
Abstract
Chronic traumatic encephalopathy (CTE) is a progressive degenerative disorder associated with repetitive traumatic brain injury. One of the primary defining neuropathological lesions in CTE, based on the first consensus conference, is the accumulation of hyperphosphorylated tau in gray matter sulcal depths. Post-mortem CTE studies have also reported myelin loss, axonal injury and white matter degeneration. Currently, the diagnosis of CTE is restricted to post-mortem neuropathological analysis. We hypothesized that high spatial resolution advanced diffusion MRI might be useful for detecting white matter microstructural changes directly adjacent to gray matter tau pathology. To test this hypothesis, formalin-fixed post-mortem tissue blocks from the superior frontal cortex of ten individuals with an established diagnosis of CTE were obtained from the Veterans Affairs-Boston University-Concussion Legacy Foundation brain bank. Advanced diffusion MRI data was acquired using an 11.74 T MRI scanner at Washington University with 250 × 250 × 500 µm3 spatial resolution. Diffusion tensor imaging, diffusion kurtosis imaging and generalized q-sampling imaging analyses were performed in a blinded fashion. Following MRI acquisition, tissue sections were tested for phosphorylated tau immunoreactivity in gray matter sulcal depths. Axonal disruption in underlying white matter was assessed using two-dimensional Fourier transform analysis of myelin black gold staining. A robust image co-registration method was applied to accurately quantify the relationship between diffusion MRI parameters and histopathology. We found that white matter underlying sulci with high levels of tau pathology had substantially impaired myelin black gold Fourier transform power coherence, indicating axonal microstructural disruption (r = −0.55, p = 0.0015). Using diffusion tensor MRI, we found that fractional anisotropy (FA) was modestly (r = 0.53) but significantly (p = 0.0012) correlated with axonal disruption, where lower FA was associated with greater axonal disruption in white matter directly adjacent to hyperphosphorylated tau positive sulci. In summary, our findings indicate that axonal disruption and tau pathology are closely associated, and high spatial resolution ex vivo diffusion MRI has the potential to detect microstructural alterations observed in CTE tissue. Future studies will be required to determine whether this approach can be applied to living people.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disease which was originally termed ‘dementia pugilistica’ in the 1920s [36]. The first NINDS/NIBIB consensus meeting in 2015 defined pathological characteristics, including hyperphosphorylated tau (p-tau) protein in neurons and astrocytes in sulcal depths and perivascular regions as diagnostic criteria for CTE [31]. Furthermore, CTE has been preliminarily categorized by a hierarchical staging system from I to IV [13, 32, 34, 35, 48], beginning with focal perivascular p-tau and accumulation of p-tau in the sulcal depths of the frontal and temporal cortex in stage I. Through stages II–IV p-tau accumulation becomes more widespread throughout the medial temporal lobe structures, diencephalon and brainstem. The progressive p-tau pathology observed in CTE is coupled with white matter swellings and distorted axonal coherence, or organization in stage II, with progressive axonal degeneration through stages III and IV [33]. While the relationship between these pathologies remains to be determined, McKee et al. hypothesized that the axonal degeneration in CTE plays a critical role in the progressive buildup of p-tau pathology [35]. It has been shown that axonal injury is caused by the shearing of axons due to acceleration/deceleration forces after an impact to the head [46], however, the specific mechanism of injury to progressive CTE pathology is not yet known.
To date, a diagnosis of CTE can only be made on post-mortem examination. There have been initial efforts to develop in vivo diagnostic methods. Blood biomarkers in concussed professional athletes are not specific for CTE; elevated concentrations in the blood may also reflect axonal injury and astrogliosis [39–41, 44]. Neuroimaging methods have been used to detect axonal injury in vivo. CT scans have showed small hemorrhages and contusions associated with axonal injury in some cases [7], and conventional MRI has shown greater sensitivity to detect subtle hemorrhages and contusions which may not be apparent on CT [27], however, most concussive brain injury patients have negative MRI findings [7]. Diffusion tensor imaging (DTI) reflects the microscopic movement of water molecules within the brain, providing detailed information on neuronal microstructural [3, 26]. DTI derived diffusion parameters, such as fractional anisotropy (FA), have been widely used to assess white matter microstructure [16, 17, 25], and has previously been used to detect traumatic brain injury in athletes [6, 10, 18, 49], and military personal following blast-related brain injury [29]. Most recently, Ghajari et al. reported reduced FA in sulcal depths in a large group of TBI patients, potentially consistent with neuropathological findings in CTE [12]. However, many concussive TBI patients do not have definitively abnormal DTI findings [19].
Although these studies are promising, most are limited by the necessary spatial resolution required to detect localized disruptions to white matter organization and degeneration observed in CTE, most notably to U-shaped fibers adjacent to sulcal depth gray matter. Furthermore, these studies have not examined the relationship between white matter pathology and adjacent gray matter p-tau accumulations. Consequently, there is a need to develop neuroimaging techniques which have the specificity and sensitivity to localize this hallmark pathology of CTE in vivo.
Advanced diffusion MRI permits the acquisition of high resolution data. One such technique is generalized Q-sampling imaging (GQI) which measures diffusion using a large number of directions to distinguish multiple fiber orientations within an MRI voxel [53, 54]. Additionally, diffusion kurtosis imaging (DKI) uses several different strengths of diffusion weighting, or b values, to measure variable diffusion patterns that are a result of tissue heterogeneity [20]. DTI FA, or metrics derived from these advanced sequences, could potentially prove useful in the diagnosis of CTE, however, the precise relationship between diffusion metrics and axonal microstructure is not yet clear. This requires radiological-pathological correlations, similar to those previously carried out using in vivo and ex vivo animal models of axonal injury [22, 28, 50]. Landmark based registration uses placed landmarks on both the histological image and the MR image, the histological data is then warped to fit to the MR data using linear (rigid body, affine) or nonlinear (piecewise affine) transformations, and quantified on a voxel-wise basis [14, 15, 52]. For this study, we implemented a robust method of MRI and histology image co-registration developed by Gangolli et al. [11] to acquire high resolution advanced diffusion MRI data of ex vivo brain tissue from subjects with pathologically confirmed CTE. We hypothesized that (1) the degree of axonal disruption has a direct relationship with p-tau pathology, and (2) advanced diffusion MRI has both the specificity and sensitivity to axonal disruption in ex vivo clinically confirmed cases of CTE. We tested the hypotheses using histological sections stained for axonal disruptions and p-tau pathology, which was correlated with diffusion MRI metrics using a robust co-registration method to permit direct radiological-pathological correlations.
