
Abstract
Evidence of misfolded wild-type superoxide dismutase 1 (SOD1) has been detected in spinal cords of sporadic ALS (sALS) patients, suggesting an etiological relationship to SOD1-associated familial ALS (fALS). Given that there are currently a number of promising therapies under development that target SOD1, it is of critical importance to better understand the role of misfolded SOD1 in sALS. We previously demonstrated the permissiveness of the G85R-SOD1:YFP mouse model for MND induction following injection with tissue homogenates from paralyzed transgenic mice expressing SOD1 mutations. This prompted us to examine whether WT SOD1 can self-propagate misfolding of the G85R-SOD1:YFP protein akin to what has been observed with mutant SOD1. Using the G85R-SOD1:YFP mice, we demonstrate that misfolded conformers of recombinant WT SOD1, produced in vitro, induce MND with a distinct inclusion pathology. Furthermore, the distinct pathology remains upon successive passages in the G85R-SOD1:YFP mice, strongly supporting the notion for conformation-dependent templated propagation and SOD1 strains. To determine the presence of a similar misfolded WT SOD1 conformer in sALS tissue, we screened homogenates from patients diagnosed with sALS, fALS, and non-ALS disease in an organotypic spinal cord slice culture assay. Slice cultures from G85R-SOD1:YFP mice exposed to spinal homogenates from patients diagnosed with ALS caused by the A4V mutation in SOD1 developed robust inclusion pathology, whereas spinal homogenates from more than 30 sALS cases and various controls failed. These findings suggest that mutant SOD1 has prion-like attributes that do not extend to SOD1 in sALS tissues.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) has both a sporadic (sALS) and familial (fALS) etiology and causes a progressive paralysis due to the degeneration of both upper and lower motor neurons. With no known cure and very few therapeutic options, this disease ultimately leads to difficulties swallowing and breathing, and has a life expectancy after diagnosis from about 2–5 years. Between 5 and 10 % of all ALS, cases are familial, and of these, around 10–20 % are due to mutations in the superoxide dismutase 1 (SOD1) gene, of which there are now more than 170 mutations identified. These mutations cause a gain of toxic function via a mutation-induced conformational change that causes this normally soluble, extremely stable protein, to form insoluble aggregates that accumulate within the spinal cord and brain.
Although all patients affected by ALS succumb to paralysis, there is a large heterogeneity in the clinical course of the disease, indicating a number of variables affecting disease pathogenesis. This heterogeneity in SOD1-fALS produces diverse clinical symptoms that vary dramatically in the location of symptom onset (e.g., arm(s), leg(s), or bulbar), the degree of upper vs. lower motor neuron involvement, the age of onset (typically from 25 to 70 years of age), and the survival time (1–20+ years) [32]. In a study in which a number of phenotypic and biochemical properties were examined among many of the SOD1 mutations, the only correlation observed was between the mean disease duration of a particular SOD1 mutant and its aggregation propensity [32]. A similar type of sequence specified heterogeneity is also observed in prion diseases, where rare polymorphisms in the prion gene, prnp, dictate whether patients develop Creutzfeldt–Jakob disease (CJD), fatal familial insomnia (FFI), or Gerstmann–Straussler–Scheinker disease (GSS) [21, 26]. In these diseases, the mutation-specific phenotypes are believed to arise due to prion strains [6, 15], in which tertiary conformations of misfolded prion protein (PrP) are said to “encipher” the information that produces the diverse phenotypes [29]. The concept of protein strains reveals the impact that distinct protein conformers can have on the pathogenesis of misfolded protein disorders. It is, therefore, possible that the wide variety of SOD1 conformations brought about by the more than 170 point mutations described for SOD1 may impact the course of disease in fALS patients and account for some of the heterogeneity observed among fALS-SOD1 cases.
