
Abstract
More than 20 genetic loci have been associated with risk for Alzheimer’s disease (AD), but reported genome-wide significant loci do not account for all the estimated heritability and provide little information about underlying biological mechanisms. Genetic studies using intermediate quantitative traits such as biomarkers, or endophenotypes, benefit from increased statistical power to identify variants that may not pass the stringent multiple test correction in case–control studies. Endophenotypes also contain additional information helpful for identifying variants and genes associated with other aspects of disease, such as rate of progression or onset, and provide context to interpret the results from genome-wide association studies (GWAS). We conducted GWAS of amyloid beta (Aβ42), tau, and phosphorylated tau (ptau181) levels in cerebrospinal fluid (CSF) from 3146 participants across nine studies to identify novel variants associated with AD. Five genome-wide significant loci (two novel) were associated with ptau181, including loci that have also been associated with AD risk or brain-related phenotypes. Two novel loci associated with Aβ42 near GLIS1 on 1p32.3 (β = −0.059, P = 2.08 × 10−8) and within SERPINB1 on 6p25 (β = −0.025, P = 1.72 × 10−8) were also associated with AD risk (GLIS1: OR = 1.105, P = 3.43 × 10−2), disease progression (GLIS1: β = 0.277, P = 1.92 × 10−2), and age at onset (SERPINB1: β = 0.043, P = 4.62 × 10−3). Bioinformatics indicate that the intronic SERPINB1 variant (rs316341) affects expression of SERPINB1 in various tissues, including the hippocampus, suggesting that SERPINB1 influences AD through an Aβ-associated mechanism. Analyses of known AD risk loci suggest CLU and FERMT2 may influence CSF Aβ42 (P = 0.001 and P = 0.009, respectively) and the INPP5D locus may affect ptau181 levels (P = 0.009); larger studies are necessary to verify these results. Together the findings from this study can be used to inform future AD studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
More than five million Americans suffer with Alzheimer’s disease (AD), the most common neurodegenerative disease leading to progressive cognitive decline, and this number continues to increase as there are currently no effective methods to treat or prevent disease. Several genome-wide association studies (GWAS) have identified at least 24 loci containing common variants associated with AD risk [37, 39, 48, 56]. AD is a complex disease that is highly heritable, with an estimated heritability as high as 79% in twin studies [31] and genetic variance analyses estimate >53% of the variance in AD status can be explained by common variants (minor allele frequency, MAF > 1%) [64]. Polygenic studies have illustrated the genetic complexity underlying AD; recent studies using polygenic risk scores (PRS) calculated by combining the small effects of independent SNPs associated with AD risk (P < 0.5) provided AD risk prediction accuracy, as measured by area under the receiver operating curve (AUC) > 0.74, which is near the maximum AUC (0.82) [22, 23]. These studies indicate many genetic loci combine to increase risk for AD, most of the genetic risk loci are tagged by common variants (MAF > 1%), and that these loci, individually, have small effects on disease. These findings reveal that most AD risk variants have not passed the strict significance threshold required for multiple-test correction in GWAS, even in large studies such as the landmark study by the International Genomics of Alzheimer’s Project (IGAP), involving more than 74,000 total individuals, which identified 11 novel loci associated with AD risk [48]. It is also important to note that most AD susceptibility loci identified in these GWAS are gene-dense regions and many significantly associated SNPs are non-coding (intronic or intergenic), making it difficult to determine which genes are involved or how identified variants influence these genes. Studies integrating alternative phenotypes, gene expression, and other omics data are important for understanding the underlying biology of AD.
There is significant evidence that AD pathology is present several years before the onset of clinical symptoms [25, 26, 41, 55]. Consequently, AD case–control GWAS can be confounded by the presence of preclinical “controls”. Case–control-based GWAS are also limited to identifying genetic associations for disease risk; results from these studies do not provide information about other aspects of disease such as age at onset (AAO) or disease progression, or information about underlying biological mechanisms involved in pathogenesis. Endophenotypes are quantitative traits strongly associated with disease that also share genetic architecture with disease; therefore, genetic studies of endophenotypes are a powerful approach to identify loci associated with complex traits without many of the limitations of case–control studies. Cerebrospinal fluid (CSF) amyloid-beta1-42 (Aβ42) and phosphorylated tau (ptau181) are well-established AD endophenotypes [7, 13–15]. CSF ptau181 levels are elevated in AD cases and positively correlate with the number of neurofibrillary tangles, while CSF Aβ42 levels are lower in cases and correlate negatively with plaque load [43, 59, 72]. Increased CSF ptau181 is predictive for cognitive decline and progression from mild cognitive impairment to AD [2, 16]. Some genetic variants associated with AD also influence CSF levels of ptau181, Aβ42, or both [13, 44]. We previously performed GWAS of CSF tau, ptau181, and Aβ42 on 1269 participants (591 cases, 687 controls) and identified four genome-wide significant loci associated with tau and ptau181, including a novel locus that also associated with AD risk, tangle pathology, and cognitive decline [13]. This study has been expanded more than twofold to 3146 participants across nine cohorts with CSF and genome-wide genotype data (Table 1), providing additional power to identify more novel loci associated with ptau181, Aβ42, and AD.
Methods
Ethics statement
The Institutional Review Boards of all participating institutions approved the study and research was carried out in accordance with the approved protocols. Written informed consent was obtained from participants or their family members.
Cohort descriptions
CSF tau, ptau, and Aβ42 were measured in 3146 individuals from nine different studies. There were 805 individuals (29.34% cases) enrolled in studies at the Charles F. and Joanne Knight Alzheimer’s Disease Research Center (Knight ADRC), 787 individuals (more than 71% cases) from Alzheimer’s Disease Neuroimaging Initiative (ADNI; 390 from ADNI1 and 397 from ADNI2), 184 individuals (5.43% cases) from BIOCARD: Predictors of Cognitive Decline Among Normal Individuals (BIOCARD), 105 individuals (no AD status) from Saarland University in Homburg/Saar, Germany (HB), 433 individuals (22.17% cases) from Mayo Clinic (MAYO), 293 individuals (all cases) from Skåne University Hospital, Sweden (SWEDEN), 164 (62.8% cases) from studies at Perelman School of Medicine at the University of Pennsylvania (UPENN), and 375 (33.33% cases) from studies at the University of Washington (UW). Table 1 shows the demographic data for each study. Clinical assessments, CSF collection, and proteins were measured by each site. Clinical dementia rating (CDR) was available for 86% of the total data set. The CDR is a five-point scale used to describe the overall dementia severity for each individual (no dementia = 0, very mild = 0.5, mild = 1, moderate = 2, and severe = 3). Individuals with CDR = 0 were categorized as controls, cases were defined as individuals with CDR > 0.
Genotyping and imputation
Samples were genotyped with the Illumina 610 or Omniexpress chip. Stringent quality control (QC) criteria were applied to each genotyping array separately before combining genotype data. The minimum call rate for single nucleotide polymorphisms (SNPs) and individuals was 98% and autosomal SNPs not in Hardy–Weinberg equilibrium (P < 1 × 10−6) were excluded. X-chromosome SNPs were analyzed to verify sex identification. Unanticipated duplicates and cryptic relatedness (Pihat ≥ 0.25) among samples were tested by pairwise genome-wide estimates of proportion identity-by-descent, and when a pair of identical or related samples was identified, the sample from Knight ADRC or with a higher number of variants that passed QC was prioritized. EIGENSTRAT [61] was used to calculate principal components. APOE ε2, ε3, and ε4 isoforms were determined by genotyping rs7412 and rs429358 using Taqman genotyping technology as previously described [14, 15, 44]. The 1000 Genomes Project Phase 3 data (October 2014), SHAPEIT v2.790 [18], and IMPUTE2 v2.3.2 [40] were used for phasing and imputation. Individual genotypes imputed with probability <0.90 were set to missing and imputed genotypes with probability ≥0.90 were analyzed as fully observed. Genotyped and imputed variants with MAF < 0.02 or IMPUTE2 information score <0.30 were excluded, leaving 7,358,575 variants for analyses.
Data normalization for statistical analyses
Prior to combining data for analyses, CSF levels of tau, ptau, and Aβ42 were log10-transformed to approximate a normal distribution and the mean from each data set was standardized to zero to account for the different platforms used by different studies to measure protein levels. There were no significant differences in the transformed and standardized values for the different studies. Study, age, sex, and the first two principal components were identified as confounding factors by stepwise regression analyses for each protein and corrected for in applicable analyses.