Methods
Tissue samples
A total of 10 samples of superior frontal cortex, Brodmann Area 8/9, (Supplementary Fig. 1) from patients with a confirmed neuropathological diagnosis of CTE (made by authors ACM, TDS, and VEA) were evaluated for radiological-pathological correlations. Next of kin provided written consent for participation and brain donation. Institutional review board approval for brain donation was obtained through the Boston University Medical Center and the Edith Nourse Rogers Memorial Veterans Hospital, Bedford, MA. Institutional review board approval for post-mortem clinical record review, interviews with family members, and neuropathological evaluation was obtained through Boston University School of Medicine. A detailed description of demographics is listed in Table 1, for the subjects from whom brain tissue samples were obtained. Comorbidities are reported as assessed by the diagnostic neuropathologists. All brain tissue samples were stored in periodate-lysine-paraformaldehyde (PLP) fixative.
Magnetic resonance image acquisition
Two weeks prior to scanning tissue, samples were rehydrated in 1 × phosphate buffered saline (1xPBS) solution plus 0.1% sodium azide at 4 °C. The volume ratio of tissue blocks and 1 × PBS was 1: 40, and 1 × PBS was changed every 3 days. Rehydration in 1 × PBS was used to reduce the effects of low T2 relaxation observed in formalin-fixed ex vivo tissue (T2 = 40 ms) [9] compared to in vivo (T2 = 79.9 ms) [51].
The meningeal layers and embedded blood vessels were removed by hand from the superficial cortical surface to eliminate air bubble formation, which contributes to additional noise during MRI data acquisition. Ex vivo tissue samples were then placed in an MRI safe cylinder container containing fluorinert, a proton free fluid void of MRI signal.
Diffusion MRI data was collected using an 11.74 T Agilent MRI scanner. A 2D standard spin echo sequence was optimized to produce high spatial resolution data with 250 µm × 250 µm × 500 µm voxel dimensions. The diffusion gradient duration (σ) was 5 ms and the diffusion time (Δ) was 15 ms. Following, extensive optimization the ex vivo MRI acquisition parameters were set as described below. All datasets had sufficiently high SNR, and test-retest reliability (Supplementary Figs. 2–4).
Diffusion tensor imaging (DTI) scheme
30 diffusion-weighted gradients [21], b value = 4000 s/mm2 and 4 non-diffusion-weighted images (b = 0 s/mm2), TR/TE = 1200/30 ms, with a total scan time of 1.5 h. DTI parameters, including fractional anisotropy were calculated with single tensor basis using Brain Suite, http://neuroimage.usc.edu/neuro/BrainSuite, using a weighted log-linear estimation [24].
Diffusion kurtosis imaging (DKI) scheme
30 diffusion-weighted gradients [21] across 5 imaging shells with b values at 2000, 4000, 6000, 8000, and 10,000 s/mm2, TR/TE = 2000/30 ms, total scan time of 11 h. DKI parameters were estimated using diffusion kurtosis estimator (DKE) with constrained linear weighted tensor fitting http://academicdepartments.musc.edu/cbi//dki. Background noise was removed by applying an image threshold on the non-diffusion attenuated, b0 image. The threshold value was optimized for each diffusion MR data set.
Generalized q-sampling imaging (GQI) scheme
202 diffusion-weighted gradients (http://dsi-studio.labsolver.org) with a b value max of 8000 s/mm2 and 10 b = 0 s/mm2, TR/TE = 1400/30 ms, total scan time of 11 h. GQI parameters, including generalized fractional anisotropy (gFA), were calculated using DSI studio http://dsi-studio.labsolver.org/. Background noise was removed by applying an image threshold on the non-diffusion attenuated, b0 image. The threshold value was optimized for each ex vivo diffusion MR data set. No spatial smoothing was applied to GQI datasets. GQI parameters were calculated using a balanced scheme with a 0.7 diffusion sampling length ratio, resolving three fiber orientations in a single voxel.
Following data acquisition Gibbs ringing artifact was successfully removed by applying Hamming filtering to all datasets.
Histopathology
Following MRI acquisition, tissue blocks were refixed in 10% neutral buffered formalin and refrigerated at 4 °C for 1 week. The tissue blocks were then equilibrated in 30% sucrose for an additional week before they were serially sectioned at 50-µm thickness using a freezing sliding microtome. Sectioning was carried out with the visual aid of MRI image planning to ensure that histology sections were in the same plane as MRI data. For each MRI slice, two 50-µm histological slices were analyzed separated by 250 µm.
