
Abstract
The deposition of amyloid β-protein (Aβ) in the vessel wall, i.e., cerebral amyloid angiopathy (CAA), is associated with Alzheimer’s disease (AD). Two types of CAA can be differentiated by the presence or absence of capillary Aβ-deposits. In addition, as in Alzheimer’s disease, risk for capillary CAA is associated with the apolipoprotein E (APOE) ε4-allele. Because these morphological and genetic differences between the two types of AD-related CAA exist, the question arises as to whether there exist further differences between AD cases with and without capillary CAA and, if so, whether capillary CAA can be employed to distinguish and define specific subtypes of AD. To address this question, we studied AD and control cases both with and without capillary CAA to identify the following: (1) distinguishing neuropathological features; (2) alterations in perivascular protein expression; and (3) genotype-specific associations. More widespread Aβ-plaque pathology was observed in AD cases with capillary CAA than in those without. Expression of perivascular excitatory amino acid transporter 2 (EAAT-2/GLT-1) was reduced in cortical astrocytes of AD cases with capillary CAA in contrast to those lacking capillary Aβ-deposition and controls. Genetically, AD cases with capillary CAA were strongly associated with the APOE ε4 allele compared to those lacking capillary CAA and to controls. To further validate the existence of distinct types of AD we analyzed polymorphisms in additional apoE- and cholesterol-related candidate genes. Our results revealed an association between AD cases without capillary CAA (i.e., AD cases with CAA but lacking capillary CAA and AD cases without CAA) and the T-allele of the α2macroglobulin receptor/low-density lipoprotein receptor-related protein-1 (LRP-1) C766T polymorphism as opposed to AD cases with capillary CAA and non-AD controls. Taken together, these results indicate that AD cases with capillary CAA differ significantly from other AD cases both genetically and morphologically, thereby pointing to a specific capillary CAA-related and APOE ε4-associated subtype of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Increasing evidence points to the hypothesis that amyloid β-protein (Aβ) aggregates are of major importance for the development of Alzheimer’s disease (AD) [30, 41, 85]. However, the etiology of the disease is by no means fully understood. In addition to Aβ, a number of other factors have also been implicated in the pathogenesis of AD: abnormal τ-protein [5, 9, 29], oxidative stress [15], altered cholesterol metabolism [31, 56], neuroinflammation [26, 27, 46, 47], blood brain barrier (BBB)-related or perivascular clearance dysfunction [78, 83, 88], and mitochondrial alterations [21, 79]. The apolipoprotein E (APOE) ε4 allele is widely recognized as a risk factor for sporadic AD [8, 19, 62], whereas most other candidate genes remain elusive owing to subsequent replication failure [8]. Furthermore, based on their APOE ε4 carrier status, AD patients display pharmacogenetic differences [59]. To date, it is unclear whether sporadic AD is a single, monolithic disorder with varying degrees of severity or whether it comprises multiple subtypes that possibly bear a strong resemblance to each other but differ with respect to pathophysiology, disease course, and/or treatment response.
Aβ deposition in cortical capillaries is strongly associated with the APOE ε4-allele and permits differentiation between two types of cerebral amyloid angiopathy (CAA): capillary CAA (i.e., CAA-type 1), which is associated with the presence of the APOE ε4 allele, and CAA without capillary Aβ-deposition (i.e., CAA-type 2), which is not associated with the APOE ε4-allele [68]. CAA-type 1 was more frequently found in AD cases than CAA-type 2 [3, 69]. Capillary CAA follows a similar hierarchical sequence of expansion throughout the brain regions as CAA in arteries and veins whereby the basal ganglia, insular cortex, and the medulla oblongata are spared [69].
The goal of the present study was to determine whether capillary CAA constitutes a distinct AD subtype that differs both in Aβ and/or neurofibrillary pathology as well as in the integrity of the pericapillary neuropil from AD cases that lack capillary CAA. We examined a sample of 71 AD cases and 309 controls for the overall distribution of Aβ and neurofibrillary tangle pathology. Because the APOE ε4 allele constitutes a major risk factor for AD and CAA-type 1, we analyzed the association of the APOE ε4 allele in AD cases with and without capillary CAA. In addition, given that apoE is involved in cholesterol trafficking in the brain [80], we also analyzed the association between AD cases with and without capillary CAA and polymorphisms in cholesterol metabolism- and apoE-related candidate genes that have been reported previously to be associated with AD [40, 44, 45, 54, 55]. Moreover, capillary CAA contributes to capillary occlusion with the result that cerebral blood flow is impaired, usually without tissue necrosis [57, 66]. Likewise, aged APOE ε4-carriers develop a widespread deficiency in regional cerebral blood flow in comparison to non ε4-carriers [75], thereby reinforcing the hypothesis that APOE ε4-associated capillary CAA may lead to alterations in cerebral blood flow. It is not known whether capillary CAA and its adverse effects are associated with cellular alterations of cortical astroglia that constitute the glia limitans. Recently, however, Zhong et al. [87] reported reduced levels of the astroglial glutamate transporter EAAT-2 (excitatory amino acid transporter 2, synonymous with GLT-1/SLC1A2) expression in a mouse model for apoE4-like domain interactions. Thus, we decided to focus our attention also on the expression of the astroglial glutamate transporters EAAT-1 (excitatory amino acid transporter 1, synonymous with GLAST/SLC1A3) and EAAT-2 to investigate potential differences between perivascular astrocytes in cases with and without capillary CAA and, in so doing, to identify further differences between AD cases with and without capillary CAA. The occipital cortex was chosen to study the glutamate transporter expression in the presence or absence of capillary CAA because this region is a predilection site for capillary CAA [69].
Here, our results corroborate the hypothesis that the presence or absence of capillary CAA makes it possible to differentiate at least two types of sporadic AD supported by type-specific genetic associations and differences in the EAAT-2 expression in perivascular astrocytes.
