
Abstract
Cerebral amyloid angiopathy (CAA) consists of beta-amyloid deposition in the walls of the cerebrovasculature and is commonly associated with Alzheimer’s disease (AD). However, the association of CAA with repetitive head impacts (RHI) and with chronic traumatic encephalopathy (CTE) is unknown. We evaluated the relationship between RHI from contact sport participation, CTE, and CAA within a group of deceased contact sport athletes (n = 357), a community-based cohort (n = 209), and an AD cohort from Boston University AD Center (n = 241). Unsupervised hierarchal cluster analysis demonstrated a unique cluster (n = 11) with increased CAA in the leptomeningeal vessels compared to the intracortical vessels (p < 0.001) comprised of participants with significantly greater frequencies of CTE (7/11) and history of RHI. Overall, participants with CTE (n = 251) had more prevalent (p < 0.001) and severe (p = 0.010) CAA within the frontal leptomeningeal vessels compared to intracortical vessels. Compared to those with AD, participants with CTE had more severe CAA in frontal than parietal lobes (p < 0.001) and more severe CAA in leptomeningeal than intracortical vessels (p = 0.002). The overall frequency of CAA in participants with CTE was low, and there was no significant association between contact sport participation and the presence of CAA. However, in those with CAA, a history of contact sports was associated with increased CAA severity in the frontal leptomeningeal vessels (OR = 4.01, 95% CI 2.52–6.38, p < 0.001) adjusting for AD, APOE ε4 status, and age. Participants with CAA had increased levels of sulcal tau pathology and decreased levels of the synaptic marker PSD-95 (p’s < 0.05), and CAA was a predictor of dementia (OR = 1.75, 95% CI 1.02–2.99, p = 0.043) adjusting for age, sex, and comorbid pathology. Overall, contact sport participation and CTE were associated with more severe frontal and leptomeningeal CAA, and CAA was independently associated with worse pathological and clinical outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral amyloid angiopathy (CAA) is characterized by the deposition of beta-amyloid (Aβ) within the vessel walls of cerebral arteries, arterioles, and capillaries [10, 11]. CAA is common in older adults and has been reported in up to 98% of Alzheimer disease (AD) cases [5]. Increasing age and genetic factors impact CAA pathology such as the presence of the apolipoprotein E (APOE) allele ε4 [11, 42, 44]. CAA is primarily found within leptomeningeal vessels and superficial intracortical vessels and is generally more common in the posterior cerebral cortex (parietal and occipital lobes) than the frontal lobes [4, 43, 53,54,55,56]. Beta amyloid (Aβ) deposition in the blood vessel wall leads to progressive weakening and dysfunction of the vessel wall with rupture and subsequent intracerebral hemorrhage (ICH). CAA is a leading cause of ICH in elderly individuals [3, 26, 55]. Independent of other pathologies, CAA is associated with age as well as impaired cognitive function and dementia likely due to a combination of hemorrhage, ischemia, and synaptic damage [11, 27].
Recent studies have suggested an association between traumatic brain injury (TBI) and repetitive head impacts (RHI) and neurodegenerative diseases [1, 20, 39, 41]. RHI and contact sport participation, such as American football, boxing, and ice hockey, are associated with the development of chronic traumatic encephalopathy (CTE). In addition to concussions and severe TBI, head impacts in contact sports that do not result in clinical symptoms (subconcussive injury) may still result in neuronal injury [33, 51]. The total number of years of contact sport participation is associated with the pathological stage of CTE and with severity of tau pathology [2, 12, 34, 35, 38]. For American football players, head collisions predominantly involve the anterior aspect of the skull, and a recent analysis of a helmet-to-helmet collision in football predicted that the greatest strain occurs in the frontal convexities and at the depths of sulci [9, 17, 21]. This correlates with the anatomical areas affected earliest and most severely by tau pathology in CTE and may result in a more frontal distribution of other pathologies, including CAA [34, 49].
Previous studies examining single TBI have not shown a significant relationship to CAA [16, 30]. However, the relationship between RHI and the development of small vessel disease is largely unknown. Amateur football players were found to have blood–brain barrier (BBB) disintegrity by dynamic contrast-enhanced MRI analysis after a single season of play compared to non-contact sport athletes [61]. RHI may also predispose individuals to small vessel vascular pathology such as CAA.
We hypothesized that participants with CTE or a history of RHI would have an altered distribution of CAA as well as increased prevalence and severity of CAA. We also performed a hypothesis free approach to look at the distribution of CAA using hierarchal clustering. Additionally, we hypothesized that CAA was associated with the presence of dementia independent of other pathologies, including CTE.
Methods
A total of 807 autopsy participants were examined from three different study groups utilizing previously described procedures [1, 40]. The Understanding Neurological Injury and Traumatic Encephalopathy (UNITE) group consisted of 357 participants with a history of exposure to contact sports such as football, ice hockey, boxing, soccer, rugby, and martial arts at either the professional or amateur level [37]. For most brain donations, the next-of-kin contacted the brain bank to donate tissue at or near the time of death. The second cohort consisted of 241 participants from Boston University’s Alzheimer’s Disease Center (BUADC) with and without cognitive impairment who underwent annual cognitive evaluations using the National Alzheimer’s Disease Coordinating Center (NACC) Uniform Data Set (UDS) protocol [32]. The third cohort consisted of 209 participants from the Framingham Heart Study (FHS), a longitudinal, community-based study. Consents for brain donation and research participation were provided by donor next of kin. Institutional review boards from the Boston University Medical Center approved brain donation, post-mortem clinical record review, neuropathological evaluation, and clinical interviews with donor family members.