Phosphorylated tau immunoreactivity
Sections were assessed for immunoreactivity with antibodies specific for phosphorylated tau (AT8, Phospho-PHF-tau pSer202 + Thr205 mouse anti-human monoclonal antibody, ThermoFisher Scientific, dilution 1:1000). Free-floating sections went through 3 × 5 min washes in 1 × tris buffered saline (TBS) followed by a 10-min incubation in 3% hydrogen peroxide to block endogenous peroxidase. After washing in TBS, antigen retrieval was carried out using 5-min incubation in 70% formic acid at room temperature. To prevent non-specific binding sections were incubated for 30 min in normal goat serum (3% normal goat serum in 1 × TBS with 0.25% Triton X (TBS-X), followed by an overnight incubation in the primary AT8 antibody dilution 1:1000 in 3% normal goat serum in TBS-X. Following the overnight incubation, sections were washed in TBS for 3 × 5 min and incubated in biotinylated goat anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA), dilution 1:1000 in TBS-X for 1 h. Detection of specific immunoreactivity was carried out using 1:400 dilution of avidin–biotin horseradish peroxidase complex (VECTASTAIN Elite ABC HRP Kit, Lot # PK 6100, Vector Laboratories, Burlingame, CA) in TBS for 1 h. Sections were then washed in 1 × TBS again before visualization using 3,3′-diaminobenzidine DAB (Sigma-Aldrich USA). Mounted sections were dehydrated through a series of ethanols, cleared in xylene and cover-slipped with Cytoseal mounting media. A negative control excluding the primary antibody revealed no staining. A positive control using tissue from an Alzheimer’s disease patient revealed extensive tangles, as expected.
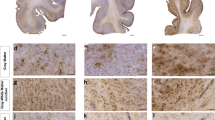
Digital images of histology sections were acquired using the Hamamatsu NanoZoomer 2.0-HT System (Hamamatsu) using a 40× optical magnification, Fig. 1a. The raw histology digital images were then converted from NDPI file format to tiff image format using ImageJ (NIH, Bethesda, MD) and the NDPI Tools plugin. The conversion factor to 10× magnification is 1 pixel = 1.09 microns isotropic, compared to 40× magnification where 1 pixel = 0.23 microns isotropic. The final image maintained optimum resolution to distinguish individual phosphorylated tau tangles and threads.

Phosphorylated-tau, myelin black gold II, and high resolution ex vivo diffusion tensor imaging in CTE tissue. a Low magnification immunohistochemistry using AT8 p-tau antibody demonstrating high tau pathology in the depth of 1 sulcus and lower tau pathology in another sulcus. b High magnification immunohistochemistry using AT8 indicating p-tau tangles and threads with the corresponding thresholded image for p-tau quantification using ImageJ. c Myelin black gold II staining in an adjacent slice from the same CTE brain tissue sample. The overlayed grid demonstrates the individual regions of interest analyzed for power coherence measurements. The arrows indicate the tau negative and tau positive sulcal depths. d High magnification myelin black gold II staining from white matter immediately adjacent to sulcal depths. Very poor fiber organization adjacent to a tau positive sulcus is illustrated in the top left panel. Highly coherent fiber organization adjacent to a tau negative sulcus is illustrated in the bottom left panel. The power coherence derived from two-dimensional Fourier transform analyses are illustrated at right. e Diffusion tensor imaging FA map from the corresponding region of the same CTE brain tissue sample. The yellow outlines indicate 0.5 mm white matter regions of interest immediately adjacent to the sulcal depths. f Diffusion tensor imaging FA map to exemplify the high spatial resolution at the gray–white junction
Myelin Black Gold II: measurement of axon disruption
Initial immunohistochemistry experiments using amyloid precursor protein (anti-APP Cat #51-2700, Zymed, Camarillo, CA) and neurofilament-L (Neurofilament-SMI-34 Cat# 835501, Biolegend, San Diego, CA) antibodies failed to clearly identify axon disruptions even after intensive attempts at optimization and excellent results in positive controls from more severely injured patient samples. Therefore, myelin black gold II (Millipore, Lot# AG105), an aurohalophosphate complex stain tailored for use on formalin-fixed tissue was used to provide high contrast, high resolution stained images of axonal fibers. 50-µm free-floating sections from all 10 ex vivo CTE tissue samples were stained for myelinated fibers by incubating in myelin black gold II solution (Black Gold II powder, resuspended in 0.9% saline solution) pre-heated to 60 °C and monitored until they had reached the optimum amount of staining, approximately 8 min. Sections were washed with MilliQ water for 2 minutes and then incubated in 1% sodium thiosulfate pre-heated to 60 °C for 3 minutes to remove excess staining. After 3 washes in 1 × TBS, stained sections were mounted on positively charged glass slides in an orientation matching that of the corresponding MRI slices.
The mounted sections were dehydrated in a series of graded ethanols, cleared in xylene and cover-slipped with cytoseal mounting media. Digital images were acquired with a Hamamatsu NanoZoomer 2.0 HT System (Hamamatsu) using a 20× optical magnification, Fig. 1a. Myelin Black Gold II images were then downsampled to 5× magnification (1 pixel = 1.84 microns isotropic) compared to the original 20× magnification (1 pixel = 0.54 microns).
Histology-MRI co-registration
Registration for each histology image to MRI space was carried out as follows;
-
1.
Concurrent histology and MRI slice images were opened to permit the placement of corresponding landmarks on both images. A minimum of 5 MRI slices per tissue block were analyzed (MRI slices with partial volume effects were not included in the analysis). Two histology slices for each stain (AT8 tau, myelin black gold II) were analyzed for each MRI slice. A minimum of 55 landmarks was placed on each pair of images at anatomical features present, particularly at the boundary between gray and white matter, in both the histology and MRI slice image.
-
2.
A custom built Matlab script [11] was then used to apply a forward nonlinear least squares transformation to transform the histology image to the MRI slice image. An inverse transform was then applied to each voxel of the transformed histology image, resulting in an MRI-based histology grid where each ROI in the grid corresponds to one MRI voxel. A more detailed description and validation of the image co-registration method is explained in Gangolli et al. (under review)
-
3.
Using the grid, the sulcal depth ROI was defined as a region extending 1 mm superficially from the deepest point of each sulci, and extending laterally on either side to the gray/white matter boundary. The resulting semi-circular ROIs specifically encompassed sulcal depth gray matter, Fig. 1a.
-
4.