Experimental design and data modeling
Studies by our group, and others, have demonstrated that when there is GWAS data available for all samples, a one-stage GWAS of combined data from both stages of a two-stage GWAS provides more power to identify genetic association than analyzing the groups separately, despite the fact that the one-stage GWAS requires a more stringent threshold to determine significance [13–15, 19, 70]. To maximize the power in our analyses, we performed a one-stage joint-GWAS. The CSF levels were measured with different platforms and at different sites, consequently the raw values could not be combined. Instead, the raw values were log10-transformed to approximate a normal distribution within each separate study and centralized by each study mean. We have used this approach in previous studies and demonstrated that it is an effective way to correct for study differences [13, 19]. We also performed analyses to ensure the results were not confounded by any study bias; to determine if the top hits were being driven by any individual study, we analyzed each dataset separately and performed meta analyses. The directions of effect for the genome-wide significant signals for Aβ42 and ptau181 were consistent across studies when analyzed separately and results from meta-analyses of the individual studies were consistent with the joint results even after removing cohort from previous study (Supplementary Figs. 1–3).
Alternative mixed model method to normalize Aβ42
Since CSF levels of Aβ42 are lower in AD cases than controls, begin decreasing prior to clinical symptom onset [25, 26, 43, 59, 72], and the studies in this dataset varied in proportion of cases to controls, we wondered if a mixture modeling approach would be more appropriate for standardizing the data between studies instead of centering on the mean of each study. This method was successfully used previously to classify AD cases in two independent cohorts with at least 94% sensitivity [17]. Mixture modeling is a statistical method for estimating subpopulations within an overall group; in this case we assumed two normally distributed subgroups within each dataset representing individuals with low Aβ42, therefore likely to be AD cases or preclinical, and with high Aβ42, likely to be cognitively normal controls. Using an expectation–maximization algorithm, we calculated estimated means, standard deviations, and subgroup proportions for each study. Based on the assumption of two univariate normal distributions within each study we obtained two estimated means (μ 1 and μ 2), two estimated standard deviations (σ 1 and σ 2), and two estimated mixing proportions (λ 1 and λ 2). We used these results to calculate the intersection of the estimated Gaussian curves using the following formula (Eq. 1):
then centered the log10-transformed Aβ42 levels to the intersection of the curves instead of the means for each study (Supplementary Table 1). CSF Aβ42 thresholds have been determined previously for both ADNI (192 pg/mL) [69] and ADRC (500 pg/mL) [25]; the calculated intersects were comparable to these values (182 and 548 pg/mL, respectively, Supplementary Table 1). The density plots of the estimated subpopulations for each study fit the overall distributions reasonably well, but after accounting for AD status the model did not appear significantly different than standardizing to the overall mean (Supplementary Figs. 4, 5). There was no difference between the two methods in a single variant analysis of the mixed model standardized CSF Aβ42 levels and the levels centered at the study mean (Supplementary Fig. 6).
The intersect was log10-transformed and subtracted from the log10-transformed values of Aβ42 (Supplementary Figs. 4, 5 and Supplementary Table 1). When the single-variant analysis was repeated using these normalized values for Aβ42, the results were comparable to those from the mean normalized values (Supplementary Fig. 6). Therefore, to be consistent, we used the mean normalized values in all analyses.
Association testing
The additive linear regression model in PLINK v1.9 [11] was used for single-variant analyses for each analyte, including study, age, sex, and the first two principal components as covariates in the default model [11]. The genomic inflation factor was λ = 1.02 for ptau181 and λ = 1.03 for tau and Aβ42 (Supplementary Fig. 7). There were no novel genetic associations identified for CSF tau levels (Supplementary Fig. 8 and Supplementary Table 2) but we did identify novel associations for ptau181 and Aβ42 (Figs. 1, 2; Table 2; Supplementary Tables 3, 4). Conditional analyses were conducted to identify additional independent signals in a locus by adding the SNP with the smallest P value as a covariate into the default regression model and testing all remaining regional SNPs for association (Supplementary Figs. 9, 10). AD status, CDR, APOE alleles, APOE ε4 carrier status, Aβ42, or ptau181 levels were corrected for in additional analyses to determine the effects of these phenotypes on the genetic associations (Supplementary Fig. 11 and Supplementary Tables 5–8). The combined dataset was stratified by AD status and cases and controls were analyzed separately for single-variant associations (Supplementary Table 8). Statistical significance for the single-variant analyses was based on the commonly used threshold from Bonferroni correction of the likely number of independent tests in genome-wide analyses (P < 5 × 10−8). Manhattan plots and regional association plots were created using the R package qqman v0.1.2 [74] and LocusZoom v1.3 [62], respectively.

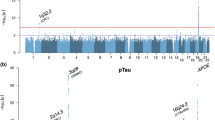
Association plots from single variant analyses of CSF ptau181 levels. a Manhattan plot shows negative log10-transformed P values from the joint analysis of ptau181. The horizontal lines represent the genome-wide significance threshold, P = 5 × 10−8 (red) and suggestive threshold, P = 1 × 10−5 (blue). Red arrows point to novel loci. The y-axis is truncated, the lowest P value on chromosome 19 was 5.30 × 10−33. b, c Regional association plots of novel loci are shown for SNPs associated with ptau181 near PCDH8 (a) and between NFATC1 and CTDP1 (b). The SNPs labeled on each regional plot had the lowest P value at each locus and are represented by a purple diamond. Each dot represents a SNP and dot colors indicate LD with the labeled SNP. Blue vertical lines show recombination rate marked on the right-hand y-axis of each regional plot. Plots for previously reported loci are in Supplementary Fig. 7

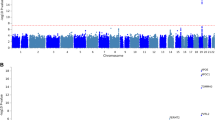
Association plots from single variant analyses of CSF Aβ42. a Manhattan plot shows negative log10-transformed P values from the joint analysis of Aβ42. The horizontal lines represent the genome-wide significance threshold, P = 5 × 10−8 (red) and suggestive threshold, P = 1 × 10−5 (blue). Red arrows point to novel loci. The y-axis is truncated, the lowest P value on chromosome 19 was 4.78 × 10−94. b, c Regional association plots of novel loci are shown for SNPs associated with Aβ42 near GLIS1 (b) and within SERPINB1 (c). The SNPs labeled on each regional plot had the lowest P value at each locus and are represented by a purple diamond. Each dot represents a SNP and dot colors indicate LD with the labeled SNP. Blue vertical lines show recombination rate marked on the right-hand y-axis of each regional plot. Plots for previously reported loci are in Supplementary Fig. 7
Meta-analyses
To test for potential systematic differences between the datasets, each study was analyzed separately for the most significant SNPs from the joint analyses. Covariates were age, sex, and the first two principal components. Meta-analyses of the results from the separate datasets were performed using METAL (version released 2011-03-25) [80]. The METAL default analysis scheme was used with sample size and beta for each SNP taken into account when combining P values across studies. For the genome-wide significant signals, the nine studies showed consistent direction of effect individually, and meta-analysis results were consistent with the joint results (Supplementary Figs. 1, 2). After removing the samples that comprised the previously published study [13], the meta-analysis results remained consistent with the joint results (Supplementary Fig. 3). Forest plots were generated using the R package rmeta v2.16.
Association with AD risk, progression, AAO
Results from independent analyses of different cohorts for AD risk [48], AAO (personal communication: Huang & Goate), and disease progression were analyzed to determine whether loci associated with CSF tau, ptau181, and Aβ42 were also associated with other AD phenotypes. Results for the most significantly associated SNPs for CSF tau, ptau181, and Aβ42 are reported here from the largest previously published two-stage meta-analysis of GWAS for AD risk consisting of a total 25,580 cases and 48,466 controls [48], and a recently published genome-wide survival analysis of AAO consisting of 39,855 individuals (personal communication: Huang & Goate). To determine disease progression in an independent cohort of 1530 individuals, we utilized the CDR Sum of Boxes (CDR-SB) which has been demonstrated to accurately stage dementia severity [57, 58]. Overall CDR is derived from scores in six individual categories (boxes) of memory, orientation, problem solving, community involvement, involvement in home and hobbies, and personal care; CDR-SB is a sum of the six boxes which provides a semi-continuous measure of symptomatic AD dementia from 0 (cognitively normal) to 18 (the most severe dementia). Disease progression from longitudinal studies at ADNI (n = 728) and Knight ADRC (n = 802) was modeled as the change in CDR-SB per year, adjusting for age, sex, baseline CDR, follow-up time, level of education, site, and PCs (Supplementary Table 9). Samples with ≥3 clinical assessments over 1.5 years after being diagnosed with AD were selected for the analysis and a mixed-model repeated measure framework was used to account for correlation between repeated measures in the same individual. We selected the appropriate optimal variance–covariance structure that minimizes the Akaike Information Criterion for testing the null model AR1 [14].