Materials and methods
Neuropathology and human sample characterization
A sample of 380 non-selected autopsy cases aged ≥50 years was investigated. This sample included 71 AD and 309 control cases (comprising 259 non-demented and 50 demented non-AD cases, Tables 1, 2). All autopsy brains were collected from individuals, who died in university or municipal hospitals in the European Union (Bonn, Offenbach am Main, Ulm, Germany, Enschede, The Netherlands, and Vienna, Austria) and in the USA (Little Rock, AR, USA) and all were obtained with local legal and ethical committee approval. Demented as well as non-demented patients were examined 1–4 weeks prior to death using standardized protocols for routine examination of patients, including neurological status, upon admission to hospital. These protocols included assessment of cognitive function and the ability to perform activities of daily living: grooming and dressing oneself, meal preparation, bladder and bowel continence, speech patterns, reading and writing skills, short-term and long-term memory, and orientation within a hospital setting. The Clinical Dementia Rating (CDR) score [33] was observed retrospectively for 47 AD cases. These data were used to determine whether individuals clinically fulfilled the DSM-IV criteria for dementia [2]. In 24 AD cases, the clinical records noted the diagnosis of dementia according to DSM-IV. AD was diagnosed when dementia was observed and when the degree of AD-related neuropathology indicated at least a moderate likelihood for AD according to acknowledged criteria [76]. Causes of death were heterogeneous including anaphylactic reaction (0.3%), brain disorders (8.6%), cancer (15.5%), heart disease (28.2%), gastrointestinal disorders (2.2%), infectious diseases (including pneumonia, 17.7%), metabolic disorders (2%), respiratory failure (22%), vascular disorders (except coronary heart disease and cerebrovascular disorders, 2.5%), and trauma (1%). In the event that brain disorders other than AD were diagnosed, this information appears in Table 2. Pathological determination of the cause of death was not possible in 21 cases as only brain autopsy was permitted, and, therefore, were not included in the cause of death statistic.
The brains were fixed in a 4% aqueous formaldehyde solution for at least 3 weeks and underwent neuropathological screening. Presence or absence of gross infarction, hemorrhage, tumor, and other findings were recorded. Blocks from the medial temporal lobe (MTL) were excised at the levels of the (1) anterior limit of the dentate gyrus, (2) lateral geniculate body, and (3) posterior limit of the dentate gyrus [35]. All tissue blocks were embedded in polyethylene glycol (PEG, Merck–Schuchardt, Hohenbrunn, Germany) [11] and/or in paraffin. Tissue blocks from the occipital cortex (Brodmann areas 17, 18, 19) were embedded in paraffin. The PEG-blocks were sectioned on a sliding microtome at 100 μm, whereas the paraffin sections were sectioned at 10 μm.
Neurofibrillary changes were detected using the Gallyas silver-staining method and/or by immunostaining with an antibody directed against abnormal phosphorylated τ-protein (AT-8, s. immunohistochemistry) [10, 11, 36]. Neuritic plaques were diagnosed either in Gallyas- or Bielschowsky-stained sections or in sections immunostained with an antibody directed against hyperphosphorylated τ-protein (AT-8, s. immunohistochemistry). The presence of amyloid deposition was assessed using the Campbell–Switzer silver impregnation method [11, 36] and/or immunohistochemistry (4G8, s. immunohistochemistry). Both methods for the detection of amyloid plaques (including diffuse plaques and fleecy amyloid) produce nearly identical results and are interchangeable [72, 73]. CAA was studied in anti-Aβ immunostained sections.
Diagnosis of the stages in the development of neurofibrillary changes (Braak NFT-stage) and the semiquantitative assessment of neuritic plaques (CERAD-score) were performed in accordance with published and recommended criteria [10, 12, 49, 76]. For staging of Aβ-pathology, we used a previously published protocol for four phases of β-amyloidosis in the MTL [72]. This hierarchically based procedure facilitates study of the topographic distribution pattern of Aβ-deposition in additional brain regions [71, 72]: Phase 1 represents Aβ-deposition that is restricted to the temporal neocortex. Phase 2 is characterized by the presence of additional Aβ-plaques in the entorhinal cortex and/or in the subiculum-CA1 region. The third phase of β-amyloidosis is marked by the presence of Aβ-plaques in the outer zone of the molecular layer of the fascia dentata, subpial band-like amyloid, and/or presubicular “lake-like” amyloid. The existence of further Aβ-plaques in CA4 and/or the pre-α layer of the entorhinal cortex characterize the fourth and final phase of Aβ-deposition in the MTL.
CAA was diagnosed whenever vascular Aβ-deposition was observed. For subclassification of CAA types, we noted whether capillary Aβ-deposits were present. Because the subiculum-CA1 region, the entorhinal cortex, and the occipital cortex (Brodmann areas 17, 18, and 19) are predilection sites for capillary CAA [69], these regions were screened for capillary CAA. In the event that capillary Aβ-deposition was observed even in a single capillary in one of the above-mentioned regions, cases were classified as CAA-type 1 (capillary CAA), whereas those lacking capillary Aβ but having Aβ-deposits in arteries or veins were referred to as CAA-type 2 [68].
Reference pathology for all cases was performed by one and the same neuropathologist (DRT).
In 18 AD and 70 control cases, we rated the overall CAA distribution according to CAA-stages [67]. For this purpose, CAA was also studied in anti-Aβ17–24- or Campbell–Switzer-stained sections of the basal ganglia, thalamus, and cerebellum.
Genotyping
Genomic DNA was extracted either from unfixed frozen brain tissue or from the paraffin-embedded brain tissue [24]. For high-quality genomic DNA templates, APOE genotyping was performed by PCR followed by digestion with the restriction enzyme HhaI [32]. For DNA templates from formaldehyde-fixed specimens, a reliable semi-nested PCR protocol was employed to achieve high yields of PCR-product [24].