The MRI-based histology grid was then overlayed on histology images to assess the quality of the co-registration. The grid was also used to guide manual drawing of ROI masks in white matter which extended 0.5 mm from the gray/white matter boundary, to isolate the U-shaped fibers potentially susceptible to axonal injury, Fig. 1c, e. The gray/white boundary was delineated by hand by an expert neuroanatomist (LH).
Quantification of tau pathology
The % area of p-tau in gray matter was quantified using an initial thresholding within ImageJ based on the intensity of DAB staining (which had slight variations based on tissue sample). Size inclusion was set at 50–50,000 pixels to exclude smaller and larger non-specifically stained structures. The smaller nonspecific staining was typically punctate DAB deposition with no discernible anatomically relevant pattern. The larger nonspecific staining was typically found around the edges of the tissue sections. The size criterion used to quantify the % area of p-tau was kept fixed in the analyses of all tissue sections to isolate the pathological target: phosphorylated tau neurofibrillary tangles, threads, and astrocytes Fig. 1a, b.
Validation of automated p-tau quantification
Stereological analysis was performed using StereoInvestigator software version 8.2 (MBF Bioscience, Williston, VT). The optical fractionator method was used to count a randomized sample of positively stained p-tau neurofibrillary tangles, threads, and astrocytes in five tissue blocks. In each tissue block, four histological slices spaced 250 µm apart were quantified. ROIs were drawn at 4× magnification, starting at the gray/white matter boundary and ending at the deepest point of the sulcus, where two gyri connected. Counting was performed at 10× magnification over sites randomly chosen by the StereoInvestigator software, using a 300 × 300 μm counting frame with a dissector height of 15 μm and a guard zone of 5 μm. P-tau reactive sites were only counted if the cell bodies were in focus and within the counting frame.
The total volume of the sampled regions was calculated as previously outlined [28] by multiplying the counting frame area by the region thickness and then multiplying by the number of counting frames. Estimation of the number of p-tau immunoreactive sites per cubic millimeter was determined by dividing the total counts within each ROI by the ROI volume.
The % area of p-tau positive staining was also calculated using the ImageJ based quantification method described above. A correlation between the stereology count of p-tau sites per area volume and the automated % area of p-tau staining obtained using the ImageJ based workflow, was carried as a validation of the automated method applied, (r 2 = 0.9507, p < 0.0001), (Supplementary Fig. 5).
Assessing axon disruption
To assess the degree of white matter axon disruption, fiber directionality was derived from the digital myelin black gold II images using a custom written ImageJ macro, [11], which applied a two dimensional discrete Fourier transformation within every ROI in the MRI-based grid to calculate the axon directionality and power coherence (Fig. 1d). Power coherence was extracted by fitting two ellipses to the power spectrum, where each ellipse represented a fiber population. The degree of coherency of each fiber population was calculated as the ratio of the radial to preferential axes to obtain a measure between 0 and 1, where a higher coherency measure represents higher fiber integrity. White matter power coherence was then directly correlated to the degree of immunoreactive p-tau in the adjacent gray matter.
Power coherence extracted from white matter ROI’s was then directly correlated with diffusion MRI parameters to determine the relationship between each imaging parameter and the degree of white matter axon disruption.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism 7.02. To determine the relationship between phospho-tau pathology in gray matter sulcal depths and adjacent white matter fiber integrity, a two-tailed Spearman rank-based correlation was performed because the relationship between the variables was not linear. To determine the relationships between diffusion MRI parameters and white matter fiber coherence, Pearson product moment correlation analyses were used. The residual plots were normally distributed (Shapiro–Wilk’s normality test p > 0.05 for all variables). These analyses were adjusted by dividing by the means across each of the 2–4 sulci in each of the 10 tissue blocks to account for potential systematic variability potentially due to post-mortem events. p values for these correlations have been reported without correction for multiple comparisons, but only considered statistically significant if p < 0.05/9.
Results
CTE phosphorylated p-tau immunoreactivity
Neurofibrillary tangles and neurites positive for AT8 were found in all samples. The percent area of phospho-tau immunoreactivity was variable across samples and across sulci within samples. In 2 of the CTE stage IV tissue samples there was widespread distribution of p-tau pathology across all gray matter regions (CTE tissue samples 8 and 10). In the other 8 samples, the p-tau staining was patchy, with some sulcal depths heavily stained and others less intensely stained (Table 1). Leukoencephalopathy and cerebrovascular pathologies reported by the diagnostic neuropathologists based on analysis of multiple brain regions were not apparent in the cortical and superficial white matter tissue samples analyzed for this study.
We performed immunohistochemical staining using amyloid precursor protein and compacted neurofilament, markers that are useful for early stage detection of traumatic axonal injury. Positive controls worked well, but no staining was detected in any of the CTE samples. This could indicate that axonal injury was either absent, or no longer marked by these indicators due to the long interval between injuries and death. We next turned to myelin black gold II staining to assess fiber orientation. We observed disrupted white matter axons directly adjacent to p-tau accumulations in the depths of gray matter sulci. These disrupted axons showed no clear fiber orientation, whereas most of the fibers in the white matter underlying p-tau negative sulci appeared to be parallel, Fig. 1d. Within each histological ROI corresponding to an MRI voxel, we analyzed the power coherence, i.e., the extent to which all of the myelinated fibers were running in the same direction or directions, quantified using two-dimensional Fourier transform analysis in each ROI voxel. There was a modest, yet statistically significant relationship between power coherence and quantified % area of p-tau, that is higher power coherence was associated with lower levels of p-tau in directly adjacent gray matter sulci (Spearman’s rho cc = −0.55, p = 0.002), Fig. 2a.