Clinical assessment
For UNITE study participants, information pertaining to RHI history, athletic or military service history, history of cognitive, mood, and behavior changes, and clinical status prior to death were all collected via post-mortem interviews with informants through online surveys and review of medical records as described previously [37]. All interviews were performed by neurologists and neuropsychologists trained to assess for RHI exposure and neurodegenerative diseases. In addition, medical records were examined to provide a comprehensive determination of clinical symptoms. For the FHS participants, an athletic history assessment identical to UNITE was performed with the donor’s next-of-kin [21]. Athletic history was not available for BU ADC participants. For all studies, the presence of dementia was determined in a subset of participants by a panel of clinicians after review of medical and study records. A diagnosis of dementia was determined based on modified Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision criteria and included reported evidence of neurobehavioral dysfunction and functional impairment. All interviews were conducted independently and blinded to the results of neuropathological examination.
Pathological assessment
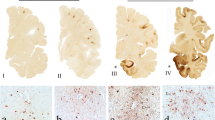
Neuropathological assessment was performed following procedures and criteria previously established for the UNITE study [37, 58]. The neuropathological diagnosis for CTE was made using only pathologic criteria as established by the National Institute of Neurological Disorders and Stroke consensus [32, 35]. Alzheimer’s disease was diagnosed based on the National Institute of Aging Reagan criteria and included intermediate or high probability [40]. CAA was assessed in four cortical regions: dorsolateral frontal (Brodmann area 6, 8), superior temporal (BA 20, 21, 22), inferior parietal (BA 39, 40), and calcarine (BA 17, 18) cortices. CAA evaluation and scoring were performed as described by Vonsattel et al. on a semi-quantitative 0–3 scale determined by the extent of Aβ deposition within a blood vessel using Aβ immunohistochemistry [59]. If no Aβ-positive vessels were seen within a given region it was given a score of 0. The presence of Aβ within smooth muscle cells in a normal vessel was classified as mild CAA and given a score of 1. Replacement of the tunica media by Aβ without evidence of hemorrhage was classified as moderate CAA and given a score of 2. Vessel wall deposition of Aβ with evidence of blood leakage or vessel wall fragmentation was defined as severe CAA and given a score of 3 [59]. Examples of each CAA score are shown in Supplemental Fig. 1. Leptomeningeal and intracortical vessels were scored separately. A global CAA severity score was determined using NIA-AA guidelines [40]. In brief, no CAA was scored as 0, mild CAA or scattered Aβ-positive vessels = 1; moderate CAA in multiple brain regions = 2; severe CAA in widespread brain regions = 3.

Distribution of cerebral amyloid angiopathy by hierarchical cluster algorithm. a An unbiased hierarchical cluster heatmap shows the distribution and severity of CAA in all participants with CAA and without missing data (n = 222). The orange color scale indicates the semi-quantitative severity of CAA (0–3). The four major clusters are indicated by the shaded gray bars to the right of the dendrogram. Pathologic diagnoses are shown in the adjacent green (No AD/CTE), blue (AD), red (CTE), and black (AD & CTE) colored column and in panel C. b The mean severity of CAA across all regions is shown for each cluster in both the leptomeningeal (red) and cortical (blue) vessel compartments. Analysis of the clusters using a mixed linear mixed effects model showed that cluster 2 had significantly more severe disease in the leptomeninges across all regions compared to the cortex (**p < 0.001). Cluster 4 and cluster 1 also showed significant differences (*p = 0.001) but had much smaller mean differences than cluster 2. c The composition of the clusters differed significantly by pathology (p < 0.001; χ2 analysis). The proportion of individuals with CTE was significantly higher in Cluster 2 compared to cluster 1 and cluster 4 (p < 0.001). Cluster 1 and cluster 4 had a significantly higher percentage of individuals with AD than cluster 2 (p < 0.001). There was no difference in the proportion of No CTE/No AD individuals throughout the clusters and the only significant difference in CTE & AD individuals was between clusters 1 and 3 (p < 0.001)
Immunohistochemistry
Tissue was fixed in periodate–lysine–paraformaldehyde, tissue blocks were paraffin-embedded, and sections were cut at 10 µm for immunohistochemistry. Antigen retrieval for Aβ was performed with formic acid treatment for 2 min. Sections were incubated overnight at 4 °C with antibodies to phosphorylated PHF-tau (AT8; Pierce Endogen, Rockford IL; 1:2000) and Aβ (4G8; BioLegend, San Diego, CA; 1:100,000). Sections were washed three times with PBS (pH 7.4), and subsequently treated with biotinylated secondary antibody and labeled with a 3-amino-9-ethylcarbazol HRP substrate kit (Vector Laboratories). The sections were then counterstained with Gill’s Hematoxylin (Vector Laboratories H-3401) and coverslipped using Permount mounting medium. For tau pathology quantification, slides immunostained for ptau (AT8) were scanned at 20× magnification with a Leica Aperio Scanscope (Leica Biosystems, Richmond, IL) as previously described [13]. ImageScope (Leica Biosystems) was used to highlight the gray matter at the bottom third of the sulcus. Leica’s image analysis and automated counting software (Aperio positive pixel count, Version 9) was calibrated for staining intensity to detect AT8-immunoreactivity within the region of interest. Counts were normalized to the area measured and are presented as positive pixels per mm2 within the sulcal depth.
Biochemical pathology measurements
Frozen tissue homogenate was prepared using ice cold 5 M guanidine hydrochloride in Tris-buffered saline (20 mM Tris–HCl, 150 mM NaCl, pH 7.4 TBS) as previously described [50]. The lysate was diluted with 1% Blocker A (Meso Scale Discovery (MSD), Rockville, MD, #R93BA-4) in wash buffer according to the assays for Aβ 1–40 and 1–42 (MSD #K15200E-2) and PSD-95 (MSD #K250QND). Standards with known concentrations were used for Aβ. For PSD-95, arbitrary values were assigned to a reference brain lysate, which was run as a standard curve with every plate. All standards and samples were run in duplicate. Sulfo-tag conjugated anti-mouse secondary antibody was used for signal detection by the MSD SECTOR Imager 2400. Measurements were made on participants with available tissue (No CTE/AD: No CAA, n = 73, CAA, n = 110; CTE: No CAA, n = 59; CAA, n = 36).