The polymorphisms in apoE- and cholesterol metabolism-related genes listed in Table 3 and discussed as AD risk factors [40, 44, 45, 54, 55] were analyzed for potential association with the CAA types in relation to AD. Owing to the common low-quality retrieval of genomic from formaldehyde-fixed archival tissue samples, PCR protocols that yield shorter products were adapted to genotype both cholesterol 25-hydroxylase gene (CH25H) polymorphisms: A list of these primers and restriction enzymes is supplied in Table 4.
Immunohistochemistry
Abnormal phosphorylated τ-protein was visualized with a monoclonal antibody (AT-8, Innogenetics, Belgium, 1/1,000) and Aβ-plaques using an antibody raised against Aβ17–24 (Covance, Emeryville, CA, USA: 4G8 [42], 1/5,000, formic acid pretreatment). EAAT-1 (Novocastra, Newcastle, UK: 10D4 [60], 1/100, 24 h at 22°C, microwave pretreatment), EAAT-2 (Novocastra, Newcastle, UK: 1H8 [48], 1/40, 24 h at 22°C, microwave pretreatment), and apoE (Covance: D6E10 [70], 1/500, 24 h at 22°C, microwave and formic acid pretreatment) were detected using monoclonal antibodies. The primary antibodies were detected with a biotinylated secondary antibody and the ABC complex (Vectastain: Vector Laboratories, Burlingame, CA, USA), and this reaction was subsequently visualized with 3,3-diaminobenzidine (DAB). Immunolabeled paraffin sections were counterstained with hematoxylin. PEG-sections were not counterstained. Positive and negative controls were included.
In selected cases, double immunolabeling was performed to demonstrate the spatial relationship between EAAT-2, glial fibrillary acidic protein (GFAP) expression, and vascular Aβ-deposition. EAAT-2 was visualized with the monoclonal mouse IgG-antibody 1H8, GFAP with a polyclonal rabbit IgG-antibody (1/1,000, DAKO, 24 h at 22°C) or with a monoclonal antibody (1/20, G-A-5, Boehringer–Mannheim, 24 h at 22°C), and Aβ with a polyclonal antibody directed against AβN1D (1/100, [58], 24 h at 22°C, microwave and formic acid pretreatment). Antibodies directed against the N-terminus of Aβ have been shown to stain all vascular Aβ-deposits just as C-terminus-specific anti-Aβ-antibodies [64, 68]. One primary antibody was detected with a carbocyanine 2-labeled secondary antibody specifically directed against either mouse or rabbit IgG (Dianova, Hamburg, Germany), whereas the second primary antibody was detected using a carbocyanine 3-labeled secondary antibody specifically directed against either mouse or rabbit IgG (Dianova, Hamburg, Germany). All tissue sections were viewed with a Leica DMLB fluorescence microscope. Digital photographs were obtained with a Leica DC 500 camera and were edited for publication layout with the assistance of CorelPhotopaint® software, release 12.0.
Morphological analysis
11 AD and 14 control cases were studied for the astroglial expression of EAAT-2 in association with CAA-type 1 and 2. The selection of eight CAA-type 1 (n = 2 controls, 6 AD), nine CAA-type 2 cases (n = 4 controls, 5 AD), and eight cases without CAA was necessary as the EAAT-1 and EAAT-2 antibodies are ineffective on long-time formalin-fixed material (data not shown) but do function on short-time fixed material [48, 65]. Therefore, we used cases that were fixed no longer than 3 weeks prior to paraffin embedding. To ensure a similar staining quality, only cases from a single center (Offenbach am Main, Germany) were used to study EAAT-1 and EAAT-2 expression. For semiquantitative estimation of the relationship between EAAT-2-expressing astrocytes and blood vessels in the occipital cortex, 20 cortical vessels in layers II and III were studied for association with EAAT-2 immunopositive astrocytic processes. For vessel selection, a region of interest (ROI) in Brodmann area 17 was chosen randomly. All blood vessels found there in layers II–III were consecutively examined until a total of 20 vessels were evaluated. Association of EAAT-2-positive astrocytes with a blood vessel was recorded when EAAT-2-positive processes had contact with the perivascular glia limitans of a given vessel. Four degrees were differentiated: 0 = no visible association; 1 = association with less than 33.3% of the vessel wall circumference showing EAAT-2-positive processes; 2 = association with 33.3–66.6% of the vessel wall circumference showing EAAT-2-positive processes; 3 = association with more than 66.6% of the vessel wall circumference showing EAAT-2-positive processes. The degree of association between EAAT-2-positive astrocytes and cortical blood vessels was obtained for each of the 20 vessels, and the scores were summed into one representative semiquantitative score.
Statistical analysis
Binary logistic regression analysis was used to estimate the association of genetic variables with CAA pathology and with distinct groups of cases controlling age and gender as covariates except otherwise mentioned [1]. Odds-ratios (OR) were obtained with 95% confidence intervals (CI). Computations were performed with the help of the SPSS® software, release 16.0.1.
Results
Our major finding was the distinction between two groups of AD cases based on morphological and genetic criteria. The morphological criteria were (1) presence or absence of capillary Aβ-deposition, (2) more versus less widespread senile plaque pathology, and (3) reduction or no reduction of perivascular EAAT-2 expression in the Brodmann area 17 (occipital cortex). Genetically, capillary CAA-related AD was associated with the APOE ε4 allele, whereas other AD cases exhibited an increased frequency of the LRP-1 C766T T-allele. Detailed statistical analyses are shown in Tables 3 and 5.