Interestingly, the symptoms associated with fALS are clinically indistinguishable from sALS patients, indicating the potential for a common pathogenic mechanism. Several recent studies have emerged, suggesting that WT SOD1 can undergo conformational changes, producing a toxic protein that can propagate the disease-causing conformation between cells, and when expressed in mice, can produce a phenotype characteristic of ALS. Oxidized and metal-free WT SOD1 have been demonstrated to acquire aggregation properties similar to mutant SOD1, and in addition, once administered to cells, these aggregates have displayed toxicity [12, 16]. In addition, in post-mortem tissue from a number of non-SOD1-linked sALS patients, the conformational SOD1 antibody C4F6, which preferentially binds to mutant over WT SOD1, revealed neuronal immunoreactivity [8]; however, more recent studies have raised questions as to the usefulness of this antibody to detect misfolded WT SOD1 due to the antibody’s complex epitope [4, 9]. Other conformational-specific SOD1 antibodies have been reported to detect immunoreactivity indicative of misfolded protein in all [17, 22, 30] or none of the cases they examined [25]. Despite these contradictory reports, WT SOD1 has been implicated as potentially being able to acquire the toxic characteristics of mutant SOD1 in studies in which mice overexpressing WT SOD1 were mated to mice expressing mutant SOD1 below the threshold level required for disease. Bigenic offspring of these crosses develops ALS symptoms, whereas mice expressing only the mutant protein do not [14, 24, 31]. Finally, studies by Graffmo et al. have shown that mating the high expressing Gur1 WT-SOD1 mice to homozygosity will produce ALS-like phenotypes [23]. Collectively, these studies reveal the possibility for WT SOD1 to adopt a toxic conformation similar to that observed for mutant SOD1 and imply the potential for WT SOD1 to play a pathogenic role in sALS.
We have previously demonstrated that injecting spinal cord homogenate prepared from paralyzed mutant SOD1 transgenic mice can accelerate motor neuron disease (MND) in transgenic mice expressing the G85R variant of SOD1 fused to YFP (G85R-SOD1:YFP) [2]. Accelerated disease progression was also observed in mice expressing untagged G85R SOD1 when young animals were similarly injected with spinal homogenates from paralyzed mutant SOD1 animals [7]. Here, we have advanced upon these studies in several ways. First, we identify two strains of mice expressing truncation mutants of SOD1 in which the disease course can be dramatically accelerated by injecting spinal homogenates from paralyzed mice into the spinal cords of young transgenic mice. Second, we demonstrate that aggregates of purified WT SOD1 induce MND in the G85R-SOD1:YFP mouse model with a distinct inclusion pathology. The distinct pathology is preserved by serial passage to naïve G85R-SOD1:YFP mice, suggesting that distinct strains of misfolded SOD1 conformers may produce distinct inclusion pathology. We have also developed an organotypic slice culture system prepared from the spinal cords of G85R-SOD1:YFP mice that support the propagation of misfolded SOD1 strains. Utilizing this ex vivo assay, we screened >30 post-mortem spinal cord homogenates obtained from patients clinically diagnosed with sALS, along with 5 cases from fALS patients (2 SOD1 and 3 non-SOD1), and 10 non-ALS diseased patients. Of the more than 50 different tissue preparations that were used, the induced misfolding of G85R-SOD1:YFP only occurred in cultures exposed to spinal homogenates from mutant SOD1-linked fALS cases. We conclude that although WT SOD1 can be misfolded in vitro into a conformation that can induce the propagation of inclusion pathology, any misfolded WT SOD1 conformers present in post-mortem spinal cord tissues from human sporadic ALS lack the same prion-like potential seen with homogenates from SOD1 mutant fALS cases.
Methods
Mice
All studies involving mice were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Florida in accordance with the NIH guidelines. All animals were housed one to five to a cage and maintained on ad libitum food and water with a 14-h light and 10-h dark cycle. All the strains of transgenic mice used in this study have previously been described: G85R-SOD1:YFP mice [37], L126Z hSOD1 Line45 mice [38], the V103Z hSOD1 Line D14 mice [39], the α-synuclein M83 mice [19], and the tau JNPL3 mice [27]. The G85R-SOD1:YFP mice are maintained on the FVB/NJ background, the JNPL3 on Swiss Webster, and the L45, D14, and M83 on a hybrid background of C57Bl/6 J and C3H/HeJ. For identification of genotype, DNA was extracted from mouse tail biopsies and analyzed by PCR as previously described [39].
Subjects
Tissues from human subjects were collected with patient consent and handled under protocols approved by the University of Florida’s, Johns Hopkins, and Emory University’s Institutional Review Boards. All samples were coded and de-identified. The total number of samples obtained and their clinical diagnosis is summarized in Table 2.
Preparation of homogenates for inoculum
Homogenates to produce inoculum were prepared as previously described [2]. Briefly, spinal cords from the lines of SOD1 transgenic mice as indicated in the text were homogenized in PBS to produce a 10 % homogenate (w/v), containing 1:100 v/v protease inhibitor cocktail (Sigma, St. Louis, MO, USA) by sonication four times for 20 s each. Homogenates were then clarified by a low-speed spin at ~800×g for 10 min and the supernatants were aliquoted and placed at −80 °C.