APOE genotyping
DNA was extracted from brain tissue and APOE genotype determined using two single nucleotide polymorphisms (National Center for Biotechnology Information SNPs rs429358 and rs7412) as described previously [50].
Statistical analysis
Statistical analysis was performed using SPSS version 20.0 (IBM Corp, Armonk, NY), Prism v6 and v7 (GraphPad Software, La Jolla, CA), and SAS version 9.4 (SAS Institute Inc., Cary, NC). To determine interrater reliability for CAA scoring between the three rating pathologists (ACM, BRH, and TDS), three participants with each score (0–3) as determined by one pathologist were blindly rated by the other two pathologists and an intraclass correlation coefficient statistic was performed to determine consistency among raters. To cluster similar distributions of CAA according to individual semi-quantitative scores in selected regions, an unbiased hierarchical cluster analysis was performed using the unbiased cluster function in R-Studio 3.4.1 (The R Foundation for Statistical Computing). The dendrogram illustrates this clustering with participants on the same branch with similar distributions and those further away with increasingly dissimilar distributions. Mean differences by cluster were analyzed using a linear mixed effects model allowing for subject-specific and location-specific random effects. For the analysis of variation between subject groups and analysis of prevalence data, we used a Chi-square test for proportions and ANOVA with Bonferroni correction for continuous variables. Age-adjusted estimated marginal means were determined for immunoassay measurements of Aβ and PSD-95. PSD-95 values that were outside 1.5× the interquartile range were eliminated as outliers and included No CTE/AD with CAA (n = 2), CTE without CAA (n = 2), and CTE with CAA (n = 1). Quantitative protein measurements and location severity were analyzed by two-way ANOVA with Holm–Sidak correction for multiple comparisons (GraphPad Prism 8). The relationships between CAA, CTE, contact sport exposure, and dementia were analyzed using logistic regression (dementia or CAA as binary outcomes) or cumulative logit proportional odds (CAA severity as ordinal outcomes) using SPSS (v.24, IBM). To account for differences between autopsy groups, propensity scores based on age, AD, and APOE ε4 status for the exposed and unexposed group were calculated in SAS version 9.4 and logistic and ordinal regressions were weighted with the calculated propensity scores.
Results
Participant groups
Participants were grouped based on the presence or absence of CTE or AD pathology. Pathologic groups differed in age, gender, and comorbid pathologies (Table 1). The CTE cohort was significantly younger than other cohorts, and the CTE & AD and the No CTE/No AD groups were younger than the AD group. The CTE and CTE & AD groups also had a higher composition of men than the other groups and were significantly more likely to have participated in contact sports. The prevalence of CAA was highest in the AD and CTE & AD groups. The interrater reliability for CAA scoring between raters was found to be intraclass correlation coefficient = 0.920 (95% CI 0.834–0.962, p < 0.001), which shows excellent agreement.
Hierarchical clustering shows distinct CAA distribution groups
To determine patterns of CAA distribution in an unbiased manner, a hierarchical clustering algorithm grouped participants with CAA and without missing data (n = 222) by severity of CAA within the leptomeningeal and intracortical vessels in the four cortical regions, resulting in four distinct clusters (Fig. 1a). Further analysis of these clusters showed that both cluster 2 and cluster 4 had significantly more leptomeningeal than intracortical CAA with cluster 2 showing a dramatic predominance of CAA in the leptomeninges compared to the cortex (Fig. 1b, p’s < 0.001). The frequency of CTE was greatest in cluster 2: 64% of individuals in cluster 2 had CTE, and 100% had a history of contact sport participation (p’s < 0.001, Table 2, RHI data were missing in one participant). Cluster 2 did not include any individuals with exclusively AD and CAA, despite that pathology comprising 40.5% of the overall cohort. (Fig. 1c, Table 2). Overall, CTE and a history of RHI were overrepresented in cluster 2 which had a predominance of leptomeningeal CAA. However, the small sample size of cluster 2 (n = 11) limits the conclusions that can be made from this analysis. We, therefore, next set out to directly test hypotheses about altered CAA with RHI and CTE.
Distribution by pathology
We hypothesized that the distribution of CAA would be altered in CTE such that there would be greater involvement of the frontal lobe compared to an aging AD population. The unbiased hierarchical clustering analysis further suggested increased leptomeningeal disease in CTE. In participants with AD, intracortical (p < 0.001) and leptomeningeal (p = 0.012) CAA were more frequent in the parietal lobe compared to the frontal lobe (Fig. 2a). In addition, CAA severity scores were significantly greater in the parietal lobe compared to the frontal lobe in AD (cortical p < 0.001; leptomeningeal p = 0.006, Fig. 2b). In AD, comparison of CAA between the leptomeningeal and intracortical vessels showed no differences in frequency and decreased CAA severity in the leptomeninges compared to the cortex. In contrast, the CTE group had increased CAA frequency in both the frontal and parietal leptomeninges (frontal p < 0.001, parietal p = 0.015) and increased CAA severity score in the frontal leptomeninges compared to both frontal and parietal cortices (p’s = 0.010). There were no significant differences between the superior temporal and calcarine cortices in participants with CTE while participants with AD had significantly increased CAA severity in the superior temporal gyrus compared to the calcarine gyrus (Supplemental Fig. 2). Subsequent analysis shows similar location distributions when analyzed by contact sport exposure history instead of pathology, such that participants with a history of contact sports, regardless of pathology, showed the same significant differences seen in the CTE group and those without a contact sport history demonstrated CAA frequency and severity similar to that of the AD group (data not shown). Direct comparison between CAA in AD and CTE further highlights the differences in distribution. The difference of CAA severity was determined for each participant between the frontal and parietal lobes (Δfrontal-parietal). Participants with CTE had a significant increase in Δfrontal-parietal CAA severity compared to those with AD in the leptomeninges (p < 0.001) and trended towards an increase in the intracortical vessels (p = 0.052; Fig. 2c). Similarly, the difference in CAA severity between the leptomeningeal and intracortical vessels (Δleptomeninges-cortex; Fig. 2d) showed a significant increase in leptomeningeal CAA in those with CTE compared to participants with AD in both the frontal and the parietal lobes (p’s = 0.002).