Morphological distinction between AD cases with and without capillary Aβ-deposition
In addition to capillary Aβ-deposition, AD cases with capillary CAA displayed more widely distributed Aβ-plaque deposits, as represented by the phase of Aβ-deposition in the medial temporal lobe (MTL), than other AD cases (p = 0.006; further information on the statistical analysis is provided in Table 5), whereas the Braak NFT-stage (p = 0.886) and the CAA-stage did not differ (p = 0.08).
Compared to controls, AD cases with capillary CAA displayed more widespread neurofibrillary tangle pathology (Braak NFT-stage: p < 0.001), neuritic plaques, as represented by the CERAD-score (p < 0.001), Aβ-plaques (p < 0.001), and CAA (p = 0.002) (Tables 5, 6). Immunohistochemistry using an antibody directed against apoE demonstrated that capillary Aβ-deposits in CAA-type 1 cases exhibited strong immunolabeling (Fig. 1a, b). Such an accumulation of apoE along capillaries was not seen in AD cases lacking capillary CAA. CAA-involved arteries and veins, however, were immunostained with anti-apoE in both CAA-type 1 as well as CAA-type 2 cases regardless of the apoE genotype.

a Capillary Aβ-deposition (arrow) in the temporal neocortex (Staining: anti-Aβ17-24). b In a subsequent section of the temporal neocortex of the same case apoE was found in vascular Aβ-deposits: in cortical arteries (arrowhead) and cortical capillaries (arrows) (Staining: anti-human apoE). c Astrocytes expressing the glutamate transporter EAAT-2 in a control case free of any Aβ or τ-pathology show a circular (arrows) or mostly circular (arrowhead) perivascular expression in the cortex (Staining: anti-EAAT-2). d EAAT-1 is diffusely expressed in the cortex (Staining: anti-EAAT-1; control case free of Aβ and τ-pathology). e–g Perivascular EAAT-2 expression in astrocytes is seen in association with cortical blood vessels of an AD case without capillary CAA (f). The astroglial nature of the EAAT-2-positive cells is confirmed by the colocalization of GFAP (e, arrows in e–g) (Staining: anti-GFAP–anti-EAAT-2). h–j In an AD case with capillary CAA, capillary Aβ-deposits (arrow in h, j) were not associated with EAAT-2-positive astrocytes (j) whereas distant from the affected capillary EAAT-2-positive astrocytes were visible (arrowhead in i, j) (Staining: anti-AβN1D–anti-EAAT-2). k–m Double label immunofluorescence demonstrates that perivascular astrocytes (arrowhead) near capillary Aβ-deposits (arrow) still express GFAP indicating the loss of EAAT-2 expression but not that of astrocytic processes (Staining: anti-AβN1D–anti-GFAP). Calibration bar corresponds to 40 μm
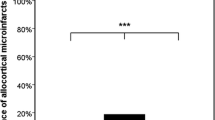
Our analysis of astroglial EAAT-1 and EAAT-2 expression in the occipital cortex revealed no changes in EAAT-1 expression either in AD cases with and without CAA-type 1 or controls (Fig. 1d). In control individuals, cortical blood vessels were frequently rimmed by EAAT-2-positive processes of perivascular astrocytes (Fig. 1c). AD cases with capillary CAA, i.e., CAA-type 1, as well as controls with CAA-type 1 exhibited reduced numbers of EAAT-2-positive cell processes around blood vessels in the occipital cortex compared to controls and CAA-type 2 AD cases (Mann–Whitney U test: p = 0.047) (Fig. 2). Double label immunohistochemistry showed the colocalization of EAAT-2 and GFAP in perivascular astrocytes of controls and in AD cases with CAA-type 2 (Fig. 1e–g), whereas astrocytes surrounding CAA-affected capillaries in AD cases with CAA-type 1 expressed GFAP but often failed to express EAAT-2 (Fig. 1h–m).

Perivascular processes of cortical astrocytes less frequently exhibit the glutamate transporter EAAT-2 in cases with capillary CAA (CAA-type 1) than in those without as indicated by a lower semiquantitative perivascular EAAT-2 expression score in cases with capillary CAA than in those without CAA or with CAA-type 2 (Asterisk represent Mann–Whitney U test: p < 0.05)
Compared to controls, AD cases that lacked capillary CAA showed more widespread neurofibrillary tangle pathology, neuritic plaques, as represented by the CERAD-score, and Aβ-plaques (all: p < 0.001) but did not differ significantly from controls with regard to the distribution of CAA-affected vessels (CAA-stage; p = 0.116) (Table 5). This group of AD cases contained cases with CAA-type 2 and cases without CAA.
Genetic distinction between AD cases with and without capillary Aβ-deposition
AD cases with capillary CAA were strongly associated with the APOE ε4-allele in comparison with other AD cases (Logistic regression analysis: OR = 4.751, CI = 1.551–14.555, p = 0.006, n = 47). In addition to APOE, the LRP-1 C766T polymorphism made it possible to distinguish between AD cases with and without capillary CAA (Logistic regression analysis: OR = 0.101, CI = 0.018–0.613, p = 0.012, n = 38). The frequency of the T-allele was higher in AD cases lacking capillary CAA than in AD cases with capillary CAA (p = 0.012) and in control individuals (p = 0.037, Table 3). The distribution of the APOE ε4-allele was similar for AD cases lacking capillary CAA and for controls (Table 3). On the other hand, the LRP-1 C766T polymorphism was not associated with AD in the presence of capillary CAA (Table 3).
Finally, the CH25H*2 T-allele was associated with the occurrence of CAA-type 1 in general (p = 0.013, Table 3) but failed to reach significance when AD cases with and without capillary CAA were compared (Logistic regression analysis: p = 0.064, n = 40). A summary of further results is supplied in Table 3.