Recombinant SOD1 purification and fibrillization
Recombinant hSOD1 proteins were expressed and purified as previously described [36]. Recombinant SOD1 was fibrillized using 50 μM of protein in 20-mM potassium phosphate, pH 7.2, with the addition of 10 mM TCEP‚ and for those samples used for screening fibril formation 4-μM thioflavin T was also added. Two hundred microliter of the protein solutions were incubated in a 96-well plate with the addition of a Teflon ball (1/8-in diameter) at 37 °C with constant agitation in a Synergy HT plate reader (BIO-TEK, Winooski, VT). Fluorescence measurements were recorded every 15 min using a λ ex = 440/30 filter to excite and a λ em = 485/20 filter to detect emission using the Gen5 software (v1.10.8).
Filter trap assay
Cellulose acetate membrane (0.22 μm; Sterlitech Corp., Kent, WA, USA) was pre-wet with 0.1 % SDS in PBS and sealed within a dot blot apparatus (Bio-Rad, Hercules, CA, USA). Following three rinses with 0.1 % SDS/PBS, 1 μg of recombinant protein in PBS was added to the membrane and removed by vacuum filtration. The wells were washed with 0.1 % SDS/PBS three more times, the membrane was removed from the apparatus, rinsed in PBS-T for 5 min, and then blocked in 5 % milk/PBS-T for 30 min. The membrane was then immunoblotted using the hSOD1 antibody at 1:2500 and imaged using the Pxi blot imaging system (Syngene, Frederick, MD, USA).
Electron microscopy
To ensure fibril formation, following in vitro fibrillization, SOD1-containing samples were absorbed to 300-mesh carbon-coated copper grids, washed, and stained with 1 % uranyl acetate. Using a Hitachi H7600 transmission electron microscope (Hitachi, Tokyo, Japan), images were captured at 100,000× magnification.
Animal inoculations
Spinal injections in neonatal mouse pups were performed as previously described [2]. Briefly, P0 neonatal pups were covered in aluminum foil and surrounded in ice until all movements stop and the skin color changes from pink to purple (5–10 min). Cryoanesthetized neonates were injected using a 10-μl syringe equipped with a 1-inch, 30-gauge needle with 30° bevel. The needle was inserted through the skin at midline, around 5 mm from the base of the tail. The needle was inserted into the vertebral column and 1 μl of the inoculum was slowly injected. After injections, pups were allowed to completely recover on a warming blanket and then returned to the home cage. After the procedure, mice were monitored to ensure full mobility and no signs of impairment.
Organotypic spinal cord slice cultures
G85R-SOD1:YFP mouse pups at 7 days old were euthanized by CO2 asphyxiation followed by immediate decapitation. The bodies were immediately placed in Hank’s Balanced Salt Solution (HBSS) supplemented with 6.4-mg/ml glucose. Spinal cords were dissected, cut to 350 μm sections using a McIlwain Tissue Chopper, then five spinal cord sections were placed onto the surface of each 0.4-μm Millipore Millicell-CM membrane insert. Inserts were placed into the wells of 6-well plates containing 1 mL of incubation medium (50 % (vol/vol) minimal essential medium—25 mM Hepes/25 % (vol/vol) heat-inactivated horse serum/25 % (vol/vol) HBSS) supplemented with 6.4-mg/ml glucose and 2-mM glutamine. Cultures were incubated at 37 °C in a 5 % CO2/95 % air humidified environment. Media were changed twice weekly. To test seeding ability, 1 μl of SOD1-containing preparations were added directly to the top of each spinal cord section following 1 week of incubation. To assess the titer of the seeding component in the spinal homogenates prepared from disease mice, we treated the slice cultures with twofold serial dilutions of the homogenates. The seeding dose (SD50) was calculated according to the method of Reed and Muench [35]. To image spinal cord sections, incubation medium was removed and 4 % paraformaldehyde was added and incubated at room temperature for 4 h. The sections were then rinsed in PBS put directly onto slides, and coverslipped in Vectastain mounting media-containing DAPI (Vector, Bulingame, CA, USA).
Tissue collection
Mice were anesthetized with isoflurane and perfused transcardially with 20 ml of PBS followed by 20 ml of 4 % paraformaldehyde. The spinal cord and brain were immediately removed and placed in 4 % paraformaldehyde for 24–48 h at 4 °C prior to paraffin processing.
Immunohistochemistry
Seven microliter paraffin sections were deparaffinized and incubated in 95 % formic acid for 10 min. Following PBS washes, sections were blocked of endogenous peroxidases by immersion in 0.3 % H2O2 in PBS for 20 min. Blocking of non-specific staining with normal goat serum in PBS-T was followed by overnight incubation at 4 °C with a 1:500 dilution of the C4F6 antibody (Medimabs, Montreal, Quebec, Canada) in PBS-T with 3 % normal goat serum. The sections were then incubated with a biotinylated secondary anti-mouse antibody (Vector Laboratories, Burlingame, CA) diluted 1:500 in PBS-T with 3 % normal goat serum followed by incubation with the ABC-horseradish peroxidase staining kit (Vector Laboratories). Sections were developed using the DAB staining kit (KPL, Gaithersburg, MD) and counterstained with hematoxylin. Images were taken using an Olympus BX60 microscope.