Functional annotation
All SNPs below the suggestive significance threshold (P = 1 × 10−5) were taken forward for functional annotation using ANNOVAR version 2015-06-17 [77] and examined for potential regulatory functions using RegulomeDB v1.1 [8] and HaploReg v4.1 [78]. The search tools on the Genotype-Tissue Expression (GTEx) Analysis Release V6, dbGaP Accession phs000424.v6.p1 portal [33], data from the Brain eQTL Almanac (Braineac) [73] analyzed with the R package MatrixEQTL [68], and the Blood eQTL browser [79] were utilized to determine if genome-wide significant SNPs were reported eQTLs. The Brain RNA-Seq database (http://web.stanford.edu/group/barres_lab/brainseqMariko/brainseq2.html) was mined to determine if genes of interest were expressed in the brain and in which cell types [84].
Summary data-based mendelian randomization
To prioritize the putative causal variant from the ptau181 and Aβ42 associated variants, we used the Summary data-based Mendelian Randomization (SMR) method which tests the functional association between gene expression levels (measured by probes) and a trait (such as CSF protein levels) through the regression of estimated effect sizes [85]. Based on the assumptions of Mendelian randomization, any gene–trait association identified in this analysis should be free of confounding from non-genetic factors. To distinguish causality of a single variant on both gene expression and the trait vs linkage of two distinct genetic variants in LD with one affecting expression and one affecting the trait, the SMR method uses a heterogeneity (HEIDI) test. For the SMR analysis, we utilized the estimates of SNP effects on gene expression from summary data of a large-scale eQTL study with gene expression measured in peripheral blood (Blood eQTL browser) [79] and gene expression data from Cardiogenics measured in macrophages [35]. There were 3000 SNPs present in both the blood eQTL data and the GWAS results so the statistical significance threshold was defined (based on Bonferroni correction) as P < 1.67 × 10−5 for the associations between eQTL in blood and CSF GWAS loci. Focusing on the SERPINB1 gene region (from the 6p terminal to 10 Mb after the defined SERPINB1 transcription region) in the macrophage eQTL data, there were 4336 SNPs; therefore, the statistical significance threshold was defined as P < 1.15 × 10−5. The HEIDI threshold was set at P > 0.05 to be conservative; since the null hypothesis is that there is only one causal variant, a P > 0.05 indicates the variant that passed the SMR test is the causal variant.
Genetic variance estimation
The Genome-wide Complex Trait Analysis (GCTA) v1.25.2 tool [82] was used to estimate the proportion of phenotypic variance explained by the common (MAF > 0.02) imputed and genotyped autosomal variants. The restricted maximum likelihood (REML) analysis was performed on the log10-transformed standardized analyte values adjusted for age and gender with the first two principal components as covariates. Results are reported in Supplementary Table 10.
Since it was reported that estimated h 2 may be biased if causal variants are enriched in areas with lower or higher LD than average [81], we also used GCTA to calculate segment-based LD scores (segment length = 200 kb) for all SNPs in the REML analysis and plotted the number of SNPs from the single-variant analyses of Aβ42 and ptau181 with P < 1 × 10−5 (Supplementary Fig. 12). Since the most significantly associated SNPs showed LD heterogeneity, and the method can be applied to imputed GWAS data, we used the LD- and MAF-stratified genomic-REML (GREML-LDMS) method [81] in GCTA to estimate h 2 for each LD quartile and calculate a total h 2 estimate (Supplementary Table 10). The GCTA-GREML power calculator (http://cnsgenomics.com/shiny/gctaPower) [76] was used to calculate the power of the REML and GREML-LDMS analyses with the actual sample sizes, estimated h 2, α = 0.05, and genetic variance = 2 × 10−5 as parameters (Supplementary Table 10).
Polygenic risk score
PRS were calculated using a weighted sum of the AD risk alleles reported by IGAP [48]. Weights for SNPs outside the APOE region were calculated by transforming the reported odds ratios by a base-2 logarithm. Proxy SNPs were utilized if the reported SNPs were unavailable in our data or did not pass QC; proxies were selected with the highest R2 and D′ values to the reported IGAP SNP in our genetic data and in 1000 Genomes. Since APOE has a large effect on AD risk and CSF protein levels, we calculated a default PRS without APOE. The effects of APOE genotype on AD risk are not additive, so APOE genotypes were weighted by the effects reported previously for each genotype (ε2/ε2 OR = 0.6, ε2/ε3 OR = 0.6, ε2/ε4 OR = 2.6, ε3/ε4 OR = 3.2, ε4/ε4 OR = 14.9) [29]. The SNPs that composed the PRS are listed in Supplementary Table 11. The PRS were calculated (with and without APOE genotype) using the score function in PLINK v1.90b3.42 [11], including the no-mean-imputation option to ensure scores would not be imputed for missing genetic data. The resulting mean score per allele was multiplied by the allele count to generate a total PRS.
Results
Reproduction of previously reported associations with CSF Aβ42, tau, and ptau181
As reported previously, the most significant variant associated with CSF levels of Aβ42, tau, and ptau181 was a proxy SNP for APOE ε4 (r 2 = 0.726, D′ = 1), rs769449[A] (Aβ42 β = −0.117, P = 9.02 × 10−47; tau β = 0.082, P = 1.95 × 10−16; ptau181 β = 0.091, P = 2.56 × 10−18) [13]. In the current analyses, the effects were similar to what was previously reported with more significant P values due to the larger sample size (Aβ42 β = −0.101, P = 4.78 × 10−94; tau β = 0.078, P = 4.05 × 10−29; ptau181 β = 0.081, P = 9.51 × 10−35). While there were no other loci associated with Aβ42 in the previous GWAS, two loci outside the APOE locus were identified to be associated with CSF tau and ptau181 [13]. We also replicated the previously reported loci for ptau 181 on 3q28 (rs9877502[A] near GMNC, β = 0.044, P = 1.68 × 10−7) and on 9p24.2 (rs514716[C] on GLIS3, β = −0.072, P = 3.22 × 10−9) were both genome-wide significant in this larger study (rs9877502[A], β = 0.032, P = 6.35 × 10−9; rs514716[C], β = −0.049, P = 2.94 × 10−8) (Table 2, Supplementary Fig. 13; see Supplementary Table 4 for all loci with P < 1 × 10−5) [13].
A small GWAS of AD CSF biomarkers from 374 ADNI participants (102 controls) identified variants in EPC2 associated with CSF levels of tau and the Tau/Aβ42 ratio [46]. In our current analyses, there were no genome-wide significant, or suggestive, associations with the EPC2 locus (tau: β = 0.005, P = 0.428; Tau/Aβ42 ratio β = 0.072, P = 0.017), but interestingly the strongest association was for Aβ42 (β = −0.016, P = 3.77 × 10−4; Supplementary Fig. 14). Another early GWAS of CSF levels from 410 ADNI participants (119 controls) did not identify any genome-wide significant variants for CSF Aβ42, ptau181, or tau in cases, but found three genome-wide significant signals for Aβ42 in controls (CYP19A1, NCAM2, and ARL5B) [36]; none of these loci were associated with Aβ42 in our current analyses of the joint dataset, cases-only, or controls-only (P > 0.1). A recent GWAS with only AD cases (N = 363) reported that SNPs located in the SUCLG2 region were associated with CSF Aβ42 levels [63] but this region was not associated with Aβ42 in any of the current analyses of the joint dataset, cases-only, or controls-only (P > 0.1). FRA10AC1 variants were associated with CSF Aβ42 levels in a two-stage GWAS of data from ADNI (two discovery sets: N = 391 and N = 385; replication set N = 204), and although there were no genome-wide significant signals within the FRA10AC1 locus in the current analyses, there was a near suggestive association between Aβ42 and indel rs143151810[-] (β = −0.033, P = 8.13 × 10−5; Supplementary Fig. 14), which is in high LD with the SNP identified in the other study, rs10509663[G] (r 2 = 0.987, D′ = 0.997), and both associations showed the same direction of effect on Aβ42 levels [51].