Distribution of CAA-type 1 and CAA-type 2 in AD and non-AD cases
As previously shown [69], 56.3% of the AD cases showed CAA-type 1, 36.6% CAA-type 2 and only 7.1% did not exhibit any CAA pathology (AD cases n = 71). In contrast, 57.3% of the age-related non-demented patients were free of CAA. Only 27.2% had CAA-type 2 and CAA-type 1 was seen even less frequently in only 15.5% (non-AD cases n = 309) (Tables 1, 2).
Demented non-AD cases with mixed dementia, vascular dementia, argyrophilic grain disease, Parkinson’s disease, and other causes of dementia had distributions of CAA and its subtypes that were similar to controls with CAA-type 1 in 20%, CAA-type 2 in 30%, and no CAA in 50% of cases.
No association of a distinct CAA-type with brain infarction or hemorrhage
No association existed between a given CAA-type and the presence or absence of brain infarction or cerebral hemorrhage when comparing CAA-type 1 and 2 cases in the entire sample (hemorrhage: p = 0.935; infarction: p = 0.483) as well as restricted to the group of the AD cases (hemorrhage: p = 0.999; infarction: p = 0.487).
Discussion
Here, we demonstrate that the previously reported two types of CAA [68] differentiate not only between CAA cases but also between at least two types of “sporadic” AD (Fig. 3): AD cases with capillary CAA constitute a specific subtype that can be distinguished from other AD cases lacking capillary CAA. This subtype is further characterized by (1) a more widespread expansion of Aβ-plaque pathology as represented by the phase of Aβ-plaque distribution in the MTL, (2) a capillary CAA-related alteration of EAAT-2 expression in perivascular astrocytes of the occipital cortex, and (3) a robust association with the APOE ε4-allele as previously reported for capillary CAA regardless of AD [68]. Intracellular Aβ accumulation is also associated with the APOE ε4-allele [16].

Morphological and genetic characteristics of the two subtypes of sporadic AD. a AD with capillary CAA: Perivascular astrocytes lack EAAT-2 expression. CAA is not only seen in arteries and veins but also occurs in cortical capillaries and leads to capillary occlusion. Aβ-plaques are more widely distributed throughout the entire brain than in other AD cases (indicated by the number of fields showing Aβ-plaques and representing different brain regions). NFT-pathology is widely distributed and constitutes the diagnosis of AD. There is a strong association with the APOE ε4-allele. b AD without capillary CAA: This second group of AD cases does not show astroglial alterations or capillary CAA. Aβ-plaques are less widely distributed than in APOE ε4-related AD cases (indicated by a single field displaying no plaques representing a brain region that contains Aβ-plaques in AD cases with capillary CAA but not in those without capillary CAA). The neurofibrillary pathology as indicated by the Braak NFT-stage constitutes the diagnosis of AD and does not differ significantly from that of AD cases with capillary CAA. Genetically, AD cases lacking capillary CAA did not show an association with the APOE ε4 allele but with the T-allele of the LRP-1 C766T polymorphism. It is not clear whether this group of AD cases that includes cases with and without CAA can be assigned as one group of cases or whether further subtypes can be identified in the future covering cases of this group
Our second group of AD cases that lacked capillary Aβ-deposition and alterations of perivascular astrocytes, as indicated by EAAT-2 expression failure, included AD cases with CAA-type 2 and AD cases without CAA, and was associated with the presence of the T-allele of the LRP-1 C766T polymorphism instead of with that of the APOE ε4-allele. Moreover, a distinction between APOE ε4-AD cases and non-ε4 AD cases appears to be important for treatment efficacy as well [59]. Aβ-targeting treatment outcomes (Phase II bapineuzumab study) differ based on APOE ε4 status, whereby APOE ε4-negative AD patients responded more favorably to treatment than APOE ε4 carriers [59]. These findings are consistent with the idea that APOE ε4 carriers frequently develop a distinct subtype of AD that is substantially different from other AD cases.
Alternatively, one could assume that capillary CAA and widespread Aβ-plaque-deposition in APOE ε4-carriers may represent the most severe degree of AD-pathology rather than a distinct type of AD. Were this assumption to be true, APOE ε4-carriers should not differ from other cases in earlier AD stages morphologically and genetically apart from their APOE ε4 status. Nonetheless, three counterarguments strongly contradict this viewpoint: (1) In our sample, capillary CAA in APOE ε4-carriers was already evident in early disease stages (Aβ-phase 1) [68, 69], thereby indicating that capillary CAA represents a distinct type of pathology rather than simply the end stage of Aβ-deposition. (2) One explanation for the development of capillary CAA is provided by different properties of apoE2, apoE3, and apoE4 in the transendothelial clearance of apoE-Aβ-complexes at the capillary segment of the BBB [20]. Here, endothelial transcytosis of apoE4-Aβ complexes was reduced when compared to that of apoE2-Aβ and apoE3-Aβ complexes [20]. Thus, it is reasonable to hypothesize that apoE4-Aβ complexes are less sufficiently cleared into the blood and, therefore, accumulate in the capillary vessel wall resulting in capillary CAA. (3) AD cases that lack capillary CAA differ genetically from AD cases having capillary CAA and from controls with respect to the frequency of the LRP-1 C766T T-allele. This, too, points to the greater likelihood that AD cases without capillary CAA, and frequently not carrying an APOE ε4-allele but the T-allele of the LRP-1 C766T polymorphism, constitute a genetically distinct class of individuals. Thus, our data strongly point to the existence of at least two specific AD subtypes related to CAA.