Power coherence in white matter ROI’s directly adjacent to sulcal depths and the corresponding diffusion MR parameter correlations. a Scatterplot of the mean (with standard error bars) power coherence and % area of p-tau for each white matter ROI and adjacent gray matter ROI for all 10 ex vivo CTE samples (Spearman’s rho cc = −0.55, p = 0.002). Individual data points represents the mean ROI value from each post-mortem tissue sample. b FA in white matter ROI’s had a positive correlation with power coherence, where high FA was associated with regions with higher power coherence (r = 0.53, p = 0.001 uncorrected, p = 0.009 corrected). FA was the only diffusion MRI parameter to have a significant correlation after correction for multiple comparisons. The diffusion MRI parameters were adjusted across all ten CTE tissue samples by dividing them by the mean of each parameter in each specific sample to correct for possible between-sample differences arising from post-mortem effects
To determine whether diffusion MRI could potentially detect axon disruptions in CTE brains, we performed voxel-by-voxel radiological–pathological correlation analyses with histological power coherence and all diffusion MRI parameters. Because of substantial brain-to-brain variability, we adjusted each diffusion MRI parameter by the mean for that parameter across all analyzed voxels in that brain sample. We found that white matter power coherence and FA were significantly correlated (r = 0.53, p = 0.009 corrected) Fig. 2b, after correction for multiple comparisons. No other diffusion MRI parameter had a significant correlation with white matter power coherence, as shown in Table 2. This result indicated that as expected, lower FA in white matter is associated with increased axon disruption, as reflected by reduced power coherence.
Because reduced FA was associated with increased axon disruption, which was coupled with increased p-tau pathology, we next asked whether reduced FA in white matter could be used to predict p-tau pathology in adjacent gray matter. However, we found that there was no statistically significant relationship between FA in white matter and % area tau pathology in overlaying gray matter sulcal depth ROI’s, for unadjusted (R = −0.019, p = 0.91) Fig. 3a, and adjusted analyses (R = −0.23, p = 0.22) Fig. 3b. Furthermore, there was no statistically significant relationship between the remaining diffusion MRI metrics from white matter ROI’s and % area of p-tau from overlaying gray matter sulcal depth ROI’s, Fig. 4.

No correlation between FA in white matter and tau pathology in adjacent gray matter sulcal depths. Unadjusted analyses (R = −0.019, p = 0.91) (a), and adjusted analyses (R = −0.23, p = 0.22) (b), results of the Spearman rho’s correlation analysis of mean FA from white matter ROI’s and mean % area of p-tau for adjacent gray matter ROI’s. a shows the mean with standard error bars of raw FA values across all white matter ROI’s. For adjusted analyses, FA was normalized by the mean FA across ROIs in each tissue block to account for possible variation in post-mortem events

No correlation between additional diffusion MRI parameters in white matter and tau pathology in adjacent gray matter sulcal depths. Spearman’s rho results show that there was no correlation between white matter diffusion MRI parameters and the % area of p-tau from adjacent gray matter
Discussion
In summary, we found reduced axon integrity in CTE which is related to the degree of p-tau pathology in directly adjacent gray matter. Furthermore, we found that FA, derived from high spatial resolution diffusion MRI data, is significantly correlated with histological evidence of axon disruption. However, the remaining diffusion MRI parameters derived from DTI, DKI and GQI datasets did not correlate with the percent area of p-tau pathology in the ex vivo CTE tissue examined.
To our knowledge, this is the first study to carry out a robust quantitative radiological-pathological correlation in brain samples from subjects with neuropathologically confirmed CTE. Technical advances relevant to our approach included the use of high spatial resolution ex vivo diffusion MRI datasets with high SNR; a novel co-registration method; and fully quantitative pathological analyses.
A major finding was that power coherence derived from myelin black gold II stain showed axon disruption in white matter directly adjacent to gray matter sulcal depths with substantial tau deposition. Axonal damage or tau positive axonal varicosities and neuropil threads have been previously reported as a characteristic feature of CTE in subcortical and deep white matter [33], but the spatial relationship between axon pathology and gray matter tau pathology has not been previously described. Increased loss of myelinated fibers and axons in white matter is associated with stage IV CTE [35, 47], which is consistent with the findings reported here where axon disruption is linked to the density of tau pathology in the sulcal depths. Furthermore, the results reported here are consistent with FA reductions in sulcal regions as opposed to gyral regions in a large group of TBI patients [12]. The same study used computational modelling to accurately predict patterns of brain tissue deformation in the sulcal depths, consistent with characteristic CTE pathology [12], though it is not clear whether any of the patients in the Ghajari et al. study actually had CTE.
Although we report a relationship between axonal disruption and tau pathology, the precise mechanisms underlying this relationship has not yet been established; specifically, the order of causality cannot be determined based on the cross-sectional data presented here. It has been hypothesized that phosphorylated tau at sites in the microtubule binding domain causes microtubule instability, resulting in interrupted axonal transport and degeneration of white matter tracts [30]. Thus, the white matter disruption could be secondary to tau pathology. On the other hand, diffuse axonal injury is a common pathological finding after traumatic brain injury, most commonly at the gray/white matter boundary [1, 2] due to shearing and stress on axonal fibers that cause cytoskeletal disruptions and axonal breakdown [45]. After axonal injury, tau accumulation due to impaired axonal transport or an aberrant response to traumatically disrupted microtubules could result in the histopathologically detected tau pathology in the cell bodies or dendrites. While the direct mechanism is not well understood, the tau pathology could be a consequence of axonal injury in some cases, as in the recent report by Shively et al. [42]. Finally, the tau pathology and axonal injury could both be due to the same third as-yet-uncharacterized mechanism. It will be of great interest in the future to determine whether there are cases of focal traumatic axonal injury at the gray/white matter boundary that do not result in tau pathology or CTE. Clearly, further investigation will be required to elucidate the pathophysiological processes underlying the relationship between tau pathology and white matter disruption.