APOE locus significantly influences CSF levels of ptau181 and tau independently of Aβ42
As we reported previously, the APOE region was still significantly associated with ptau181 after including CSF Aβ42 levels in the analysis (rs769449[A]: default, β SNP = 0.079, P = 5.30 × 10−33; adjusted for Aβ42 levels, β SNP = 0.046, P = 2.08 × 10−11), and in the current analysis the association between rs769449[A] and ptau181 remained genome-wide significant after including the interaction between Aβ42 levels and APOE genotype in the model (β SNP = 0.042, P = 1.65 × 10−8), suggesting APOE may influence tau pathology independently of Aβ42 and supporting our previous findings (Supplementary Fig. 11 and Supplementary Table 5) [13]. Similar results were observed with CSF tau as well (default, β SNP = 0.077, P = 6.75 × 10−28; adjusted for Aβ42 levels, β SNP = 0.048, P = 4.11 × 10−11; Aβ42 and APOE genotype interaction, β SNP = 0.045, P = 1.04 × 10−8). Low Aβ42 levels (ADRC < 500 pg/mL and ADNI < 192 pg/mL) have been associated with amyloid positron emission tomography (PET-PIB) evidence of Aβ deposition [25, 69]. To determine if the possible presence of Aβ pathology influenced the effect of the APOE locus on ptau181 levels as we reported previously [13], we stratified the data from ADRC, ADNI1, and ADNI2 by high and low levels of Aβ42 and found the association between APOE locus and ptau181 levels in both groups with a higher effect size in the individuals with low Aβ42 (β = 0.055, P = 2.12 × 10−7) than those with high Aβ42 (β = 0.037, P = 1.05 × 10−2).
We wanted to determine if the signal in the APOE locus was driven entirely by APOE genotype (APOE ε2, ε3, and ε4), or if there was an independent signal influencing CSF levels of ptau181 and Aβ42, so we performed conditional analyses on APOE genotype accounting for both ε2 and ε4 effects. The APOE genotype showed the strongest association with CSF levels of ptau181 (β = 0.042, P = 3.13 × 10−40) and Aβ42 (β = −0.053, P = 8.88 × 10−114) after correcting for age, sex, study, and two principal components. The association between the top hit in the APOE locus (rs769449) and ptau181 or Aβ42, remained significant, but not genome-wide significant, after adding APOE genotype to the model (ptau181: β = 0.034, P = 1.07 × 10−3; Aβ42: β = -0.036, P = 1.65 × 10−6) suggesting that there may be a signal in this region independent of APOE ε2, ε3, and ε4 (Supplementary Table 12). To further explore this finding, we conditioned on the most significant SNP (rs769449), which is in high LD for the APOE ε4 allele (rs429358[C], D′ = 1, r 2 = 0.726). We found that although the associations between APOE genotype and ptau181 and Aβ42 decreased, they remained genome-wide significant (conditioned: β = 0.029, P = 5.91 × 10−9 and β = −0.040, P = 2.28 × 10−28, respectively) (Supplementary Table 12). Together, these results suggest that most of the signal in this region is driven by APOE genotype, but additional independent SNPs in this region may influence CSF levels of both ptau181 and Aβ42.
Novel associations in single-variant regression analyses for Aβ42 and ptau181
The genomic inflation was minimal in all analyses suggesting no evidence of confounding by systematic biases (default model λ = 1.03 for Aβ42 and tau, 1.02 for ptau181; Supplementary Fig. 7). In addition to the loci reported previously, two novel genetic associations with CSF ptau181 were identified on 13q21.1 (rs9527039[C] near PCDH8, β = −0.061, P = 5.95 × 10−9) and 18q23 (rs12961169[T] near CTDP1, β = 0.050, P = 5.12 × 10−10) (Fig. 1; Table 2). We also identified, for the first time, two genome-wide significant loci outside of the APOE region associated with CSF Aβ42 on 1p32.3 (rs185031519[G] near GLIS1, β = −0.059, P = 2.08 × 10−8) and on 6p25 (rs316341[G] within SERPINB1, β = −0.025, P = 1.72 × 10−8) (Fig. 2; Table 2; see Supplementary Table 3 for all loci with P < 1 × 10−5). Conditioning on the most significant SNPs in each of these identified loci did not reveal any additional genome-wide significant signals (Supplementary Figs. 9, 10).
When clinical dementia rating (CDR) or clinical status were included in the model for either Aβ42 or ptau181, the results for the top loci were not significantly different than the default model (Supplementary Table 7), and when the analyses were stratified by AD status, the betas were similar for cases and controls (Supplementary Table 8). When individuals were stratified by high or low CSF Aβ42 levels (Aβ42 threshold: ADRC = 500 pg/mL [25], ADNI = 192 pg/mL [69]), the betas for the top loci were similar between the two groups (Supplementary Table 8). These results suggest that all of the individuals in this study contributed to the associations with CSF Aβ42 and ptau181 levels, independent of status or amyloid pathology.
Effects of associated genetic loci on other AD phenotypes
Since the purpose of studying these AD endophenotypes was to identify genetic factors associated with AD, we tested the genome-wide significant loci for associations with AD risk [48], rate of AD progression [58], or AAO (personal communication: Huang & Goate) in independent cohorts. The loci associated with Aβ42 were also associated with risk, AAO, and/or progression (Table 3). The GLIS1 locus was associated with lower CSF Aβ42 levels (rs185031519[G], β = −0.059, P = 2.08 × 10−8), increased AD risk (rs114122417[A], OR = 1.105, P = 0.034) [48], and faster disease progression (rs185031519[G], β = 0.277, P = 0.019) (Table 3). The intronic SERPINB1 variant, rs316341[G], was associated with earlier AAO (β = 0.043, P = 4.62 × 10−3) as well as lower Aβ42 (β = −0.025, P = 1.72 × 10−8) (Table 3). Although the loci associated with ptau181 that we reported previously were associated with AD risk and AAO [13], we did not find evidence that the novel loci were associated with risk, AAO, or progression (Table 3). We were unable to test other AD phenotypes such as brain atrophy or neuropathology. However, both the MAPT locus on 17q21, which is associated with CSF tau levels in the presence of Aβ deposition [45], and the GMNC locus, which was associated with CSF levels of tau (β = 0.040, P = 3.07 × 10−11) and ptau181 (β = 0.035, P = 7.62 × 10−10), as well as AD risk (OR = 1.044, P = 9.08 × 10−3), tangle pathology (P = 0.039, reported previously) and cognitive decline (P = 4.86 × 10−5, reported previously) [13], have recently been associated with total brain volume in a meta-analysis of 26,577 individuals of European descent [1], suggesting variants associated with ptau181 may also be associated with other brain-related or neurodegenerative phenotypes.
Bioinformatics annotation
None of the genotyped or imputed SNPs in the genome-wide significant loci for Aβ42 or ptau181 were coding variants (R 2 > 0.5, Supplementary Tables 3, 4). In an effort to pinpoint functional genes influencing CSF protein levels, we searched for SNPs in the genome-wide significant loci with cis expression quantitative trait locus (eQTL) effects in human tissues. The top SNPs associated with Aβ42 on 6p25 have eQTL effects for SERPINB1 in transformed fibroblasts (rs316341[G]: β = 0.24, P = 1.3 × 10−7) and whole blood (rs316339[A]: Z score = 28.96, P = 2.2 × 10−184), and rs316339 had the strongest eQTL effect on SERPINB1 in the hippocampus (β = 0.30, P = 3.90 × 10−5) (Table 4). To determine if the putative causal variant is the same for SERPINB1 expression and Aβ42 levels, we utilized Summary data-based Mendelian Randomization (SMR) [85] to test the Westra whole blood expression data [79]. One SERPINB1 variant, rs316339, which is in LD with rs316341 (D’ = 1, r 2 = 0.993; CSF Aβ42 β = −0.025, P = 1.76 × 10−8), passed the SMR analysis (P = 2.95 × 10−8) and HEIDI test (P = 0.258). We performed the same test on macrophage expression data obtained from Cardiogenics and rs316341 passed the SMR analysis (P = 1.23 × 10−7) and HEIDI test (P = 0.240). This suggests that the locus associated with CSF Aβ42 is the same locus that affects expression of SERPINB1 in blood and macrophages.