In cases with capillary CAA, cortical perivascular astrocytes often lack EAAT-2 expression, especially around capillaries with Aβ-deposits. Whether hypoperfusion traceable to CAA-related capillary occlusion impairs these astrocytes [66] or whether apoE-Aβ complexes directly impact on these cells during clearance is not yet known. However, Arg-61 apoE mice, a transgenic mouse model mimicking human apoE4 function by exhibiting similar interactions between apoE protein domains as apoE4, showed synaptic pathology as well as reduced levels of EAAT-2 (GLT-1) expression in astrocytes [87]. This apoE4-like domain interaction was also associated with a higher vulnerability to endoplasmatic reticulum (ER)-stress [86]. Thus, an apoE4-related effect may provide an alternative explanation for the alteration of perivascular astrocytes in AD cases with capillary CAA. This hypothesis is supported by the finding that astrocytes with a reduced EAAT-2 expression exhibit increased levels of cholesterol 24S-hydroxylase indicating alterations in the cerebral cholesterol metabolism [77] possibly influenced by apoE4-related alterations in cholesterol transport. Whether the reduction of EAAT-2 expression in perivascular astrocytes results in a general dysfunction of these cells or in excitotoxicity is currently unclear. One could speculate that dysfunction of perivascular astrocytes that constitute the glia limitans, which is the border between the perivascular space and the brain parenchyma, may contribute to BBB alterations. An argument favoring astroglia-related BBB alterations in capillary CAA cases is the finding that CAA reduces the expression of water (aquaporin 4) and potassium channels (Kir. 4) in astrocytic glia limitans end feet [84], presumably resulting in imbalances of the water and potassium distribution. Since the glutamate concentration in the AD brain declines [34], it seems less likely that reduced glutamate transporter expression has a major impact on excitotoxicity. An additional aspect in support of this hypothesis is that no differences exist in the NFT-distribution in the brain between AD cases with and without capillary CAA, although excitotoxicity is capable of influencing τ-pathology [34, 61] that represents the molecular basis of NFTs.
AD cases without capillary CAA were not associated with the APOE ε4-allele but with the T-allele of the LRP-1 C766T polymorphism. This polymorphism has been discussed previously as being associated with AD [40], but other authors could not replicate that group’s findings [7, 8]. Inasmuch as the polymorphism was only associated with a distinct AD subtype, heterogeneity of study samples in previous analyses may explain the conflicting results. The T-allele of the LRP-1 C766T polymorphism was related to the severity of CAA in arteries and veins but not to that in capillaries [17], thereby providing a possible explanation for its association with AD cases lacking capillary CAA. The possession of an LRP-1 C766T T-allele leads to an increased expression of LRP-1 [39] and to a reduction of Aβ-plaques in the brain. A possible explanation for this scenario is increased clearance of Aβ through this receptor either in its apoE-bound form [20, 43] or in its α2macroglobulin-bound form [52]. Because LRP-1 is expressed in perivascular astrocytes of the glia limitans [28, 78], a presumably increased LRP-1-related clearance of Aβ into the perivascular space [6, 81, 82] by T-allele carriers may lead to CAA formation. As such, non-capillary vessels would be the major target because capillary CAA formation can be prevented by transendothelial clearance of apoE2-Aβ and apoE3-Aβ complexes at the capillary segment [20]. Only APOE ε4-carriers with a less effective transendothelial clearance [20] are at high risk for capillary CAA. These results are also in line with our finding here of less advanced Aβ-plaque pathology in AD cases without capillary CAA and overrepresenting the LRP-1 C766T T-allele than in those with capillary CAA. However, AD cases with and without capillary CAA displayed similar levels of NFT-pathology, thus indicating that possession of an LRP-1 C766T T-allele may be beneficial for reducing amyloid deposition while, at the same time, permitting the development of neurofibrillary pathology. Since LRP-1 is a neuronal receptor [50] its increased expression in T-allele carriers [39] may support an influx of LRP-1 ligands, including Aβ, into neurons with hypothetic effects on τ-phosphorylation [53, 63]. Further detailed studies are required to clarify the pathogenesis of non-APOE ε4 AD without capillary CAA.
Other apoE and cholesterol-related genes studied here did not show a significant association with either type of AD. However, CAA-type 1 was associated with the T-allele of the CH25H*2 polymorphism, thereby supporting the view that, for capillary CAA, alterations in cholesterol transport (APOE) and metabolism (CH25H) are important contributors to the pathogenetic events. Notably, the pathogenesis of AD lacking capillary CAA can also be related to cholesterol transport and metabolism because the genetic association with the LRP-1 C766T polymorphism points to the involvement of the apoE receptor LRP-1 in this subtype of AD.
Neither cerebral hemorrhage nor infarction was specifically associated with a distinct type of AD or even with CAA generally. These results confirmed those of a recently published autopsy study [4]. Thus, CAA-related hemorrhage and infarction cannot account for the differences between the two distinct AD subtypes, i.e., those with and without capillary CAA. Nor did the CDR-scores from both AD subtypes differ, thereby pointing to a very similar clinical phenotype.
Finally, we classified all demented cases with at least Braak NFT-stage III which exhibited significant levels of cerebrovascular or non-AD neurodegenerative pathology (e.g., argyrophilic grain disease, Parkinson’s disease) as “mixed dementia” [37] or argyrophilic grain disease cases [13, 74]. We categorized these cases as well as cases with pure vascular dementia or dementia with Lewy bodies as demented non-AD cases. Because dementia results from overall brain damage, cerebrovascular and other neurodegenerative changes can lower the threshold for AD-related pathology so that these factors taken together result in dementia [22, 38, 74]. Such AD-related types of mixed dementia should be distinguished from “pure” AD cases with and without capillary CAA. In the event of multiple microinfarctions or argyrophilic grain disease, the clinical differentiation from pure AD cases may be very difficult, if not impossible. The inclusion of AD-related mixed dementia cases, especially in studies with large numbers of non-autopsy proven cases that fulfill clinical criteria for AD, may explain the existence of heterogeneous results from genetic association studies because the “demented non-AD” cases, clinically misdiagnosed as AD, exhibited CAA and its subtypes in a similar distribution pattern as control cases and were, therefore, indistinguishable from controls. In our sample of non-selected autopsy cases, there were 71 AD and 50 demented non-AD cases. This fact highlights the importance of the second group which although difficult to distinguish clinically from true AD cases, nevertheless represented 41% of our clinically demented cases.