Distribution of CAA by pathology group. a Participants with AD had a significantly increased frequency of CAA in the parietal lobe in both the cortex and the leptomeninges as compared to the frontal lobe. In contrast, participants with CTE had increased CAA frequency in the leptomeninges in both the frontal and parietal lobes compared to the cortex; *p < 0.05, ***p < 0.001, χ2 tests. b Participants with AD had increased CAA severity in the parietal lobe (including in both the cortex and leptomeninges) compared to the frontal lobe compartments. In contrast, participants with CTE had increased CAA severity in the leptomeninges of the frontal lobe compared to the frontal or parietal cortices. c CAA severity was significantly increased in the frontal lobe of participants with CTE compared to AD in the leptomeninges (p < 0.001) and trended towards increased CAA in the frontal cortex (p = 0.052). d CAA severity was significantly increased in the leptomeninges of participants with CTE compared to those with AD in both the frontal and the parietal lobes (p’s = 0.002). For b–d, *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA with Holm–Sidak correction for multiple comparisons
To test whether differences were also present between cohorts, we compared the FHS and UNITE groups (ADC was excluded due to almost unanimous CAA prevalence), adjusting for age, APOE ε4, and AD. Logistic regressions showed that compared to the FHS cohort, the UNITE group was less likely to develop CAA overall (OR = 0.49, 95% CI 0.28–0.84, p = 0.010), but was more likely to develop CAA in the frontal leptomeninges (OR = 2.12, 95% CI 1.21–3.71, p = 0.009).
Contact sport participation and CAA severity
To test the relationship between contact sport participation and CAA in the frontal leptomeninges, logistic (CAA presence) and cumulative logit proportional odds (CAA severity ordinal outcomes) were conducted controlling for age, APOE ε4, and AD in a pooled group of participants where contact sport history was ascertained from the FHS and UNITE studies. Due to differences in the groups used for this pooled analysis, we generated propensity scores by matching individuals by age, AD, and APOE ε4 status. The sample size was 469 for these analyses due to missing data for APOE genotype. To distinguish effects for the presence of CAA (yes/no) versus the severity of CAA (1–3 severity score) we examined each separately. A logistic regression demonstrated that the presence of CAA in the frontal leptomeninges was not significantly associated with a history of contact sport participation (OR = 0.79, 95% CI 0.60–1.04, p = 0.089) when the model was weighted by the calculated propensity scores. However, within those participants with CAA, a weighted cumulative logit proportional odds model showed that a history of contact sport participation was associated with increased odds of more severe CAA in the frontal leptomeninges (OR = 4.01, p < 0.001, Table 3). Years of contact sport participation was also associated with increased odds of more severe CAA in the frontal leptomeninges (OR = 1.06 per year of play, p < 0.001). Sensitivity analyses showed that adjusting for CTE stage gave similar results (contact sport play OR = 3.67, 95% CI 1.72–7.82, p < 0.001), suggesting that the association between contact sports and CAA severity is not due in increasing CTE severity.
CAA was associated with increased neurodegenerative proteins and decreased PSD-95 density
We next quantified levels of Aβ1-40, Aβ1-42, and PSD-95 in participants with and without CTE and with frozen tissue available (NoCTE/AD: n = 183; CTE: n = 95; Fig. 3). As expected, Aβ1-40, a major component of the amyloid in CAA was significantly increased in those with CAA within the CTE group when comparing age-adjusted means (p < 0.001, Fig. 3a). In addition, Aβ1-42 was increased in participants with CAA with and without CTE (p’s = 0.037, Fig. 3b). Age-adjusted AT8 density, a marker of ptau burden, was significantly increased within the sulcus (p = 0.033) in those with CTE and CAA as compared to only CTE (Fig. 3c). Given this increased pathology in participants with CAA, we hypothesized that PSD-95 (a marker for synaptic density) would be decreased in those with CAA. In fact, the age-adjusted mean of PSD-95 density was significantly decreased in those with CAA compared to those without CAA with and without CTE (p’s < 0.05, Fig. 3d). For PSD-95, mean values were similar, but less significant when outliers were included in the analysis (adjusted p = 0.072).

CAA was associated with altered Aβ, sulcal tau pathology, and synaptic density. Aβ1-40, Aβ1-42, tau pathology (AT8 density), and PSD-95, a marker of synaptic density, were measured by immunoassay in the dorsolateral frontal cortex, and age-adjusted means are shown. a Multiple comparison testing (Holm–Sidak) showed that Aβ1-40 was significantly increased in those with CTE and CAA compared to only CTE. b Aβ1-42 was significantly increased in those with CTE and CAA and in those without CTE or AD. c Tau pathology as measured by AT8 density was significantly increased within the sulcal depth in participants with CTE and CAA compared to those with CTE alone. d CAA was associated with a significant decrease in the age-adjusted means of PSD-95 density in participants without CTE or AD as well as in those with CTE. Age-adjusted means with standard error are shown (*p < 0.05, ***p < 0.001, ANOVA with Holm–Sidak correction for multiple comparisons)
Pathological associations with dementia
We next tested the association of CAA with dementia adjusting for comorbid pathology. A logistic regression demonstrated that moderate to severe CAA was significantly associated with increased odds for ante-mortem dementia (OR 1.75, p = 0.043) adjusting for age, sex, CTE stage (stages 0-IV), neocortical Lewy body disease (LBD), AD, and cohort (Table 4). A sensitivity analysis demonstrated a similar effect of CAA on dementia (OR 1.84, CI 1.04–3.26, p = 0.037) when adjusting for the presence of cortical microinfarcts.