Fluorescence microscopy
Tissue to be examined for direct fluorescence tissue was impregnated with 30 % sucrose in PBS, mounted in OCT media (Sakura, The Netherlands), and sectioned to 30 μm using a cryostat, placed in a dish-containing anti-freeze solution (100-mM sodium acetate, 250-mM polyvinylpyrrolidone, and 40 % ethylene glycol) at pH 6.5, and stored at 4 °C. Sections were then mounted onto slides, air-dried overnight, and coverslipped in mounting media-containing DAPI (Vector, Bulingame, CA). Fluorescence images were either visualized on an epifluorescence Olympus BX60 microscope or imaged by confocal microscopy on a Nikon A1RMPsi-STORM4.0.
Quantification of inclusion pathology
Fluorescence images (n = 3) from three animals per injection cohort were taken and used for quantification of inclusion pathology. To reveal the difference in pathologies, the images were first opened in ImageJ (v1.8.0_73) and thresholded. Using the ‘analyze particle’ function, each of the inclusions was assigned a circularity value based on a built-in algorithm (4π × [area]/[perimeter]2) ranging from 0 (elongated polygon) to 1 (perfect circle).
Statistical analysis
Differences in the circularity of G85R-SOD1:YFP inclusions were analyzed by two-way ANOVA and Sidak’s multiple comparisons test. All statistical analyses were performed using Prism 7.0 (GraphPad Software) with p < 0.05 considered statistically significant. The significance values in figures were indicated as follows: * (p ≤ 0.05), ** (p ≤ 0.01), and *** (p ≤ 0.001).
Results
Recombinant WT SOD1 fibrils induce MND in the G85R:SOD1-YFP mouse model
We previously demonstrated the ability to induce MND through the exogenous administration of spinal homogenates from paralyzed SOD1 mice into the spinal cords of G85R-SOD1:YFP mice at postnatal day P0 [2]. This study revealed that some factor(s) were contained within these preparations that could induce the misfolding and aggregation of the G85R-SOD1:YFP protein. Although it is tempting to point to SOD1 as the factor, there may be a number of other molecules that could impact disease induction. Utilizing this injection paradigm, we injected a number of control inoculum to better characterize the inducing component contained within these homogenates. In addition to increasing the number of mice injected with PBS and homogenates from non-transgenic mice (first described in [2]), we also injected a cohort of G85R-SOD1:YFP mice with homogenate prepared from an asymptomatic, heterozygous G85R-SOD1:YFP mouse. All of these cohorts were aged to ~20 months, displaying no clinical signs of MND or inclusion pathology upon examination of their spinal cords (Table 1). In addition, as disease controls, we prepared spinal cord homogenates from two transgenic lines of mice that develop a similar progressive paralytic phenotype as observed in SOD1 transgenic mouse lines. These mouse lines included the transgenic M83 line, which express human alpha-synuclein containing the A53T mutation [19], and the JNPL3 line, which expresses human tau containing the P301L mutation [27]. Homogenates were prepared from a paralyzed M83 mouse and a paralyzed JNPL3 mouse and injected into the spinal cords of P0 G85R-SOD1:YFP mice. The mice are currently 13–15 months old and have displayed no signs of MND (Table 1). One mouse from the M83 injected cohort (12.5 months) and two mice from the JNPL3 injected cohort (4.1 and 7.2 months) had to be euthanized for non-MND-related reasons, and upon examination of their spinal cords, no G85R-SOD1:YFP inclusions were observed. These control injections support the idea that the misfolded SOD1 protein in the spinal homogenates of paralyzed mutant SOD1 mice is the main factor that induces SOD1 pathology and MND when injected into the spinal cord of P0 G85R-SOD1:YFP mice.