In conclusion, a large cohort of autopsy cases provides a high phenotype resolution that permits identification of at least two specific types of AD which are not only morphologically distinct but also exhibit genetic and pharmacogenetic differences [59]. These differences include a distinction between APOE ε4 associated AD cases with capillary CAA versus LRP-1 C766T T-allele-associated AD cases lacking capillary CAA. These results also imply that subtype specific treatment strategies must be developed, as suggested already by Salloway et al. [59].
References
Agresti A (2002) Categorical data analysis, 2nd edn. Wiley, Hoboken
American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders. American Psychiatric Association, Washington DC
Attems J, Jellinger KA (2004) Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol (Berl) 107:83–90
Attems J, Lauda F, Jellinger KA (2008) Unexpectedly low prevalence of intracerebral hemorrhages in sporadic cerebral amyloid angiopathy: an autopsy study. J Neurol 255:70–76
Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM (1989) Tau and ubiquitin immunoreactivity at different stages of formation of Alzheimer neurofibrillary tangles. Prog Clin Biol Res 317:837–848
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118:103–113
Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE (2008) Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet 83:623–632
Bertram L, Tanzi RE (2008) Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 9:768–778
Braak E, Braak H, Mandelkow EM (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87:554–567
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404
Braak H, Braak E (1991) Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1:213–216
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Braak E (1998) Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm 105:801–819
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Castellani RJ, Lee HG, Zhu X, Perry G, Smith MA (2008) Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol 67:523–531
Christensen DZ, Schneider-Axmann T, Lucassen PJ, Bayer TA, Wirths O (2010) Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol 119:555–566
Christoforidis M, Schober R, Krohn K (2005) Genetic-morphologic association study: association between the low density lipoprotein-receptor related protein (LRP) and cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 31:11–19
Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H (2004) The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci 1019:24–28
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV (2008) apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest 118:4002–4013
Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14:1097–1105
Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD (1999) Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet 354:919–920
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278:1349–1356
Ghebremedhin E, Braak H, Braak E, Sahm J (1998) Improved method facilitates reliable APOE genotyping of genomic DNA extracted from formaldehyde-fixed pathology specimens. J Neurosci Methods 79:229–231
Ghebremedhin E, Schultz C, Thal DR, Del Tredici K, Rub U, Braak H (2002) Genetic association of argyrophilic grain disease with polymorphisms in alpha-2 macroglobulin and low-density lipoprotein receptor-related protein genes. Neuropathol Appl Neurobiol 28:308–313
Griffin WS, Liu L, Li Y, Mrak RE, Barger SW (2006) Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation 3:5
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL 3rd, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86:7611–7615
Grinberg LT, Thal DR (2010) Vascular pathology in the aged human brain. Acta Neuropathol 119:277–290
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8:101–112
Hartmann T, Kuchenbecker J, Grimm MO (2007) Alzheimer’s disease: the lipid connection. J Neurochem 103(Suppl 1):159–170
Hixson JE, Vernier DT (1990) Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 31:545–548
Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL (1982) A new clinical scale for the staging of dementia. Br J Psychiatry 140:566–572
Hyman BT, Van Hoesen GW, Damasio AR (1987) Alzheimer’s disease: glutamate depletion in the hippocampal perforant pathway zone. Ann Neurol 22:37–40
Insausti R, Amaral DG (2004) Hippocampal Formation. In: Paxinos G, Mai JK (eds) The human nervous system. Elsevier, London, pp 872–914
Iqbal K, Braak H, Braak E, Grundke-Iqbal I (1993) Silver labeling of Alzheimer neurofibrillary changes and brain beta-amyloid. J Histotechnol 16:335–342
Jellinger KA, Attems J (2007) Neuropathological evaluation of mixed dementia. J Neurol Sci 257:80–87
Josephs KA, Whitwell JL, Parisi JE, Knopman DS, Boeve BF, Geda YE, Jack CR Jr, Petersen RC, Dickson DW (2008) Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol Aging 29:566–573
Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH (2000) Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest 106:1159–1166
Kang DE, Saitoh T, Chen X, Xia Y, Masliah E, Hansen LA, Thomas RG, Thal LJ, Katzman R (1997) Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer’s disease. Neurology 49:56–61
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489
Kim KS, Miller DL, Sapienza VJ, Chen C-MJ, Bai C, Grundke-Iqbal I, Currie JR, Wisniewski HM (1988) Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun 2:121–130
Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10:719–726
Kölsch H, Lüthjohann D, Jessen F, Popp J, Hentschel F, Kelemen P, Schmitz S, Maier W, Heun R (2009) CYP46A1 variants influence Alzheimer’s disease risk and brain cholesterol metabolism. Eur Psychiatry 24:183–190
Kölsch H, Lütjohann D, Ludwig M, Schulte A, Ptok U, Jessen F, von Bergmann K, Rao ML, Maier W, Heun R (2002) Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer’s disease. Mol Psychiatry 7:899–902
Li Y, Liu L, Barger SW, Griffin WS (2003) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 23:1605–1611
McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107:341–346
Milton ID, Banner SJ, Ince PG, Piggott NH, Fray AE, Thatcher N, Horne CH, Shaw PJ (1997) Expression of the glial glutamate transporter EAAT2 in the human CNS: an immunohistochemical study. Brain Res Mol Brain Res 52:17–31
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The consortium to establish a registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Moestrup SK, Gliemann J, Pallesen G (1992) Distribution of the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res 269:375–382
Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L (2001) Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol 58:397–405
Narita M, Holtzman DM, Schwartz AL, Bu G (1997) Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J Neurochem 69:1904–1911
Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43:321–332
Papassotiropoulos A, Lambert JC, Wavrant-De Vrieze F, Wollmer MA, von der Kammer H, Streffer JR, Maddalena A, Huynh KD, Wolleb S, Lütjohann D, Schneider B, Thal DR, Grimaldi LM, Tsolaki M, Kapaki E, Ravid R, Konietzko U, Hegi T, Pasch T, Jung H, Braak H, Amouyel P, Rogaev EI, Hardy J, Hock C, Nitsch RM (2005) Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer’s disease. Neurodegener Dis 2:233–241
Papassotiropoulos A, Streffer JR, Tsolaki M, Schmid S, Thal D, Nicosia F, Iakovidou V, Maddalena A, Lütjohann D, Ghebremedhin E, Hegi T, Pasch T, Traxler M, Bruhl A, Benussi L, Binetti G, Braak H, Nitsch RM, Hock C (2003) Increased brain beta-amyloid load, phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol 60:29–35
Reid PC, Urano Y, Kodama T, Hamakubo T (2007) Alzheimer’s disease: cholesterol, membrane rafts, isoprenoids and statins. J Cell Mol Med 11:383–392
Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ (1993) beta-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA 90:10836–10840
Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S (1995) Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 14:457–466
Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M (2009) A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73:2061–2070
Sasaki S, Warita H, Abe K, Komori T, Iwata M (2001) EAAT1 and EAAT2 immunoreactivity in transgenic mice with a G93A mutant SOD1 gene. Neuroreport 12:1359–1362
Sautiere PE, Sindou P, Couratier P, Hugon J, Wattez A, Delacourte A (1992) Tau antigenic changes induced by glutamate in rat primary culture model: a biochemical approach. Neurosci Lett 140:206–210
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981
Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK (2008) Co-occurrence of Alzheimer’s disease beta-amyloid and tau pathologies at synapses. Neurobiol Aging 31(7):1145–1152
Tekirian TL, Saido TC, Markesbery WR, Russell MJ, Wekstein DR, Patel E, Geddes JW (1998) N-terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits. J Neuropathol Exp Neurol 57:76–94
Thal DR (2002) Excitatory amino acid transporter EAAT-2 in tangle-bearing neurons in Alzheimer’s disease. Brain Pathol 12:405–411
Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N (2009) Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging 30:1936–1948
Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer’s disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62:1287–1301
Thal DR, Ghebremedhin E, Rüb U, Yamaguchi H, Del Tredici K, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293
Thal DR, Griffin WST, De Vos RAI, Ghebremedhin E (2008) Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol 115:599–609
Thal DR, Larionov S, Abramowski D, Wiederhold KH, Van Dooren T, Yamaguchi H, Haass C, Van Leuven F, Staufenbiel M, Capetillo-Zarate E (2007) Occurrence and co-localization of amyloid beta-protein and apolipoprotein E in perivascular drainage channels of wild-type and APP-transgenic mice. Neurobiol Aging 28:1221–1230
Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of Abeta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Thal DR, Rüb U, Schultz C, Sassin I, Ghebremedhin E, Del Tredici K, Braak E, Braak H (2000) Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol 59:733–748
Thal DR, Sassin I, Schultz C, Haass C, Braak E, Braak H (1999) Fleecy amyloid deposits in the internal layers of the human entorhinal cortex are comprised of N-terminal truncated fragments of Abeta. J Neuropathol Exp Neurol 58:210–216
Thal DR, Schultz C, Botez G, Del Tredici K, Mrak RE, Griffin WS, Wiestler OD, Braak H, Ghebremedhin E (2005) The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer’s disease-related pathology. Neuropathol Appl Neurobiol 31:270–279
Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM (2010) APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol 67:93–98
The National Institute on Aging (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 18:S1–S2
Tian G, Kong Q, Lai L, Ray-Chaudhury A, Lin CL (2010) Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer’s disease. J Neurochem 113:978–989
Utter S, Tamboli IY, Walter J, Rijal Upadhaya A, Birkenmeier G, Pietrzik CU, Ghebremedhin E, Thal DR (2008) Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes. J Neuropathol Exp Neurol 67:842–856
Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA 105:19318–19323
Wang Y, Muneton S, Sjovall J, Jovanovic JN, Griffiths WJ (2008) The effect of 24S-hydroxycholesterol on cholesterol homeostasis in neurons: quantitative changes to the cortical neuron proteome. J Proteome Res 7:1606–1614
Weller RO (1998) Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol 57:885–894
Weller RO, Djuanda E, Yow HY, Carare RO (2009) Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol 117:1–14
Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 153:725–733
Wilcock DM, Vitek MP, Colton CA (2009) Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 159:1055–1069
Yankner BA, Duffy LK, Kirschner DA (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250:279–282
Zhong N, Ramaswamy G, Weisgraber KH (2009) Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. J Biol Chem 284:27273–27280
Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH (2008) Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement 4:179–192
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201
Acknowledgments
The authors thank Irina Lungrin, Heike Korff, Sue Woodward, Uta Enderlein, Nicole Kolosnjaji, Katrin Pruy, Alice Yeates, and Claudia Berchtold for technical help. The authors thank Drs. Peter Müller, Mario Orantes (Offenbach am Main), and Rob A.I. de Vos (Enschede) for providing autopsy tissue.
Conflict of interest statement
This study was supported by DFG-grants TH624/4-1 and TH624/4-2 (DRT), GH12/2-2 (EG), and NIH-NIA-grant AG12411 (WSTG). Heike Kölsch is an employee of IQWIG (Institut für Qualität und Wirtschaftlichkeit im Gesundsheitswesen) Cologne, Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thal, D.R., Papassotiropoulos, A., Saido, T.C. et al. Capillary cerebral amyloid angiopathy identifies a distinct APOE ε4-associated subtype of sporadic Alzheimer’s disease. Acta Neuropathol 120, 169–183 (2010). https://doi.org/10.1007/s00401-010-0707-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0707-9