Discussion
This study examined the relationships between neuropathological diagnoses, RHI exposure from contact sport participation, and CAA. Unbiased hierarchical cluster analysis showed a unique cluster of individuals with leptomeningeal dominant CAA, most of whom had CTE and all of whom had a history of contact sport participation. Subsequent analysis by pathology showed that those with CTE had more CAA in leptomeningeal vessels than in intracortical vessels and had greater involvement of the frontal lobe. In contrast, participants with AD showed more CAA in the parietal lobe compared to the frontal lobe in both leptomeningeal and intracortical vessels. Ordinal logistic regression analysis showed that a history of contact sport play was not significantly associated with the overall presence of CAA, but was associated with more severe CAA in the frontal leptomeninges, adjusting for age, APOE ε4, and AD. The presence of CAA, in turn, was associated with increased levels of neurodegenerative proteins, decreased levels of PSD-95, and the presence of dementia. Overall, contact sport participation did not appear to affect the risk of developing CAA, but did modulate the severity and distribution of CAA when present.
Accumulating evidence suggests that prolonged contact sport participation and exposure to RHI are associated with multiple neurodegenerative conditions [1, 34, 38, 39, 50, 60]. However, the role of RHI in the development of small vessel disease is largely unknown. Human post-mortem analyses of individuals exposed to a single TBI demonstrated disruption of the BBB by the presence of extravasated serum proteins in brain tissue, even years after the injury [24, 51] and similar results have been described in a case of CTE [19]. Another study found that Aβ burden was increased in individuals with a history of TBI [47]. Animal models have shown an association between TBI and damage to the meningeal cerebrovasculature, including intravascular leakage into the CSF after TBI [15, 31, 46] and marked disruption of the BBB following single and repetitive blast and concussion exposure [22, 51]. A study of two separate and pooled autopsy cohorts did not find an association between TBI with or without loss of consciousness and the presence of CAA [16]. However, the distribution and severity of CAA was not delineated in this study.
The distribution of CAA has been characterized in multiple aging populations, largely focusing on the topographical distribution between different cortical regions [4, 55, 56]. In AD, CAA predominates in the parietal and occipital lobes [4, 43, 53,54,55,56] and may favor the leptomeningeal over the intracortical vessels [4, 6]. We also found CAA significantly increased in the parietal lobe compared to the frontal lobe in AD. In contrast, CAA in the CTE group was more severe in frontal than parietal lobes. Moreover, CAA in the RHI and CTE groups was predominantly leptomeningeal. The leptomeningeal vessels may be particularly susceptible to tissue strain associated with RHI as they are tethered to the pia mater and travel within sulci [21]. Cortical capillary involvement by CAA is a distinct subtype that is related to APOEε4 [25, 52]. In addition, dysphoric CAA has been related to tau pathology [45]. Although CAA in participants with RHI and CTE predominantly involved the leptomeningeal vessels and dysphoric angiopathy was not a prominent feature, further study of capillary CAA and dysphoric angiopathy in association with RHI and CTE is warranted.
One functional consequence of CAA may be the loss of vessel elasticity and the subsequent inhibition of perivascular clearance of solutes out of the brain parenchyma into CSF [18, 23, 48, 62]. Thus, CAA may contribute to the buildup of both Aβ and tau proteins within the brain parenchyma. We found that within participants with CTE, the presence of CAA was associated with significantly increased Aβ1-40 and Aβ1-42, sulcal tau pathology, and decreased PSD-95 (synaptic density marker) (Fig. 3). Although CAA may precipitate tau accumulation via vessel dysfunction, it is also possible that tau accumulation may lead to vessel damage and subsequent CAA [36]. Future studies examining the temporal and spatial relationship between CAA and tau accumulation in CTE are necessary.
Consistent with the association between CAA and decreased PSD-95, CAA was a significant predictor of dementia even when adjusting for AD, LBD, CTE, and study group (Table 4). Similarly, a previous study showed that CAA significantly increased risk of AD dementia controlling for AD pathology such as plaques and tangles, suggesting an independent contribution to clinical dementia [8]. As CAA is associated with increased risk for spontaneous hemorrhage, dementia may be partially due to hemorrhagic tissue damage [7, 29]; however, CAA may also independently predict dementia as cognitive decline often precedes overt hemorrhage in subjects with CAA [14, 57]. Our findings show an association between CAA and dementia when adjusting for a history of microinfarcts supporting a role for other mechanisms in CAA-associated dementia such as persistent inflammation, vasoconstriction-related ischemia [28, 63], and decreased synaptic density.
Limitations
There were several limitations to this study. Autopsy groups differed in composition and recruitment. Individuals in the UNITE group were largely self-selected or referred by the next-of-kin after death and do not represent all individuals who have a history of contact sport play. The FHS brain cohort is a subset of the larger FHS cohort, and although it is a community-based study population, autopsy-based selection bias was present as evidenced by high disease prevalence. Although the three cohorts were distinct, data were carefully harmonized. Neuropathological evaluation was performed for all three studies by the same experienced neuropathologists using standard research protocols, and dementia diagnoses were obtained by expert clinicians using established guidelines. Methods for determination of RHI exposure were standardized across FHS and UNITE; however, the data depended on retrospective review that might have introduced bias. In attempts to account for differences in age, sex, pathology, and autopsy group, we adjusted for these variables when appropriate and performed a propensity analysis to appropriately group comparisons when testing for the association of contact sport participation on the development and severity of CAA. Future studies within larger community-based aging cohorts with RHI history as well as prospective studies will be necessary to confirm and expand these findings. Although we used PSD-95 as a marker for synaptic density, there are more precise ways to measure synaptic density such as electron microscopy and super-resolution microscopy, which warrant future study. In addition, the mechanisms underlying the putative vascular injury due to RHI remain to be elucidated.