To further investigate the seeding component, we tested whether recombinant SOD1 could be aggregated in vitro into a conformation that could similarly induce MND in this model. It has been commonly demonstrated that when shaken and incubated at 37 °C, recombinant SOD1 forms thioflavin positive aggregates that are fibrillar structures upon EM examination [5, 12]. We, therefore, fibrillized recombinant WT SOD1 (recWT) in a thioflavin T assay and ensured its fibrillar structure by both EM and filter trap assays (Fig. 1a–c). A 50-μM recWT sample, not containing thiovlavin T, was run in parallel and used as inoculum in our G85R-SOD1:YFP mouse model for SOD1 seeding. Of the six P0 G85R-SOD1:YFP mice we injected within the spinal cord with these recWT fibrils, five developed MND, with an average time to end-stage of 10.1 ± 1.3 months (Fig. 1d; Table 2). When examining the spinal cords from these mice, we observed a unique pathology that appeared as mainly fibrillar deposits in the neuropil, and within a subset of cells, including motor neurons, a distinctive skein-like pathology was observed (Fig. 1e). As negative controls, we also injected mice with unfibrillized recWT SOD1, or with PBS containing the reducing agent (tris(2-carboxyethyl)phosphine (TCEP)) used to aid in the fibrillation of WT SOD1. Of the six animals injected with unfibrillized recWT SOD1, three were harvested at 6.2–7.8 months of age due to unrelated health issues and lacked evidence of G85R-SOD1:YFP inclusion pathology, while the other three were euthanized at the experimental end-point of 16 months and displayed no symptoms or inclusion pathology (Fig. 1d; Table 1). One of the four mice injected with PBS and TCEP developed paralysis at 10.5 months of age, and upon examination, the spinal cord displayed sparse fibrillar G85R-SOD1:YFP inclusions. The other three animals within this cohort were aged to 16.8 months, never displayed symptoms, and contained no inclusion pathology within their spinal cords. These findings demonstrate the effectiveness of recombinant misfolded SOD1 to act as a seed and induce MND and inclusion pathology in the G85R-SOD1:YFP mouse model. Furthermore, it supports the idea that misfolded forms of WT SOD1 can induce inclusion formation and cause a pathogenic cascade.

In vitro fibrillized recombinant WT SOD1 induces MND in G85R-SOD1:YFP injected mice. a Thioflavin T fluorescence of recombinant WT SOD1 while shaking at 37°C. n = 2; mean ± SEM. b Representative image from the EM analysis of WT SOD1 fibrils (n = 3). c Representative image from filter trap assay revealing HMW species formed in recombinant WT SOD1 samples (n = 3). d Kaplan–Meier survival curves show MND disease induction in G85R-SOD1:YFP mice injected within the spinal cord at P0 with WT SOD1 fibrils and one animal that developed disease following a control injection. e Representative image of inclusion pathology induced in the spinal cord of a diseased G85R-SOD1:YFP mouse that had been injected with WT SOD1 fibrils (n = 5 mice, 16 sections per mouse)
Serial passage of misfolded SOD1 conformers reveals distinct strain characteristics
We have previously demonstrated that the induction of MND and G85R-SOD1:YFP misfolding can be serially passaged, similar to that observed for prion strains [2]. For clarity purposes, diseased G85R-SOD1:YFP mice will be referred to by the initial inoculum injected and the number of passages. For example, G85R-SOD1:YFP mice that develop disease following injection with G93A homogenate will be referred to as G85R-YFPG93A-P1, whereas passaging of homogenate from these diseased G85R-SOD1:YFP mice back into naïve G85R-SOD1:YFP mice will be referred to as G85R-YFPG93A-P2. To better understand the effects of serial passaging on the disease course, we passaged homogenates from diseased G85R-SOD1:YFP mice that had been induced with homogenate from a variety of SOD1 transgenic mouse lines (G93A, G37R, and L126Z). In every case examined, the second passage of homogenates resulted in an earlier onset of paralysis and an increase of incidence to almost 100 % (Table 2). Similarly, when we passaged homogenate from paralyzed G85R-SOD1:YFP mice that had been injected with recWT fibrils (G85R-YFPrecWT-P1) into naïve mice (G85R-YFPrecWT-P2), 14 of the 14 mice injected at P0 developed paralysis at 3.9 ± 0.1 months, which was a significant acceleration over the first passage of WT SOD1 fibrils (9.1 ± 1.3 months) (Table 2). These decreases in incubation period and increases in incidence observed upon second passage of these homogenates are similar to prion host adaptation and are thought to occur by the elimination of amino-acid sequence differences between the initial inducing prion conformer and the host [1, 10]. An additional explanation for these findings could be a difference in the transmission titer of the injected homogenates. To address this variable, we developed an organotypic spinal cord slice culture system prepared from the spinal cords of G85R-SOD1:YFP mice to detect the seeded misfolding of G85R-SOD1:YFP in a system that is less expensive and time-consuming as an end-point titration bioassay performed in animals. We found that the G85R-YFPG93A-P1 and G85R-YFPG37R-P1 homogenates, which resulted in significantly different incubation periods when injected into G85R-SOD1:YFP mice, had relatively similar seeding doses (SD50) of 101.7/μl and 101.6/μl, respectively (Table 2). In regard to the second passage homogenates, the G85R-YFPG37R-P2 homogenate, which was associated with a significantly slower incubation period than G85R-YFPG93A-P2, had a slightly higher SD50 when compared with the G85R-YFPG93A-P2 homogenate (102.4/μl versus 102.0/μl) (Table 2). This finding suggests that the cause for the disparate incubation periods in the mice was not due to differing levels of transmissible seeds in these tissues.