The relationship between FA and axon disruption, reported here, suggests that diffusion MRI has the potential to detect regions of white matter damage that is associated with gray matter phosphorylated tau pathology. FA was the only diffusion MRI parameter to show a statistically significant relationship with axon disruption. This suggests that in white matter closely underlying cortical sulci, DTI is sensitive to axon disruption in CTE. Still, it would be premature to conclude that DTI will be useful in living humans. The spatial resolution of the ex vivo data acquired was much higher than that currently available for in vivo MRI sequences. Here, the spatial resolution was 250 × 250 µm with a slice thickness of 500 µm, whereas current in vivo DTI is typically performed with isotropic voxel sizes in the range of 1.25–2.5 mm3. The larger voxel sizes result in partial volume effects, particularly in white matter regions on the border with gray matter. It has been shown previously that DTI tractography results in ex vivo rhesus macaque is greatly dependent on the spatial resolution and diffusion sampling scheme applied; the authors advocated for a balanced consideration between these two major acquisition variables to optimize tractography accuracy [4]. Our results indicate that increasing spatial resolution is a necessary requirement to detect focal microstructural alterations. However, our analyses was limited to white matter directly adjacent to gray matter sulcal depths and did not include regions of dense crossing fibers such as the centrum semiovale where FA cannot give an accurate estimate of white matter organization.
Limitations of this and all ex vivo human diffusion MRI data include the effects of post-mortem degeneration, fixation and resulting dehydration. The fixation process augments the changes in the microstructure, above that caused by cell death. A study examining the effects of brain tissue decomposition on diffusion MRI parameters reported a steady and significant decrease in FA and ADC, which is most evident between days 0 and 1 postmortem interval, after which the effect of delayed fixation becomes less significant [8]. The postmortem interval of the ex vivo CTE samples used in this study ranged from 2 to 92 h, however, histopathology and myelin black gold II results indicate preservation of tau staining and axon fiber orientation in all samples regardless of postmortem interval. An additional step to overcome this limitation was to perform a corrected correlation analysis to moderate any variance derived from different postmortem interval times. However, additional studies using ex vivo CTE tissue with more uniform and short postmortem intervals is essential to fully understand the relationship between tau pathology and diffusion MRI parameters. Furthermore, CTE is often associated with comorbidities, such as leukoencephalopathy and vascular pathology observed in 8 of the 10 CTE cases included in this study. White matter degeneration as a result of the reported comorbidities cannot be directly differentiated from the axonal disruption pertaining to CTE. However, an association between gray matter hyperphosphorylated tau pathology and axonal microstructural disruption was observed in 2 cases which had no reported comorbidities. In addition, we did not observe any areas of white matter pallor on myelin black gold II staining, indicating that there were no infarcts or areas of severe leukoencephalopathy in the cortical samples included in this study. Although comorbidities are common in CTE postmortem cases, additional analysis on tissue with an exclusive diagnosis would be necessary to further differentiated the extent of white matter disruption specifically related to CTE.
This analysis was limited to radiological-pathological correlations in ex vivo tissue samples with stage III and stage IV CTE. Analysis of stage I and II tissue is necessary to determine if this detection remains consistent with the findings reported here. If so, this would make diffusion MRI in theory useful for early detection of CTE prior to major neurodegenerative changes, however, as noted above this would require significant improvements in the spatial resolution of current in vivo diffusion MRI studies. Future studies including non-CTE tauopathies and other control cases are also necessary to determine whether the relationship between axonal injury and tau pathology is unique to CTE. Additional limitations include the relatively restricted set of cases examined which were all retired professional contact sport athletes from a single center. The post-mortem intervals were variable and often not fully known. The sample locations were not fully standardized. The extent to which two-dimensional sampling of fractional area using a single immunohistochemical marker of tau pathology may have biased the results remains to be determined. Further work will be required involving assessments of other disease controls using the same methods. Thus, selection bias and generalizability cannot be determined at present.
While our results suggest a relationship between white matter diffusion MRI parameters and histopathology, additional analysis is necessary to determine if neuroinflammatory markers of CTE, such as changes in microglia and astrocytes, result in cytoarchitectural changes that can be detected using imaging methods applied here. Shively et al. recently reported significant astroglial scarring at the gray–white matter junction in ex vivo human brain tissue samples following blast exposure [43]. Increased positive immunolabeled microglial has also been reported in a repetitive mild traumatic brain injury mouse model [38], however, it is becoming more evident that microglia are highly dynamic [23] and inclusion of fractal analysis to assess microglia morphology, similar to that carried out by Namjoshi et al. [37] may provide a more accurate description of neuroinflammation in CTE, similar to that presented by Cherry et al. [5].
Conclusion and implications
Undoubtedly, a non-invasive method to detect CTE related pathology in vivo would be a major medical contribution benefitting those who are at risk of developing CTE. Here we have shown that high spatial resolution advanced diffusion MRI data has the potential to play a role for in vivo CTE diagnosis, especially with regard to assessment of disrupted white matter microstructural integrity adjacent to sulci with tau pathology. However, to bring high spatial resolution diffusion MRI to a clinical useful setting would require substantial further improvements in clinical diffusion MRI acquisition methods.
