prion-like seeding of misfolded alpha-synuclein in the brains of Dementia with Lewy Body patients in RT-QUIC. Mol Neurobiol 55:3916–3930
Schaffert LN, Carter WG (2020) Do post-translational modifications influence protein aggregation in neurodegenerative diseases: a systematic review. Brain Sci 10:232
Scheltens P, Hallikainen M, Grimmer T, Duning T, Gouw AA, Teunissen CE et al (2018) Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res Ther 10:107
Schilling S, Hoffmann T, Wermann M, Heiser U, Wasternack C, Demuth HU (2002) Continuous spectrometric assays for glutaminyl cyclase activity. Anal Biochem 303:49–56
Schilling S, Manhart S, Hoffmann T, Ludwig HH, Wasternack C, Demuth HU (2003) Substrate specificity of glutaminyl cyclases from plants and animals. Biol Chem 384:1583–1592
Schilling S, Hoffmann T, Manhart S, Hoffmann M, Demuth HU (2004) Glutaminyl cyclases unfold glutamyl cyclase activity under mild acid conditions. FEBS Lett 563:191–196
Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A et al (2008) Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nature Med 14:1106–1111
Schlenzig D, Rönicke R, Cynis H, Ludwig HH, Scheel E, Reymann K et al (2012) N-Terminal pyroglutamate formation of Abeta38 and Abeta40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J Neurochem 121:774–784
Schulze A, Wermann M, Demuth HU, Yoshimoto T, Ramsbeck D, Schlenzig D et al (2018) Continuous assays for meprin alpha and beta using prolyl tripeptidyl aminopeptidase (PtP) from porphyromonas gingivalis. Anal Biochem 559:11–16
Schulz-Schaeffer WJ (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120:131–143
Seifert F, Schulz K, Koch B, Manhart S, Demuth HU, Schilling S (2009) Glutaminyl cyclases display significant catalytic proficiency for glutamyl substrates. Biochemistry 48:11831–11833
Sorrentino ZA, Giasson BI, Chakrabarty P (2019) α-Synuclein and astrocytes: tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol 138:1–21
Sorrentino ZA, Giasson BI (2020) The emerging role of α-synuclein truncation in aggregation and disease. J Biol Chem 295:10224–10244
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251:205–208
Sung JY, Park SM, Lee CH, Um JW, Lee HJ, Kim J et al (2005) Proteolytic cleavage of extracellular secreted alpha-synuclein via matrix metalloproteinases. J Biol Chem 280:25216–25224
Tagliaferro P, Burke RE (2016) Retrograde axonal degeneration in Parkinson disease. J Parkinsons Dis 6:1–15
Tian C, Liu G, Gao L, Soltys D, Pan C, Stewart T et al (2019) Erythrocytic alpha-synuclein as a potential biomarker for Parkinson’s disease. Transl Neurodegener 8:15
Vicente Miranda H, Szego ÉM, Oliveira LMA, Breda C, Darendelioglu E, de Oliveira RM et al (2017) Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 140:1399–1419
Watts JC (2019) Calling α-synuclein a prion is scientifically justifiable. Acta Neuropathol 138:505–508
Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S et al (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci USA 108:4194–4199
Yamakado H, Moriwaki Y, Yamasaki N, Miyakawa T, Kurisu J, Uemura K et al (2012) α-Synuclein BAC transgenic mice as a model for Parkinson’s disease manifested decreased anxiety-like behavior and hyperlocomotion. Neurosci Res 73:173–177
Xiang W, Schlachetzki JCM, Helling S, Bussmann JC, Berlinghof M, Schäffer TE et al (2013) Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci 54:71–83
Zhang J, Li X, Li JD (2019) The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front Neurosci 13:381
Acknowledgements
We are grateful to the Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program for the provision of human brain tissue. The Program has been supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson's Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. Aspects of this work were supported by the German Research Foundation (DFG grant #RO2226/13-1 to SR), by the Alzheimer Forschungsinitiative e.V. (AFI #16004 to SR), and by the German Federal Department of Education, Science, and Technology, BMBF (grant #01ED1501B to SR and grant #01ED1501C to SvH) within the European Union Joint Program for Neurodegenerative Disease (JPND) Research, Project CrossSeeds. We thank Angelika Hähnel and Jessica Klehm (Fraunhofer IMWS Halle/S., Germany) for electron microscopic analysis. AB received a Pre-doc Award funded by the Research Academy of Leipzig University.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MHR designed and supervised the study, performed the histological processing, pathological examination and quantification of human brain tissue and was a major contributor in writing the manuscript. AB and SM validated antibody specificity and performed immunohistochemical labellings and histological examination of mouse brain tissue. JK and AS performed enzymatic activity and mass spectrometry assays and electron microscopy. LM and AS expressed and purified α-synuclein variants and performed aggregation assays. MS established and performed cell culture and toxicity assays. IH and MH planned and analyzed biochemical and cell biological experiments. FR provided experimental tools for and scientific advice on animal experiments and was involved in interpretation of data. RC performed histological processing and pathological examination of human brain tissue. GES and TGB performed clinico-pathological classification of human subjects and contributed to writing the manuscript. WX and MW established and performed SEC experiments and WX provided research tools. SvH, AS, SS and WX designed and supervised the study, analyzed data and participated in writing the manuscript. SR initiated, designed and supervised the study and was a major contributor in writing the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Ethics approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All experiments carried out were in agreement with European regulations on the protection of animals used for scientific purposes (Directive 2010/63/EU), and they were also approved by the Ethical Committee for Animal Research of Landesdirektion Sachsen, license numbers T28/16 and the local ethical board of the District Government of Middle Franconia, Bavaria, Germany; approval # 55.2-DMS 2532-2-218.
Consent for publication
All the authors have approved publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hartlage-Rübsamen, M., Bluhm, A., Moceri, S. et al. A glutaminyl cyclase-catalyzed α-synuclein modification identified in human synucleinopathies. Acta Neuropathol 142, 399–421 (2021). https://doi.org/10.1007/s00401-021-02349-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02349-5