One of the properties used to define a particular prion strain is a distinct neuropathology that is consistent upon repeated passages in naïve hosts. While analyzing tissue from the first passage of homogenates into G85R-SOD1:YFP mice, we began to observe differences in the SOD1:YFP inclusions dependent on the inoculum injected. The most striking differences were found between those animals injected with homogenate from paralyzed animals versus those injected with recWT SOD1 fibrils. G85R-SOD1G93A-P1 mice displayed punctate G85R-SOD1:YFP inclusions throughout the spinal cord that appeared both within the neuropil and within cell bodies, with occasional fibrillar inclusions detected in the neuropil (Fig. 2a). In comparison, the paralyzed G85R-YFPrecWT-P1 mice displayed mainly fibrillar inclusions within the neuropil and also revealed distinctive skein-like inclusions within cell bodies (Fig. 2a). Surprisingly, passaging homogenates containing these distinct G85R-SOD1:YFP inclusions produced identical inclusions in the spinal cords of naïve injected G85R-SOD1:YFP mice (Fig. 2a). To quantify these distinct pathologies, we analyzed the circularity of G85R-SOD1:YFP inclusions using algorithms contained in the ImageJ to define the thresholded inclusions from 0.0 (elongated polygon) to 1.0 (perfect circle) (Supplemental Fig. 1). Using this method, we observed a greatly skewed histogram towards 1.0 for the inclusions produced in G85R-SOD1:YFPG93A-P2 mice, indicating that the majority of inclusions were mainly circular (Fig. 2b, c). In comparison, the inclusions from G85R-SOD1:YFPrecWT-P2 mice displayed a more evenly distributed histogram, thereby supporting our observations that the inclusions from these mice were more fibrillar than the round, punctate inclusions observed following the G93A passages (Fig. 2b). Second-passage G85R-SOD1:YFPG37R-P2 and G85R-SOD1:YFPL126Z-P2 mice displayed more of mixture of fibrillary and punctate inclusions, and yet, each could be distinguished from the other by the frequency of round inclusions (Fig. 2c). In addition, the survival plots for G85R-SOD1:YFP in these second-passage experiments revealed that the time to paralysis varied according to mutant SOD1 used in the initial passage, providing additional evidence that distinct features of the mutant SOD1 used in the initial induction were retained in the second passage (Fig. 2d).

Distinct pathologies of G85R-SOD1:YFP inclusions suggest SOD1 prion-like properties. a Representative images of spinal cords from diseased G85R-SOD1:YFP mice injected within their spinal cords at P0 with either the first or second passages of the indicated homogenates. First and second passages of recWT fibrils produced distinct skein-like inclusion (arrows and insets). b Circularity of the G85R-SOD1:YFP inclusions produced from the second passage of homogenates was quantified. n = 3 images per animal and three animals per cohort; mean ± SEM. c Based on the circularity, the inclusions were grouped into three categories: fibrillar (0.0–0.35), intermediate (0.35–0.65), and round (0.65–1.0). n = 3 images per animal and three animals per cohort; mean ± SEM; Two-way ANOVA with Sidak’s multiple comparisons test: *p ≤ 0.05, ** ≤0.01, and *** ≤0.001. d Kaplan–Meier survival curves show MND induction from the second passages of the indicated homogenates in G85R-SOD1:YFP mice. e Representative image of inclusion pathology induced in G85R-SOD1:YFP spinal cord slices following incubation with the indicated second-passage homogenates (n = 15 slices per homogenate). In the slices that have inclusions, the background fluorescence of non-aggregated G85R-SOD1:YFP is not visible when exposures are optimized for image capture of the inclusions. Nuclei are stained blue with DAPI
To augment the in vivo animal studies, we utilized the G85R-SOD1:YFP organotypic spinal cord slice culture system as described above. We observed that after adding homogenates prepared from paralyzed mice of various SOD1 lines to these cultures, inclusions were induced that increased in abundance over time (Supplemental Fig. 2). When we treated these cultures with homogenates prepared from both the first and second passages of G93A or recWT fibrils, we observed the same distinct pathologies that were observed in vivo, primarily round inclusions upon treatment with G93A homogenate and primarily fibrillar pathology upon treatment with recWT homogenate (Fig. 2e). Although the inclusions induced in vivo following injection with the second passages of G37R and L126Z homogenates produced a more heterogeneous population of inclusions, treatment of the slice cultures with these samples mainly produced fibrillar inclusions (Fig. 2e). This finding may be a consequence of changes in tissue architecture in these ex vivo slice cultures. Despite the potential impact of culturing, we have confidence that the misfolded G93A SOD1 (in tissue homogenates) and recWT fibrils are capable of inducing distinct strains of misfolded G85R-SOD1:YFP and that these strains are preserved upon second passage in naïve G85R-SOD1:YFP mice.