The other genetic loci for Aβ42 and ptau181 were not as enriched for significant eQTL effects as SERPINB1, but there were suggestive results for 1p32.3 (near GLIS1) and 18q23 (near CTDP1). The signal near GLIS1 associated with Aβ42 (1p32.3) had an eQTL effect on SLC1A7 throughout the brain (rs185031519[G]: P = 8.8 × 10−5); however, overall expression of SLC1A7 was reported to be relatively low in the human brain, within the 33rd percentile of all gene expression in the temporal cortex, primarily in endothelial cells [84] (Supplementary Table 13). The locus on 18q23 associated with ptau181, between CTDP1 and NFATC1, may have eQTL effects on both genes in the frontal cortex (rs12961169[T]: CTDP1, β = −0.319, P = 3.85 × 10−5; NFATC1, β = −0.290, P = 1.71 × 10−5). Both NFATC1 and CTDP1 are expressed in the human temporal cortex (NFATC1 = 58th percentile; CTDP1 = 37th percentile; Supplementary Table 13).
Effect of AD risk loci on CSF levels
We wanted to determine whether known loci for AD risk are also associated with CSF levels of Aβ42 or ptau181. AD risk variants identified in the IGAP study [48] that were most significantly associated with Aβ42 were located in the CLU (β = 0.014, P = 0.001) and FERMT2 (β = −0.018, P = 0.009) gene regions, and SNPs in the CELF1 and ABCA7 regions had P < 0.05 (Supplementary Table 11). For CSF ptau181 levels, the most significant association was in the INPP5D region (β = 0.014, P = 0.009) and the CR1, PICALM, and FERMT2 regions had P < 0.05 (Supplementary Table 11). These results suggest that the risk variant in the CLU locus (rs11136000[T]) may increase risk for AD through an Aβ-associated mechanism and the INPP5D locus by a ptau-associated mechanism. Other loci like APOE or FERMT2 may act through both Aβ- and ptau-associated pathways to affect AD risk, and still other risk loci may act through alternate mechanisms such as neuronal survival, apoptosis, or homeostasis.
Although the individual AD risk variants were not strongly associated with Aβ42 or ptau181, we decided to analyze the potential overlap in the genetic architecture of AD risk and these endophenotype levels by determining whether PRS (with or without the effect of APOE genotype) calculated from the genome-wide significant hits for AD risk are also associated with CSF levels. We found not only a strong association between the non-APOE PRS and Aβ42 (β = −0.033, P = 5.01 × 10−7), but also tau (β = 0.049, P = 1.38 × 10−7) and ptau181 (β = 0.049, P = 1.81 × 10−8) (Supplementary Table 11). The strength of the association with the non-APOE PRS was greater than any of the individual SNPs composing the PRS. The addition of APOE genotype significantly increased the PRS association with CSF levels (Aβ42: β = −0.065, P = 5.01 × 10−88; tau: β = 0.051, P = 1.38 × 10−31; and ptau181: β = 0.044, P = 1.81 × 10−31) (Supplementary Table 11).
Estimation of CSF level variance explained by associated genetic loci
To determine the proportion of phenotypic variance (h 2) explained by the genetic loci identified for Aβ42 and ptau181, we analyzed all of the tested genotyped and imputed autosomal common variants (MAF > 0.02). It was recently demonstrated that estimated h 2 may be biased if causal variants are enriched in areas with lower or higher LD than average [81], so we used the GCTA tool to calculate segment-based LD scores (segment length = 200 kb) for all SNPs and plotted the number of SNPs with P < 1 × 10−5 for Aβ42 and ptau181 (Supplementary Fig. 12). Since we observed LD heterogeneity in the associated variants, and the LDMS method can be applied to imputed GWAS data, we used the GCTA LDMS method to test all SNPs in our genetic data [81]. After correcting for age, sex, and two principal components, approximately 35.5% of the variability in Aβ42 and 24.9% in ptau181 levels were explained by common variants; the respective SNPs associated with CSF Aβ42 and ptau181 with P < 1 × 10−5 only accounted for 3.5% (2.9% from chromosome 19) of the variability in Aβ42 levels and 3.2% (1.4% from chromosome 19) in ptau181 levels, corresponding to 10 and 13% of the estimated h 2 for CSF Aβ42 and ptau181, respectively. These results suggest many genetic variants have yet to be discovered.
Discussion
Genetic studies using disease endophenotypes as quantitative traits provide power to identify loci associated with disease risk with smaller sample sizes, and endophenotypes provide biological context to help identify loci associated with other disease phenotypes such as AAO and disease progression. In our previous study using CSF levels of Aβ42 and ptau181 as endophenotypes, rs9877502 (near GMNC1 on 3q28) was reported, for the first time, to be associated with ptau181 levels, AD risk, tangle pathology, and cognitive decline [13]. The ptau181 association was recently replicated in an independent cohort [63] and we confirmed the association in this much larger dataset. The GMNC1 locus was also recently reported to be associated with intracranial volume [1], suggesting that tau-associated pathology and brain volume share some genetic architecture. This larger study also revealed novel loci associated not only with Aβ42 but also with AD risk and disease progression (rs185031519[G], (rs185031519[G], P = 3.43 × 10−2 and P = 1.92 × 10−2, respectively), or AAO (rs316341[G], P = 4.62 × 10−3). The associations with AD risk and AAO were tested in independent datasets. The associations of these SNPs with risk, disease progression, and AAO may not pass stringent multiple test correction if we take into account the number of SNPs and phenotypes tested. However, it is important to note that we had a very specific hypothesis, including direction of effect, for each SNP. As expected, the alleles associated with lower CSF levels of Aβ42 were also associated with earlier disease symptom onset, increased AD risk, or faster progression. In any case, the associations with risk, disease progression, and AAO were identified in the largest datasets available to date, but additional studies will be needed to confirm the role of these loci in AD. By increasing the sample size more than twofold, we not only verified the results from our previous analyses, but also uncovered additional findings that can be used to inform future AD studies.
APOE genotype is the strongest genetic risk factor for sporadic AD, and is consistently the strongest association with CSF levels of Aβ42, tau, and ptau181 in several GWAS as well [13, 36, 46, 63]. Numerous studies have explored how APOE influences amyloid pathology in AD [67]. A few studies have also looked at the role of ApoE in tau pathology [30, 49, 52]. A recent study of brain tissue from 1056 individuals (659 AD cases) found that the APOE ε4 and ε2 alleles were not associated with tau tangle pathology in the absence of amyloid deposits [27]. As we previously reported, after accounting for CSF Aβ42, there was a strong association for APOE with CSF ptau181, although it no longer passed genome-wide significance [13]. We verified these results in the current study, and with the larger dataset the APOE signal remained genome-wide significant after accounting for Aβ42 levels. This provides additional evidence that APOE influences ptau181-associated mechanisms of AD independently of Aβ42-associated mechanisms. We also found, through conditional analyses, that although APOE genotype is driving most of the association for APOE with CSF Aβ42 and ptau181, there appears to be an additional signal within the APOE gene region that is independent of APOE ε2, ε3, and ε4.