Conclusions
Both a history of contact sport participation and years of play were significantly associated with increased severity, but not frequency, of CAA in the frontal leptomeninges. In CTE, CAA was primarily leptomeningeal with increased involvement of the frontal relative to parietal cortex compared to participants with AD. Overall, RHI may alter the distribution and severity of CAA and thus lead to increased risk of dementia through multiple pathways.
References
Adams JW, Alvarez VE, Mez J, Huber BR, Tripodis Y, Xia W, Meng G, Kubilus CA, Cormier K, Kiernan PT et al (2018) Lewy body pathology and chronic traumatic encephalopathy associated with contact sports. J Neuropathol Exp Neurol 77:757–768. https://doi.org/10.1093/jnen/nly065
Alosco ML, Mez J, Tripodis Y, Kiernan PT, Abdolmohammadi B, Murphy L, Kowall NW, Stein TD, Huber BR, Goldstein LE et al (2018) Age of first exposure to tackle football and chronic traumatic encephalopathy. Ann Neurol 83:886–901. https://doi.org/10.1002/ana.25245
Attems J, Jellinger K, Thal DR, Van Nostrand W (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37:75–93. https://doi.org/10.1111/j.1365-2990.2010.01137.x
Attems J, Jellinger KA, Lintner F (2005) Alzheimer's disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol 110:222–231. https://doi.org/10.1007/s00401-005-1064-y
Attems J, Lauda F, Jellinger KA (2008) Unexpectedly low prevalence of intracerebral hemorrhages in sporadic cerebral amyloid angiopathy: an autopsy study. J Neurol 255:70–76. https://doi.org/10.1007/s00415-008-0674-4
Attems J, Quass M, Jellinger KA, Lintner F (2007) Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. J Neurol Sci 257:49–55. https://doi.org/10.1016/j.jns.2007.01.013
Biffi A, Greenberg SM (2011) Cerebral amyloid angiopathy: a systematic review. J Clin Neurol 7:1–9. https://doi.org/10.3988/jcn.2011.7.1.1
Boyle PA, Yu L, Nag S, Leurgans S, Wilson RS, Bennett DA, Schneider JA (2015) Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 85:1930–1936. https://doi.org/10.1212/WNL.0000000000002175
Broglio SP, Sosnoff JJ, Shin S, He X, Alcaraz C, Zimmerman J (2009) Head impacts during high school football: a biomechanical assessment. J Athl Train 44:342–349. https://doi.org/10.4085/1062-6050-44.4.342
Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM (2017) Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 140:1829–1850. https://doi.org/10.1093/brain/awx047
Charidimou A, Gang Q, Werring DJ (2012) Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 83:124–137. https://doi.org/10.1136/jnnp-2011-301308
Cherry JD, Mez J, Crary JF, Tripodis Y, Alvarez VE, Mahar I, Huber BR, Alosco ML, Nicks R, Abdolmohammadi B et al (2018) Variation in TMEM106B in chronic traumatic encephalopathy. Acta Neuropathol Commun 6:115. https://doi.org/10.1186/s40478-018-0619-9
Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML et al (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4:112. https://doi.org/10.1186/s40478-016-0382-8
Cordonnier C, Leys D, Dumont F, Deramecourt V, Bordet R, Pasquier F, Henon H (2010) What are the causes of pre-existing dementia in patients with intracerebral haemorrhages? Brain 133:3281–3289. https://doi.org/10.1093/brain/awq246
Corps KN, Roth TL, McGavern DB (2015) Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol 72:355–362. https://doi.org/10.1001/jamaneurol.2014.3558
Crane PK, Gibbons LE, Dams-O'Connor K, Trittschuh E, Leverenz JB, Keene CD, Sonnen J, Montine TJ, Bennett DA, Leurgans S et al (2016) Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol 73:1062–1069. https://doi.org/10.1001/jamaneurol.2016.1948
Crisco JJ, Fiore R, Beckwith JG, Chu JJ, Brolinson PG, Duma S, McAllister TW, Duhaime AC, Greenwald RM (2010) Frequency and location of head impact exposures in individual collegiate football players. J Athl Train 45:549–559. https://doi.org/10.4085/1062-6050-45.6.549
Diem AK, Tan M, Bressloff NW, Hawkes C, Morris AW, Weller RO, Carare RO (2016) A simulation model of periarterial clearance of amyloid-beta from the brain. Front Aging Neurosci 8:18. https://doi.org/10.3389/fnagi.2016.00018
Farrell M, Aherne S, O'Riordan S, O'Keeffe E, Greene C, Campbell M (2019) Blood-brain barrier dysfunction in a boxer with chronic traumatic encephalopathy and schizophrenia. Clin Neuropathol 38:51–58. https://doi.org/10.5414/NP301130
Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K (2014) Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol 71:1490–1497. https://doi.org/10.1001/jamaneurol.2014.2668
Ghajari M, Hellyer PJ, Sharp DJ (2017) Computational modelling of traumatic brain injury predicts the location of chronic traumatic encephalopathy pathology. Brain 140:333–343. https://doi.org/10.1093/brain/aww317
Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MWet al (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4: 134ra60 Doi 10.1126/scitranslmed.3003716
Hawkes CA, Jayakody N, Johnston DA, Bechmann I, Carare RO (2014) Failure of perivascular drainage of beta-amyloid in cerebral amyloid angiopathy. Brain Pathol 24:396–403. https://doi.org/10.1111/bpa.12159
Hay JR, Johnson VE, Young AM, Smith DH, Stewart W (2015) Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 74:1147–1157. https://doi.org/10.1097/NEN.0000000000000261
Hecht M, Kramer LM, von Arnim CAF, Otto M, Thal DR (2018) Capillary cerebral amyloid angiopathy in Alzheimer's disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol 135:681–694. https://doi.org/10.1007/s00401-018-1834-y
Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T (1993) Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci 116:135–141. https://doi.org/10.1016/0022-510x(93)90317-r
Keage HA, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA, Wharton SB, Weller RO, Brayne C (2009) Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol 9:3. https://doi.org/10.1186/1471-2377-9-3
Kumar-Singh S (2008) Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav 7(Suppl 1):67–82. https://doi.org/10.1111/j.1601-183X.2007.00380.x
Lauer A, van Veluw SJ, William CM, Charidimou A, Roongpiboonsopit D, Vashkevich A, Ayres A, Martinez-Ramirez S, Gurol EM, Biessels GJ et al (2016) Microbleeds on MRI are associated with microinfarcts on autopsy in cerebral amyloid angiopathy. Neurology 87:1488–1492. https://doi.org/10.1212/WNL.0000000000003184
Leclercq PD, Murray LS, Smith C, Graham DI, Nicoll JA, Gentleman SM (2005) Cerebral amyloid angiopathy in traumatic brain injury: association with apolipoprotein E genotype. J Neurol Neurosurg Psychiatry 76:229–233. https://doi.org/10.1136/jnnp.2003.025528
Mamourian AC, Hoopes PJ, Lewis LD (2000) Visualization of intravenously administered contrast material in the CSF on fluid-attenuated inversion-recovery MR images: an in vitro and animal-model investigation. AJNR Am J Neuroradiol 21:105–111
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. https://doi.org/10.1007/s00401-015-1515-z
McKee AC, Daneshvar DH, Alvarez VE, Stein TD (2014) The neuropathology of sport. Acta Neuropathol 127:29–51. https://doi.org/10.1007/s00401-013-1230-6
McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, Alvarez VE, Lee H-S, Hall G, Wojtowicz SM, Baugh CM et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. https://doi.org/10.1093/brain/aws307
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. https://doi.org/10.1093/brain/aws307
Merlini M, Wanner D, Nitsch RM (2016) Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer's disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol 131:737–752. https://doi.org/10.1007/s00401-016-1560-2
Mez J, Solomon TM, Daneshvar DH, Murphy L, Kiernan PT, Montenigro PH, Kriegel J, Abdolmohammadi B, Fry B, Babcock KJ et al (2015) Assessing clinicopathological correlation in chronic traumatic encephalopathy: rationale and methods for the UNITE study. Alzheimers Res Ther 7:62. https://doi.org/10.1186/s13195-015-0148-8
Montenigro PH, Alosco ML, Martin BM, Daneshvar DH, Mez J, Chaisson CE, Nowinski CJ, Au R, McKee AC, Cantu RC et al (2017) Cumulative head impact exposure predicts later-life depression, apathy, executive dysfunction, and cognitive impairment in former high school and college football players. J Neurotrauma 34:328–340. https://doi.org/10.1089/neu.2016.4413
Montenigro PH, Corp DT, Stein TD, Cantu RC, Stern RA (2015) Chronic traumatic encephalopathy: historical origins and current perspective. Annu Rev Clin Psychol 11:309–330. https://doi.org/10.1146/annurev-clinpsy-032814-112814
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS et al (2012) National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. https://doi.org/10.1007/s00401-011-0910-3
Nordstrom A, Nordstrom P (2018) Traumatic brain injury and the risk of dementia diagnosis: a nationwide cohort study. PLoS Med 15:e1002496. https://doi.org/10.1371/journal.pmed.1002496
O'Donnell HC, Rosand J, Knudsen KA, Furie KL, Segal AZ, Chiu RI, Ikeda D, Greenberg SM (2000) Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med 342:240–245. https://doi.org/10.1056/NEJM200001273420403
Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58:1629–1634. https://doi.org/10.1212/wnl.58.11.1629
Rannikmae K, Kalaria RN, Greenberg SM, Chui HC, Schmitt FA, Samarasekera N, Al-Shahi Salman R, Sudlow CL (2014) APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J Neurol Neurosurg Psychiatry 85:300–305. https://doi.org/10.1136/jnnp-2013-306485
Richard E, Carrano A, Hoozemans JJ, van Horssen J, van Haastert ES, Eurelings LS, de Vries HE, Thal DR, Eikelenboom P, van Gool WA et al (2010) Characteristics of dyshoric capillary cerebral amyloid angiopathy. J Neuropathol Exp Neurol 69:1158–1167. https://doi.org/10.1097/NEN.0b013e3181fab558
Russo MV, Latour LL, McGavern DB (2018) Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat Immunol 19:442–452. https://doi.org/10.1038/s41590-018-0086-2
Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, Leech R, Greenwood RJ, Turkheimer FE, Gentleman SM et al (2016) Amyloid pathology and axonal injury after brain trauma. Neurology 86:821–828. https://doi.org/10.1212/WNL.0000000000002413
Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ, Frosch MP, Hyman BT, Moskowitz MA, Ayata C (2007) Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 130:2310–2319. https://doi.org/10.1093/brain/awm156
Stein TD, Alvarez VE, McKee AC (2014) Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther 6:4. https://doi.org/10.1186/alzrt234
Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, Daneshvar DH, Mez J, Solomon T, Meng G et al (2015) Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol 130:21–34. https://doi.org/10.1007/s00401-015-1435-y
Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, Zhang XL, Wojnarowicz MW, Casey N, Lu H, Kokiko-Cochran ON et al (2018) Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 141:422–458. https://doi.org/10.1093/brain/awx350