References
Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR (1989) Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 15:49–59
Adams JH, Doyle D, Graham DI, Lawrence AE, McLellan DR (1985) Microscopic diffuse axonal injury in cases of head injury. Med Sci Law 25:265–269
Basser PJ, Pierpaoli C (1996) Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B 111:209–219
Calabrese E, Badea A, Coe CL, Lubach GR, Styner MA, Johnson GA (2014) Investigating the tradeoffs between spatial resolution and diffusion sampling for brain mapping with diffusion tractography: time well spent? Hum Brain Mapp 35:5667–5685. doi:10.1002/hbm.22578
Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4:112. doi:10.1186/s40478-016-0382-8
Cubon VA, Putukian M, Boyer C, Dettwiler A (2011) A diffusion tensor imaging study on the white matter skeleton in individuals with sports-related concussion. J Neurotrauma 28:189–201. doi:10.1089/neu.2010.1430
Currie S, Saleem N, Straiton JA, Macmullen-Price J, Warren DJ, Craven IJ (2016) Imaging assessment of traumatic brain injury. Postgrad Med J 92:41–50. doi:10.1136/postgradmedj-2014-133211
D’Arceuil H, de Crespigny A (2007) The effects of brain tissue decomposition on diffusion tensor imaging and tractography. NeuroImage 36:64–68. doi:10.1016/j.neuroimage.2007.02.039
Dawe RJ, Bennett DA, Schneider JA, Vasireddi SK, Arfanakis K (2009) Postmortem MRI of human brain hemispheres: T2 relaxation times during formaldehyde fixation. Magn Reson Med 61:810–818. doi:10.1002/mrm.21909
Gajawelli N, Lao Y, Apuzzo ML, Romano R, Liu C, Tsao S, Hwang D, Wilkins B, Lepore N, Law M (2013) Neuroimaging changes in the brain in contact versus noncontact sport athletes using diffusion tensor imaging. World Neurosurg 80:824–828. doi:10.1016/j.wneu.2013.10.020
Gangolli M, Kim J, Holleran L, Stein T, Alvarez V, McKee A, Brody D (2016) Development of Methods to Perform Radiological-Pathological Correlations in Chronic Traumatic Encephalopathy. In: National Neurotrauma Society Symposium, Lexington, Kentucky
Ghajari M, Hellyer PJ, Sharp DJ (2017) Computational modelling of traumatic brain injury predicts the location of chronic traumatic encephalopathy pathology. Brain 140:333–343. doi:10.1093/brain/aww317
Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4:134–160. doi:10.1126/scitranslmed.3003716
Goubran M, Crukley C, de Ribaupierre S, Peters TM, Khan AR (2013) Image registration of ex vivo MRI to sparsely sectioned histology of hippocampal and neocortical temporal lobe specimens. NeuroImage 83:770–781. doi:10.1016/j.neuroimage.2013.07.053
Goubran M, de Ribaupierre S, Hammond RR, Currie C, Burneo JG, Parrent AG, Peters TM, Khan AR (2015) Registration of in vivo to ex vivo MRI of surgically resected specimens: a pipeline for histology to in vivo registration. J Neurosci Methods 241:53–65. doi:10.1016/j.jneumeth.2014.12.005
Hasan KM, Narayana PA (2003) Computation of the fractional anisotropy and mean diffusivity maps without tensor decoding and diagonalization: theoretical analysis and validation. Magn Reson Med 50:589–598. doi:10.1002/mrm.10552
Hasan KM, Narayana PA (2006) Retrospective measurement of the diffusion tensor eigenvalues from diffusion anisotropy and mean diffusivity in DTI. Magn Reson Med 56:130–137. doi:10.1002/mrm.20935
Henry LC, Tremblay J, Tremblay S, Lee A, Brun C, Lepore N, Theoret H, Ellemberg D, Lassonde M (2011) Acute and chronic changes in diffusivity measures after sports concussion. J Neurotrauma 28:2049–2059. doi:10.1089/neu.2011.1836
Ilvesmaki T, Luoto TM, Hakulinen U, Brander A, Ryymin P, Eskola H, Iverson GL, Ohman J (2014) Acute mild traumatic brain injury is not associated with white matter change on diffusion tensor imaging. Brain 137:1876–1882. doi:10.1093/brain/awu095
Jensen JH, Helpern JA, Ramani A, Lu H, Kaczynski K (2005) Diffusional kurtosis imaging: the quantification of non-gaussian water diffusion by means of magnetic resonance imaging. Magn Reson Med 53:1432–1440. doi:10.1002/mrm.20508
Jones DK, Horsfield MA, Simmons A (1999) Optimal strategies for measuring diffusion in anisotropic systems by magnetic resonance imaging. Magn Reson Med 42:515–525
Kamnaksh A, Budde MD, Kovesdi E, Long JB, Frank JA, Agoston DV (2014) Diffusion tensor imaging reveals acute subcortical changes after mild blast-induced traumatic brain injury. Sci Rep 4:4809. doi:10.1038/srep04809
Karperien A, Ahammer H, Jelinek HF (2013) Quantitating the subtleties of microglial morphology with fractal analysis. Front Cell Neurosci 7:3. doi:10.3389/fncel.2013.00003
Koay CG, Chang LC, Carew JD, Pierpaoli C, Basser PJ (2006) A unifying theoretical and algorithmic framework for least squares methods of estimation in diffusion tensor imaging. J Magn Reson 182:115–125. doi:10.1016/j.jmr.2006.06.020
Lawrenz M, Finsterbusch J (2015) Mapping measures of microscopic diffusion anisotropy in human brain white matter in vivo with double-wave-vector diffusion-weighted imaging. Magn Reson Med 73:773–783. doi:10.1002/mrm.25140
Le Bihan D, Johansen-Berg H (2012) Diffusion MRI at 25: exploring brain tissue structure and function. NeuroImage 61:324–341. doi:10.1016/j.neuroimage.2011.11.006
Lee B, Newberg A (2005) Neuroimaging in traumatic brain imaging. NeuroRx 2:372–383. doi:10.1602/neurorx.2.2.372
Mac Donald CL, Dikranian K, Song SK, Bayly PV, Holtzman DM, Brody DL (2007) Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury. Exp Neurol 205:116–131. doi:10.1016/j.expneurol.2007.01.035
Mac Donald CL, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, Snyder AZ, Raichle ME, Witherow JR, Fang R, Flaherty SF, Brody DL (2011) Detection of blast-related traumatic brain injury in US military personnel. N Engl J Med 364:2091–2100. doi:10.1056/NEJMoa1008069
Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E (2003) Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging 24:1079–1085. doi:10.1016/j.neurobiolaging.2003.04.007
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA, Group TC (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. doi:10.1007/s00401-015-1515-z
McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735. doi:10.1097/NEN.0b013e3181a9d503
McKee AC, Daneshvar DH (2015) The neuropathology of traumatic brain injury. Handb Clin Neurol 127:45–66. doi:10.1016/B978-0-444-52892-6.00004-0
McKee AC, Stein TD, Kiernan PT, Alvarez VE (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25:350–364. doi:10.1111/bpa.12248
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. doi:10.1093/brain/aws307
Millspaugh J (1937) Dementia pugilistica. US Naval Med Bull 35:297–303
Namjoshi DR, Cheng WH, McInnes KA, Martens KM, Carr M, Wilkinson A, Fan J, Robert J, Hayat A, Cripton PA, Wellington CL (2014) Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener 9:55. doi:10.1186/1750-1326-9-55
Robinson S, Berglass JB, Denson JL, Berkner J, Anstine CV, Winer JL, Maxwell JR, Qiu J, Yang Y, Sillerud LO, Meehan WP 3rd, Mannix R, Jantzie LL (2016) Microstructural and microglial changes after repetitive mild traumatic brain injury in mice. J Neurosci Res 95:1025–1035. doi:10.1002/jnr.23848
Shahim P, Linemann T, Inekci D, Karsdal MA, Blennow K, Tegner Y, Zetterberg H, Henriksen K (2016) Serum tau fragments predict return to play in concussed professional ice hockey players. J Neurotrauma 33:1995–1999. doi:10.1089/neu.2014.3741
Shahim P, Mattsson N, Macy EM, Crimmins DL, Ladenson JH, Zetterberg H, Blennow K, Tegner Y (2015) Serum visinin-like protein-1 in concussed professional ice hockey players. Brain Inj 29:872–876. doi:10.3109/02699052.2015.1018324
Shahim P, Tegner Y, Wilson DH, Randall J, Skillback T, Pazooki D, Kallberg B, Blennow K, Zetterberg H (2014) Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol 71:684–692. doi:10.1001/jamaneurol.2014.367
Shively SB, Edgerton SL, Iacono D, Purohit DP, Qu BX, Haroutunian V, Davis KL, Diaz-Arrastia R, Perl DP (2016) Localized cortical chronic traumatic encephalopathy pathology after single, severe axonal injury in human brain. Acta Neuropathol. doi:10.1007/s00401-016-1649-7
Shively SB, Horkayne-Szakaly I, Jones RV, Kelly JP, Armstrong RC, Perl DP (2016) Characterisation of interface astroglial scarring in the human brain after blast exposure: a post-mortem case series. Lancet Neurol 15:944–953. doi:10.1016/S1474-4422(16)30057-6
Siman R, Shahim P, Tegner Y, Blennow K, Zetterberg H, Smith DH (2015) Serum SNTF increases in concussed professional ice hockey players and relates to the severity of postconcussion symptoms. J Neurotrauma 32:1294–1300. doi:10.1089/neu.2014.3698
Smith DH, Hicks R, Povlishock JT (2013) Therapy development for diffuse axonal injury. J Neurotrauma 30:307–323. doi:10.1089/neu.2012.2825
Smith DH, Meaney DF, Shull WH (2003) Diffuse axonal injury in head trauma. J Head Trauma Rehabil 18:307–316
Stein TD, Alvarez VE, McKee AC (2014) Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther 6:4. doi:10.1186/alzrt234
Stein TD, Alvarez VE, McKee AC (2015) Concussion in chronic traumatic encephalopathy. Curr Pain Headache Rep 19:522. doi:10.1007/s11916-015-0522-z
Tremblay S, Henry LC, Bedetti C, Larson-Dupuis C, Gagnon JF, Evans AC, Theoret H, Lassonde M, De Beaumont L (2014) Diffuse white matter tract abnormalities in clinically normal ageing retired athletes with a history of sports-related concussions. Brain 137:2997–3011. doi:10.1093/brain/awu236
Wang Y, Sun P, Wang Q, Trinkaus K, Schmidt RE, Naismith RT, Cross AH, Song SK (2015) Differentiation and quantification of inflammation, demyelination and axon injury or loss in multiple sclerosis. Brain 138:1223–1238. doi:10.1093/brain/awv046
Wansapura JP, Holland SK, Dunn RS, Ball Jr WS (1999) NMR relaxation times in the human brain at 3.0 tesla. J Magn Reson Imaging JMRI 9:531–538
Weiss M, Alkemade A, Keuken MC, Muller-Axt C, Geyer S, Turner R, Forstmann BU (2015) Spatial normalization of ultrahigh resolution 7 T magnetic resonance imaging data of the postmortem human subthalamic nucleus: a multistage approach. Brain Struct Funct 220:1695–1703. doi:10.1007/s00429-014-0754-4
Yeh FC, Wedeen VJ, Tseng WY (2010) Generalized q-sampling imaging. IEEE Trans Med Imaging 29:1626–1635. doi:10.1109/TMI.2010.2045126
Yeh FC, Wedeen VJ, Tseng WY (2011) Estimation of fiber orientation and spin density distribution by diffusion deconvolution. NeuroImage 55:1054–1062. doi:10.1016/j.neuroimage.2010.11.087
Acknowledgements
We would like to thank the donors and their families for the generous brain donation which made this research possible. We would also like to thank the Boston University CTE Center for sample procurement. The studies presented in this work were carried out, in part, using Small-Animal MR Facility of the Mallinckrodt Institute of Radiology, and the Hope Center Alafi Neuroimaging Lab, Washington University in St. Louis. This research was funded by the National Institute of Health, and the views expressed are those of the authors. NIH UO1 NS086659-02 (Overall PI: A. McKee, Subproject 3 PI: Brody).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
NIH UO1 NS086659-02 (Overall PI: A. McKee, Subproject 3 PI: Brody).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Holleran, L., Kim, J.H., Gangolli, M. et al. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol 133, 367–380 (2017). https://doi.org/10.1007/s00401-017-1686-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1686-x