CSF Aβ42 and ptau181 are well-established AD endophenotypes with a clear common genetic association for APOE and AD risk, but the shared genetic architecture between the disease and AD biomarkers is not as well-understood [7, 13–15]. Shared heritability between two traits can be estimated using different methods to calculate genetic and phenotypic correlations by linear mixed models, LD scoring, or genome partitioning; but most methods currently available usually require sample sizes in the tens of thousands to counteract statistical noise [3, 9, 10, 12, 66]. Another method to detect shared genetic etiology between traits is to calculate a PRS from a well-characterized cohort, usually from large case–control GWAS, and regress the other trait of interest, such as CSF protein levels, on the PRS in an independent cohort [21, 24]. Small studies (N < 350) have found that PRS were negatively correlated with CSF Aβ42 but not correlated with tau or ptau181 [54, 65, 71]. Recent studies of AD cases (N = 338) or individuals with mild cognitive impairment (N = 454), reported that their PRS without APOE were not associated with Aβ42, but tau and ptau were associated with the score without the APOE effect [53, 71]. In our current study of both AD cases and controls (N = 3145), we calculated a PRS composed of genome-wide significant AD risk loci that were reported in the largest AD case–control GWAS thus far [48]. Since the APOE locus is strongly associated with CSF levels and AD risk, it is not unexpected that PRS that include APOE effect would also be significantly associated with Aβ42 and ptau181. Calculating PRS without including the APOE effect can provide information about the much smaller genetic effect of other AD risk loci, and although the individual variants were not even suggestively significant the PRS was significantly associated with both proteins. This suggests there is a genetic overlap between AD risk and the CSF biomarkers that is not apparent in single variant analyses. Since we restricted the PRS to genome-wide significant AD risk loci, we may actually be underestimating genetic overlap between AD risk and CSF Aβ42 and ptau181. Some of the AD risk loci did not appear to be associated with either biomarker, suggesting they affect AD risk through mechanisms independent of Aβ42 and ptau181. Some AD risk loci such as CLU and PERMT2 for Aβ42 and INPP5D for ptau181 may be associated with these AD endophenotypes but did not reach genome-wide significance. Reasons for this could be that multiple risk loci interact to influence CSF levels, or possibly a lack of power due to small effect size of the individual variants. For example, we estimated that at least 4500 samples would be necessary for the association for CLU with Aβ42 to pass the genome-wide significance threshold (P < 5 × 10−8), suggesting that additional signals could be identified with a larger sample size.
Genetic studies of endophenotypes not only provide enough power to identify novel associations with smaller sample sizes than case–control studies, but can also help with understanding biological mechanisms of disease. Loci identified in this study alter gene expression or protein binding, which can provide valuable information for understanding the biological basis for AD pathology. We identified here, for the first time, two genome-wide significant signals for CSF Aβ42 outside the APOE region. Of particular interest is the locus on 6p25 which is associated with lower CSF Aβ42 and earlier AAO. This may be mediated through SERPINB1, because the same SNPs affect SERPINB1 expression in blood and macrophages. SERPINB1 encodes a serine protease inhibitor that is a key regulator of neutrophil programmed cell death [28, 50]. SERPINB1 is expressed in the human brain, primarily in microglia and macrophages [84]. Recent research of transgenic mouse models for AD (5xFAD and 3xTg-AD) reported that neutrophils were present in the brain near Aβ deposits, and researchers observed neutrophil migration from blood into the brain toward amyloid plaques [4, 83]. They discovered that Aβ42 triggered the high-affinity state of integrin LFA-1, which is necessary for neutrophil infiltration of the CNS [83]. Their results, combined with our findings that genetic variants that increase expression of SERPINB1 are also associated with lower levels of Aβ42, support other studies suggesting that immune response pathways may play a key role in AD pathology [38, 75]. Our results indicate a potential role for SERPINB1 in AD and suggest that adaptive immune response mechanisms are associated with Aβ-mediated pathology. Key proteins in neuroinflammation, triggering receptor expressed on myeloid cells 2 (TREM2) and YKL-40, are promising AD biomarkers [59, 60], and TREM2 variants are also strongly associated with AD risk [6, 34, 42]. Clusterin (CLU) has been associated with AD risk in numerous studies [37, 48] and a GWAS of CSF CLU levels suggested CLU may be associated with immune response [20]. Our findings add to this growing evidence that immune response plays a key role in AD and CSF levels of Aβ42 may be representative of this role.
The associations of the GMNC locus with ptau181 levels and brain volume suggest biological mechanisms other than immune response may be associated with tau-mediated pathology in AD [1, 13, 63]. Although not well studied, GMNC (also known as GEMC1) is a necessary regulator of DNA replication [5] and recently was shown to be a key player in the differentiation of radial glial cells to multiciliated neuroepithelial cells during neurogenesis in the subventricular zone [47]. Combined with our GWAS results, it appears GMNC may influence CSF ptau181 as part of the neurogenesis process. Further research is needed to determine if GMNC is indeed the gene affecting ptau181 and what biological mechanism is involved. However, some of the loci associated with ptau181 suggest immune response may also play a role in tau-associated pathology. NFATC1 encodes the nuclear factor of activated T-cells cytoplasmic 1 protein which is important in gene transcription induced by immune response. CTDP1 encodes the RNA polymerase II subunit A C-terminal domain phosphatase which interacts with the TFIIF transcription factor. Both NFATC1 and CTDP1 are expressed in the human temporal cortex, NFATC1 (58th percentile) more so than CTDP1 (37th percentile). NFATC1 is also the more promising candidate than CTDP1 because CTDP1 is primarily expressed in fetal astrocytes and nominally in other cell types, while NFATC1 is predominantly expressed in microglia and macrophages [84] (Supplementary Table 13). We were unable to test for the putative causal variant in these regions for these eQTL effects. However, these data suggest the top loci may influence ptau181 levels by affecting expression of these genes.
In summary, by increasing the sample size more than twofold we not only verified the results from our previous analyses, but also uncovered additional findings that can be used to inform future AD studies. We identified novel associations between genetic loci and CSF levels that may provide insight into the biological mechanisms that affect protein levels, influence AD risk, AAO, and disease progression. Our findings suggest CSF Aβ42 levels may be representative of the role of immune response on Aβ-associated pathology, and that this role may influence AAO. Although immune-related genes may be associated with ptau181, our results suggest that CSF ptau181 may reflect pathways related to neurogenesis and brain volume. Although we did not identify individual AD risk variants outside the APOE region, the PRS results indicate shared genetic architecture between AD risk and these CSF biomarkers. Larger studies using AD endophenotypes will likely provide even more information to help understand the biology underlying AD pathology.
References
Adams HH, Hibar DP, Chouraki V, Stein JL, Nyquist PA, Renteria ME, Trompet S, Arias-Vasquez A, Seshadri S, Desrivieres S et al (2016) Novel genetic loci underlying human intracranial volume identified through genome-wide association. Nat Neurosci. doi:10.1038/nn.4398
Andersson C, Blennow K, Almkvist O, Andreasen N, Engfeldt P, Johansson SE, Lindau M, Eriksdotter-Jonhagen M (2008) Increasing CSF phospho-tau levels during cognitive decline and progression to dementia. Neurobiol Aging 29:1466–1473. doi:10.1016/j.neurobiolaging.2007.03.027
Anttila V, Bulik-Sullivan B, Finucane HK, Bras J, Duncan L, Escott-Price V, Falcone G, Gormley P, Malik R, Patsopoulos N, et al (2016) Analysis of shared heritability in common disorders of the brain. bioRxiv doi:10.1101/048991
Baik SH, Cha MY, Hyun YM, Cho H, Hamza B, Kim DK, Han SH, Choi H, Kim KH, Moon M et al (2014) Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol Aging 35:1286–1292. doi:10.1016/j.neurobiolaging.2014.01.003
Balestrini A, Cosentino C, Errico A, Garner E, Costanzo V (2010) GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat Cell Biol 12:484–491. doi:10.1038/ncb2050
Benitez BA, Cooper B, Pastor P, Jin SC, Lorenzo E, Cervantes S, Cruchaga C (2013) TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging 34(1711):e1715–e1717. doi:10.1016/j.neurobiolaging.2012.12.018