Thal DR, Papassotiropoulos A, Saido TC, Griffin WS, Mrak RE, Kolsch H, Del Tredici K, Attems J, Ghebremedhin E (2010) Capillary cerebral amyloid angiopathy identifies a distinct APOE epsilon4-associated subtype of sporadic Alzheimer's disease. Acta Neuropathol 120:169–183. https://doi.org/10.1007/s00401-010-0707-9
Tian J, Shi J, Bailey K, Mann DMA (2003) Negative association between amyloid plaques and cerebral amyloid angiopathy in Alzheimer's disease. Neurosci Lett 352:137–140. https://doi.org/10.1016/j.neulet.2003.08.048
Tian J, Shi J, Bailey K, Mann DMA (2004) Relationships between arteriosclerosis, cerebral amyloid angiopathy and myelin loss from cerebral cortical white matter in Alzheimer's disease. Neuropathol Appl Neurobiol 30:46–56. https://doi.org/10.1046/j.0305-1846.2003.00510.x
Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke 18:311–324. https://doi.org/10.1161/01.str.18.2.311
Vinters HV, Gilbert JJ (1983) Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 14:924–928. https://doi.org/10.1161/01.str.14.6.924
Viswanathan A, Patel P, Rahman R, Nandigam RN, Kinnecom C, Bracoud L, Rosand J, Chabriat H, Greenberg SM, Smith EE (2008) Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke 39:1988–1992. https://doi.org/10.1161/STROKEAHA.107.509091
Vonsattel JP, Del Amaya MP, Keller CE (2008) Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol 115:509–532. https://doi.org/10.1007/s00401-007-0311-9
Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–649. https://doi.org/10.1002/ana.410300503
Walt GS, Burris HM, Brady CB, Spencer KR, Alvarez VE, Huber BR, Guilderson L, Abdul Rauf N, Collins D, Singh T et al (2018) Chronic traumatic encephalopathy within an amyotrophic lateral sclerosis brain bank cohort. J Neuropathol Exp Neurol 77:1091–1100. https://doi.org/10.1093/jnen/nly092
Weissberg I, Veksler R, Kamintsky L, Saar-Ashkenazy R, Milikovsky DZ, Shelef I, Friedman A (2014) Imaging blood-brain barrier dysfunction in football players. JAMA Neurol 71:1453–1455. https://doi.org/10.1001/jamaneurol.2014.2682
Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008) Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 18:253–266. https://doi.org/10.1111/j.1750-3639.2008.00133.x
Xiong L, Boulouis G, Charidimou A, Roongpiboonsopit D, Jessel MJ, Pasi M, Reijmer YD, Fotiadis P, Ayres A, Merrill E et al (2018) Dementia incidence and predictors in cerebral amyloid angiopathy patients without intracerebral hemorrhage. J Cereb Blood Flow Metab 38:241–249. https://doi.org/10.1177/0271678X17700435
Acknowledgements
This work was supported by the Department of Veterans Affairs, Veterans Health Administration, Clinical Sciences Research and Development Merit Award (I01-CX001038); Veterans Affairs Biorepository (BX002466); Alzheimer’s Association (NIRG-305779, NIRG-362697, AARF-17-529888); National Institute of Aging (RF1AG054156, R56AG057768, R01AG057768, K23AG046377, R01AG016495, R01AG033040, K23NS102399); National Institute of Neurological Disorders and Stroke (U01NS086659, F32NS096803, R01NS017950); National Institute of Aging Boston University AD Center (P30AG13846; supplement 0572063345-5); Department of Defense, Chronic Effects of Neurotrauma Consortium (CENC) Award W81XWH-13-2-0095 and Department of Veterans Affairs CENC Award I01-CX001135; National Heart, Lung, and Blood Institute, Framingham Heart Study (N01-HC-25195, HHSN268201500001I); Concussion Legacy Foundation. IM receives funding as a Fonds de Recherche du Québec - Santé (FRQS) postdoctoral scholar. This work was also supported by unrestricted gifts from the Andlinger Foundation and WWE. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We gratefully acknowledge the use of resources and facilities at the Edith Nourse Rogers Memorial Veterans Hospital (Bedford, MA), the outreach and recruitment contributions of Dr. Christopher Nowinski from the Concussion Legacy Foundation, and all the individuals whose participation and contributions made this work possible.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Lee E. Goldstein is a paid consultant to Johnson & Johnson (New Brunswick, NJ) / Janssen Research & Development, LLC (Raritan, NJ) and Rebiscan, Inc. (Cambridge, MA). He has received funding from the WWE and Ivivi Health Sciences. Robert A. Stern has received research funding from the NFL, the NFL Players Association, and Avid Radiopharmaceuticals, Inc. (Philadelphia, PA, USA). He is a member of the Mackey-White Committee of the NFL Players Association. He is a paid consultant to Amarantus BioScience Holdings, Inc. (San Francisco, CA, USA) and Avanir Pharmaceuticals, Inc. (Aliso Viejo, CA). He receives royalties for published neuropsychological tests from Psychological Assessment Resources, Inc. (Lutz, FL, USA), as well as compensation from expert legal opinion. He has also provided consultation for Biogen (Cambridge, MA). Robert C. Cantu is a paid consultant to the NFL Head Neck and Spine Committee, NOCSAE, and Concussion Legacy Foundation, and he receives royalties from book publications and compensation from expert legal opinion.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Standring, O.J., Friedberg, J., Tripodis, Y. et al. Contact sport participation and chronic traumatic encephalopathy are associated with altered severity and distribution of cerebral amyloid angiopathy. Acta Neuropathol 138, 401–413 (2019). https://doi.org/10.1007/s00401-019-02031-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-02031-x