Induction of accelerated MND in SOD1 transgenic mice expressing truncated versions of hSOD1
Transmission of MND has now been demonstrated in both tagged and untagged mouse models of the G85R variant of SOD1 [7]. We have previously demonstrated the inability to accelerate MND in transgenic mice expressing the G37R and G93A variants of SOD1 by tissue homogenate injections [2]. To better understand the permissiveness of MND in SOD1 transgenic lines and to determine whether the phenomenon is unique to G85R-SOD1, we examined mouse lines overexpressing two truncation mutants of SOD1. The first line examined expresses the L126Z mutation, and uninjected mice normally develop paralysis at 7.5 months of age (Fig. 3a) [38]. We prepared spinal cord homogenates from paralyzed L126Z mice and injected them into the spinal cord of newborn L126Z mice and non-transgenic littermates. All of the transgenic L126Z mice that were injected (n = 10) developed paralysis significantly sooner than the uninjected control transgenic mice, with an average age of paralysis of 3.1 months of age. When compared with age-matched uninjected mice, the spinal cords of the injected L126Z mice at disease end-stage contained widespread C4F6 positive SOD1 inclusion immunoreactivity.

MND induction in untagged SOD1 transgenic mice. a Kaplan–Meier survival curves show a decrease in MND end-stage following syngeneic transmission of homogenate from a diseased mouse in the L126Z-SOD1 line of mice. b Kaplan–Meier survival curves show a decrease in MND end-stage following syngeneic transmission of homogenate from a diseased mouse in the V103Z-SOD1 line of mice. c Representative images revealing abundant C4F6 immunoreactivity in diseased mice when compared with uninjected controls (n = 3 animals, 4 sections per animal)
The same paradigm was used in another transgenic mouse line expressing the first 102 amino acids of SOD1 with mutations at histidines 46, 48, and 63 to eliminate copper binding (V103Z mice). Only ~30 % of mice from this line develop paralysis with an average age of end-point of 19.4 months [39]. However, when we injected spinal cords of newborn V103Z mice with homogenate prepared from a paralyzed V103Z mouse, all of the injected transgenic animals (n = 10) developed MND with an average age of onset of 3.7 months of age. Similar to the L126Z mice, the injected mice at disease end-stage exhibited widespread C4F6-immunoreactive inclusions within the spinal cords, whereas no inclusions were detected in uninjected age-matched V103Z mice. Together, these findings identify two additional lines of mutant SOD1 mice that are susceptible to induced misfolding.
Tissue homogenates from sALS patients lack misfolded SOD1 conformers that can template G85R-SOD1:YFP misfolding
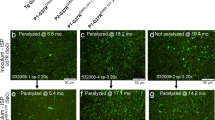
A number of studies have implicated misfolded WT SOD1 as a pathogenic factor in sALS cases [8, 23, 31, 34]. With the establishment of ex vivo slice cultures from G85R-SOD1:YFP mice as a useful model to detect the presence of misfolded SOD1 seeding conformers, we embarked on a study to screen post-mortem spinal cord homogenates prepared from sALS patients for the presence of misfolded SOD1 templating conformers. Our patient donors included 2 patients that had SOD1-linked fALS. Collectively, we tested homogenates from 49 non-SOD1-ALS samples, including those from 36 sALS cases, 3 C9orf72 fALS cases, 6 Alzheimer’s Disease cases, and 4 non-neurologic control patients (Table 2). A total of 10 individual slice cultures were exposed to each of the tissue homogenates and incubated for up to 1-month post-exposure to assess whether G85R-SOD1:YFP inclusion pathology developed. While remaining blinded to the primary diagnosis for the majority of samples, we identified only two samples that seeded G85R-SOD1:YFP inclusion pathology in the slice cultures (Fig. 4; Table 3). Both of these cases were identified as spinal cords from patients carrying the A4V SOD1 mutation, which began to develop small punctate inclusions beginning ~7 days following exposure to the tissue homogenates. The induction of G85R-SOD1:YFP inclusion pathology by the A4V tissue homogenates was very efficient, with 26 of 35 slices exposed to homogenate developing obvious inclusions (Fig. 4; Table 3). Furthermore, we determined the SD50 for the 2 A4V homogenates by end-point dilution using the slice cultures to be 100.9/μl and 101.1/μl (Table 3). We, therefore, conclude from this data that spinal cords from sALS patients do not contain the same type, or amount, of misfolded SOD1 seeding conformers that are present in tissues from A4V-SOD1-fALS cases or transgenic mice expressing ALS-mutant SOD1.