Blennow K, Hampel H, Weiner M, Zetterberg H (2010) Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 6:131–144. doi:10.1038/nrneurol.2010.4
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S et al (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22:1790–1797. doi:10.1101/gr.137323.112
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, Duncan L, ReproGen C, Psychiatric Genomics C, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control C et al (2015) An atlas of genetic correlations across human diseases and traits. Nat Genet 47:1236–1241. doi:10.1038/ng.3406
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, Daly MJ, Price AL, Neale BM, Schizophrenia Working Group of the Psychiatric Genomics C (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47:291–295. doi:10.1038/ng.3211
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi:10.1186/s13742-015-0047-8
Cross-Disorder Group of the Psychiatric Genomics C (2013) Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 45:984–994. doi:10.1038/ng.2711
Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D et al (2013) GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 78:256–268. doi:10.1016/j.neuron.2013.02.026
Cruchaga C, Kauwe JS, Mayo K, Spiegel N, Bertelsen S, Nowotny P, Shah AR, Abraham R, Hollingworth P, Harold D et al (2010) SNPs associated with cerebrospinal fluid phospho-tau levels influence rate of decline in Alzheimer’s disease. PLoS Genet 6:e1001101. doi:10.1371/journal.pgen.1001101
Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, Bertelsen S, Hinrichs A, Alzheimer’s Disease Neuroimaging I, Fagan AM et al (2012) Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet 21:4558–4571. doi:10.1093/hmg/dds296
de Leon MJ, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Segal S, Clark C, Kerkman D, DeBernardis J, Li J et al (2004) MRI and CSF studies in the early diagnosis of Alzheimer’s disease. J Intern Med 256:205–223. doi:10.1111/j.1365-2796.2004.01381.x
De Meyer G, Shapiro F, Vanderstichele H, Vanmechelen E, Engelborghs S, De Deyn PP, Coart E, Hansson O, Minthon L, Zetterberg H et al (2010) Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol 67:949–956. doi:10.1001/archneurol.2010.179
Delaneau O, Marchini J, Consortium GP (2014) Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun. doi:10.1038/ncomms4934
Deming Y, Xia J, Cai Y, Lord J, Del-Aguila JL, Fernandez MV, Carrell D, Black K, Budde J, Ma S et al (2016) Genetic studies of plasma analytes identify novel potential biomarkers for several complex traits. Scientific Reports 6:18092. doi:10.1038/srep18092
Deming Y, Xia J, Cai Y, Lord J, Holmans P, Bertelsen S, Holtzman D, Morris JC, Bales K, Pickering EH et al (2016) A potential endophenotype for Alzheimer’s disease: cerebrospinal fluid clusterin. Neurobiol Aging 37(208):e201–e209. doi:10.1016/j.neurobiolaging.2015.09.009
Dudbridge F (2016) Polygenic epidemiology. Genet Epidemiol 40:268–272. doi:10.1002/gepi.21966
Escott-Price V, Shoai M, Pither R, Williams J, Hardy J (2017) Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging 49:214-e217–214-e211. doi:10.1016/j.neurobiolaging.2016.07.018
Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, Badarinarayan N, Gerad Perades, Morgan K, Gerad/Perades, consortia I, Consortia I et al (2015) Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 138:3673–3684. doi:10.1093/brain/awv268
Euesden J, Lewis CM, O’Reilly PF (2015) PRSice: polygenic Risk Score software. Bioinformatics 31:1466–1468. doi:10.1093/bioinformatics/btu848
Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM (2009) Decreased cerebrospinal fluid Aβ(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol 65:176–183. doi:10.1002/ana.21559
Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM (2007) Cerebrospinal fluid tau/β-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64:343–349. doi:10.1001/archneur.64.3.noc60123
Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA (2016) Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol Aging 37:19–25. doi:10.1016/j.neurobiolaging.2015.09.011
Farley K, Stolley JM, Zhao P, Cooley J, Remold-O’Donnell E (2012) A serpinB1 regulatory mechanism is essential for restricting neutrophil extracellular trap generation. J Immunol 189:4574–4581. doi:10.4049/jimmunol.1201167
Farrer LA, Cupples L, Haines JL et al (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein e genotype and alzheimer disease: a meta-analysis. JAMA 278:1349–1356. doi:10.1001/jama.1997.03550160069041
Fleming LM, Weisgraber KH, Strittmatter WJ, Troncoso JC, Johnson GV (1996) Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp Neurol 138:252–260. doi:10.1006/exnr.1996.0064
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiat 63:168–174. doi:10.1001/archpsyc.63.2.168
Grundberg E, Adoue V, Kwan T, Ge B, Duan QL, Lam KCL, Koka V, Kindmark A, Weiss ST, Tantisira K et al (2011) Global analysis of the impact of environmental perturbation on cis-regulation of gene expression. PLoS Genet. doi:10.1371/journal.pgen.1001279
GTEx Consortium (2015) Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348:648–660. doi:10.1126/science.1262110
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Lupton MK et al (2013) TREM2 variants in Alzheimer’s disease. New Engl J Med 368:117–127. doi:10.1056/NEJMoa1211851
Guo H, Fortune MD, Burren OS, Schofield E, Todd JA, Wallace C (2015) Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum Mol Genet 24:3305–3313. doi:10.1093/hmg/ddv077
Han MR, Schellenberg GD, Wang LS, Alzheimer’s Disease Neuroimaging I (2010) Genome-wide association reveals genetic effects on human Aβ42 and tau protein levels in cerebrospinal fluids: a case control study. BMC Neurol 10:90. doi:10.1186/1471-2377-10-90
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A et al (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease (vol 41, pg 1088, 2009). Nat Genet 41:1156. doi:10.1038/ng1009-1156d
Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16:358–372. doi:10.1038/nrn3880
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43:429–435. doi:10.1038/ng.803
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44:955. doi:10.1038/ng.2354
Hulette CM, Welsh-Bohmer KA, Murray MG, Saunders AM, Mash DC, McIntyre LM (1998) Neuropathological and neuropsychological changes in “normal” aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol 57:1168–1174
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ et al (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. New Engl J Med 368:107–116. doi:10.1056/NEJMoa1211103
Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, Sasaki H, Abe K, Iwatsubo T, Kosaka T et al (1998) Longitudinal study of cerebrospinal fluid levels of tau, A beta1-40, and A beta1-42(43) in Alzheimer’s disease: a study in Japan. Ann Neurol 44:17–26. doi:10.1002/ana.410440108
Kauwe JS, Cruchaga C, Bertelsen S, Mayo K, Latu W, Nowotny P, Hinrichs AL, Fagan AM, Holtzman DM, Alzheimer’s Disease Neuroimaging I et al (2010) Validating predicted biological effects of Alzheimer’s disease associated SNPs using CSF biomarker levels. J Alzheimers Dis 21:833–842. doi:10.3233/JAD-2010-091711
Kauwe JS, Cruchaga C, Mayo K, Fenoglio C, Bertelsen S, Nowotny P, Galimberti D, Scarpini E, Morris JC, Fagan AM et al (2008) Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci USA 105:8050–8054. doi:10.1073/pnas.0801227105
Kim S, Swaminathan S, Shen L, Risacher SL, Nho K, Foroud T, Shaw LM, Trojanowski JQ, Potkin SG, Huentelman MJ et al (2011) Genome-wide association study of CSF biomarkers Aβ1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology 76:69–79. doi:10.1212/WNL.0b013e318204a397
Kyrousi C, Arbi M, Pilz GA, Pefani DE, Lalioti ME, Ninkovic J, Gotz M, Lygerou Z, Taraviras S (2015) Mcidas and GemC1 are key regulators for the generation of multiciliated ependymal cells in the adult neurogenic niche. Development 142:3661–3674. doi:10.1242/dev.126342
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45:1452–1458. doi:10.1038/ng.2802
Leoni V, Solomon A, Kivipelto M (2010) Links between ApoE, brain cholesterol metabolism, tau and amyloid β-peptide in patients with cognitive impairment. Biochem Soc Trans 38:1021–1025. doi:10.1042/BST0381021
Lepretre C, Tchakarska G, Blibech H, Lebon C, Torriglia A (2013) Apoptosis-inducing factor (AIF) and leukocyte elastase inhibitor/L-DNase II (LEI/LDNaseII), can interact to conduct caspase-independent cell death. Apoptosis 18:1048–1059. doi:10.1007/s10495-013-0862-2
Li QS, Parrado AR, Samtani MN, Narayan VA, Alzheimer’s Disease Neuroimaging I (2015) Variations in the FRA10AC1 Fragile Site and 15q21 Are Associated with Cerebrospinal Fluid Aβ1-42 Level. PLoS ONE 10:e0134000. doi:10.1371/journal.pone.0134000
Liraz O, Boehm-Cagan A, Michaelson DM (2013) ApoE4 induces Aβ42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol Neurodegener 8:16. doi:10.1186/1750-1326-8-16
Louwersheimer E, Wolfsgruber S, Espinosa A, Lacour A, Heilmann-Heimbach S, Alegret M, Hernandez I, Rosende-Roca M, Tarraga L, Boada M et al (2016) Alzheimer’s disease risk variants modulate endophenotypes in mild cognitive impairment. Alzheimers Dement 12:872–881. doi:10.1016/j.jalz.2016.01.006
Martiskainen H, Helisalmi S, Viswanathan J, Kurki M, Hall A, Herukka SK, Sarajarvi T, Natunen T, Kurkinen KM, Huovinen J et al (2015) Effects of Alzheimer’s disease-associated risk loci on cerebrospinal fluid biomarkers and disease progression: a polygenic risk score approach. J Alzheimers Dis 43:565–573. doi:10.3233/JAD-140777
Morris JC, Price JL (2001) Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci 17:101–118
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436–441. doi:10.1038/ng.801
O’Bryant SE, Lacritz LH, Hall J, Waring SC, Chan W, Khodr ZG, Massman PJ, Hobson V, Cullum CM (2010) Validation of the new interpretive guidelines for the clinical dementia rating scale sum of boxes score in the national Alzheimer’s coordinating center database. Arch Neurol 67:746–749. doi:10.1001/archneurol.2010.115
O’Bryant SE, Waring SC, Cullum CM, Hall J, Lacritz L, Massman PJ, Lupo PJ, Reisch JS, Doody R, Texas Alzheimer’s Research C (2008) Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer’s research consortium study. Arch Neurol 65:1091–1095. doi:10.1001/archneur.65.8.1091
Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, Holtta M, Rosen C, Olsson C, Strobel G et al (2016) CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 15:673–684. doi:10.1016/S1474-4422(16)00070-3
Piccio L, Deming Y, Del-Aguila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C (2016) Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol 131:925–933. doi:10.1007/s00401-016-1533-5
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909. doi:10.1038/ng1847
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26:2336–2337. doi:10.1093/bioinformatics/btq419
Ramirez A, van der Flier WM, Herold C, Ramonet D, Heilmann S, Lewczuk P, Popp J, Lacour A, Drichel D, Louwersheimer E et al (2014) SUCLG2 identified as both a determinator of CSF Aβ1-42 levels and an attenuator of cognitive decline in Alzheimer’s disease. Hum Mol Genet 23:6644–6658. doi:10.1093/hmg/ddu372
Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, Mayeux R, Farrer LA, Pericak-Vance MA, Schellenberg GD et al (2016) Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging 41(200):e213–e220. doi:10.1016/j.neurobiolaging.2016.02.024
Sabuncu MR, Buckner RL, Smoller JW, Lee PH, Fischl B, Sperling RA, Alzheimer’s Disease Neuroimaging I (2012) The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb Cortex 22:2653–2661. doi:10.1093/cercor/bhr348
Sampson JN, Wheeler WA, Yeager M, Panagiotou O, Wang Z, Berndt SI, Lan Q, Abnet CC, Amundadottir LT, Figueroa JD et al (2015) Analysis of heritability and shared heritability based on genome-wide association studies for thirteen cancer types. J Natl Cancer Inst 107:djv279. doi:10.1093/jnci/djv279
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595–608. doi:10.15252/emmm.201606210
Shabalin AA (2012) Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28:1353–1358. doi:10.1093/bioinformatics/bts163
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P et al (2009) Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65:403–413. doi:10.1002/ana.21610
Skol AD, Scott LJ, Abecasis GR, Boehnke M (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38:209–213. doi:10.1038/ng1706
Sleegers K, Bettens K, De Roeck A, Van Cauwenberghe C, Cuyvers E, Verheijen J, Struyfs H, Van Dongen J, Vermeulen S, Engelborghs S et al (2015) A 22-single nucleotide polymorphism Alzheimer’s disease risk score correlates with family history, onset age, and cerebrospinal fluid Aβ42. Alzheimers Dement 11:1452–1460. doi:10.1016/j.jalz.2015.02.013
Sunderland T, Linker G, Mirza N, Putnam KT, Friedman DL, Kimmel LH, Bergeson J, Manetti GJ, Zimmermann M, Tang B et al (2003) Decreased β-amyloid1-42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA 289:2094–2103. doi:10.1001/jama.289.16.2094
Trabzuni D, Ryten M, Walker R, Smith C, Imran S, Ramasamy A, Weale ME, Hardy J (2011) Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J Neurochem 119:275–282. doi:10.1111/j.1471-4159.2011.07432.x
Turner SD (2014) qqman: an R package for visualizing GWAS results using QQ and manhattan plots. bioRxiv doi:10.1101/005165
Van Eldik LJ, Carrillo MC, Cole PE, Feuerbach D, Greenberg BD, Hendrix JA, Kennedy M, Kozauer N, Margolin RA, Molinuevo JL et al (2016) The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv 2:99–109. doi:10.1016/j.trci.2016.05.001
Visscher PM, Hemani G, Vinkhuyzen AA, Chen GB, Lee SH, Wray NR, Goddard ME, Yang J (2014) Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet 10:e1004269. doi:10.1371/journal.pgen.1004269
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. doi:10.1093/nar/gkq603
Ward LD, Kellis M (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 40:D930–D934. doi:10.1093/nar/gkr917
Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE et al (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 45:1238-U1195. doi:10.1038/ng.2756
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191. doi:10.1093/bioinformatics/btq340
Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AAE, Lee SH, Robinson MR, Perry JRB, Nolte IM, van Vliet-Ostaptchouk JV et al (2015) Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet 47:1114. doi:10.1038/ng.3390
Yang JA, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88:76–82. doi:10.1016/j.ajhg.2010.11.011
Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S et al (2015) Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med 21:880–886. doi:10.1038/nm.3913
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N et al (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. doi:10.1523/JNEUROSCI.1860-14.2014
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM et al (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48:481–487. doi:10.1038/ng.3538
Acknowledgements
We thank all the participants and their families, as well as the many institutions and their staff that provided support for all studies involved in this collaboration. We also thank the Alzheimer Disease Genetic Consortium (ADGC) for genotyping and providing data for the BIOCARD, UPENN, HB, SWEDEN, and MAYO cohorts. We thank the Cardiogenics (European Project reference LSHM-CT-2006-037593) project for providing data for the eQTL analysis. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Author contributions
YD analyzed data and wrote the manuscript. ZL verified imputation with genotyped data. MK performed colocalization tests of GWAS and expression data. OH contributed conceptually to the analysis. KB performed genotyping for imputation verification. JLD-A performed disease progression analysis. DC, YC, MVF, JB, SM, BS, BH, KH, and SB prepared genetic data: performed imputation, cleaning, and calculated principal components. AMF, DMH, JCM, SK, AJS., PLDJ., MA, AM, RO, MR, RCP, KB, HZ, LM, VMVD, VM-YL, LMS, JQT, JLH, RM, MAP-V, LAF, ERP, GL, AFDN, ADNI, ADGC, JK and AG provided data. CC prepared the manuscript and supervised the project. All authors read and approved the manuscript.
Author information
Authors and Affiliations
Author notes
J. S. K. Kauwe, A. M. Goate and C. Cruchaga equally contributed to this work.
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Consortia
Corresponding author
Ethics declarations
Conflict of interest
KB and HZ are co-founders of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg, Sweden. A.M.G. serves on the SAB for Denali Therapeutics and is the inventor on a patent for MAPT mutations.
Funding
This work was supported by grants from the National Institutes of Health (R01AG044546, P01AG003991, RF1AG053303, R01AG035083, and R01NS085419), and the Alzheimer’s Association (NIRG-11-200110). This research was conducted while C.C. was a recipient of a New Investigator Award in Alzheimer’s disease from the American Federation for Aging Research. C.C. is a recipient of a BrightFocus Foundation Alzheimer’s Disease Research Grant (A2013359S). The recruitment and clinical characterization of research participants at Washington University were supported by NIH P50 AG05681, P01 AG03991, and P01 AG026276. ADGC is supported by grants from the NIH (#U01AG032984) and GERAD from the Wellcome Trust (GR082604MA) and the Medical Research Council (G0300429); additional support was provided by NCRAD (U24 AG21886), NACC (U01 AG016976), NIAGADS (U24-AG041689) and UPENN (P30 AG010124). Support for A.S. was provided by U01 AG024904, R01 AG19771, and P30 AG10133. P.D.J. received support from R01 AG048015. UW ADRC received funding from AG05136. S.K. received support from NIA R03AG050856, Alzheimer’s Association, Michael J. Fox Foundation, and ARUK Biomarkers Across Neurodegenerative Diseases (BAND). M.R. received support from the German Federal Ministry of Education and Research (BMBF) National Genome Research Network (NGFN) Grant No. 01GS08125 and through the Helmholtz Alliance for Mental Health in an Aging Society (HELMA) Grant No. Ha-15. This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders and the Departments of Neurology and Psychiatry at Washington University School of Medicine.
Additional information
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Deming, Y., Li, Z., Kapoor, M. et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol 133, 839–856 (2017). https://doi.org/10.1007/s00401-017-1685-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1685-y