Induction of G85R-SOD1:YFP pathology with human A4V SOD1-fALS tissue. Confocal images of G85R-SOD1:YFP spinal cord slices following incubation with post-mortem spinal cord tissue from a sALS or A4V SOD1-fALS case. In the slices that have inclusions, the background fluorescence of non-aggregated G85R-SOD1:YFP is not visible when exposures are optimized for image capture of the inclusions. Nuclei are stained blue with DAPI. Table 3 documents the number of slices examined for each tissue homogenate preparation
Discussion
Misfolded conformers of WT SOD1 have been implicated to have a toxic role in cases of sALS, which comprise greater than 80 % of all ALS cases [8, 23, 31, 34]. Given that several therapeutic strategies targeting SOD1 are under development, it is important to better understand the potential contribution of misfolded SOD1 in sporadic disease. Using in vitro fibrillized recombinant WT SOD1, we first demonstrated that misfolded WT SOD1 induces MND and inclusion pathology in G85R-SOD1:YFP mice and in an ex vivo organotypic spinal cord slice culture prepared from these mice. These findings support in vitro data demonstrating the ability for WT SOD1 fibrils to induce misfolding of SOD1 [13, 22] and reveal that misfolded SOD1 protein itself is capable of inducing pathology without the need of additional factors. We also demonstrate that misfolded SOD1 can produce two distinct types of inclusion pathology, spherical or fibrillar. Spinal tissues from G93A mutant SOD1 mice induce spherical inclusions, whereas the inclusions induced by fibrillized WT SOD1 and spinal tissues from G37R mutant mice are fibrillar. Tissues from L126Z mutant mice produce a mixed inclusion pathology. These distinct pathologies were retained when MND was passaged to a second cohort of G85R-SOD1:YFP mice. Second-passage studies also revealed that these strains of misfolded SOD1 differed in their interval to paralysis after injection (or incubation period when using prion terminology). We identify two new lines of SOD1 mice that are permissive to accelerated paralysis by injection of tissues from paralyzed mutant SOD1 mice. Finally, we demonstrate that spinal tissues from human SOD1-linked ALS have the same capacity to accelerate paralysis as first identified in transgenic mouse tissues.
Although we are certain that recWT SOD1 fibrils seed a distinct strain of misfolded G85R-SOD1:YFP, we cannot rule out the possibility that the method of producing the recWT fibrils did not contribute to disease induction. Notably, one of the 4 G85R-SOD1:YFP mice injected at P0 with PBS containing the reducing agent, TCEP, developed paralysis with sparse fibrillar inclusion pathology. A reducing agent, such as TCEP or dithiothreitol (DTT), is commonly added to SOD1 fibrillization assays to break the intramolecular disulfide bonds within the SOD1 molecule, thereby monomerizing the protein and aiding in fibril formation [12, 33]. To determine whether TCEP alone might have been responsible for the induction of G85R-SOD1:YFP misfolding, we exposed our slice culture models to various samples containing TCEP, including concentrations 10 times greater than that used to prepare the WT SOD1 fibrils (100 mM), and found no evidence of induced misfolding (Supplemental Fig. 3). We currently view the one case of induced MND by TCEP injection as an inexplicable outlier. Importantly, even if the TCEP in the recWT fibril preparations had some influence on the initial induction, the fact remains that recWT fibrils containing TCEP produced a distinct strain-like pathology that was heritable upon subsequent passages in G85R-SOD1:YFP mice. This finding provides strong evidence that misfolded WT SOD1 seeded a novel misfolded conformation that could be propagated in a fashion that resembles templated misfolding.
We previously demonstrated the ability to induce MND through the exogenous administration of spinal homogenates from paralyzed SOD1 mice into the spinal cords of G85R-SOD1:YFP mice [2]. In contrast, we now demonstrate that spinal homogenates prepared from two other mouse models (mutant a-synuclein and mutant tau), which both exhibit progressive motor paralysis and extensive spinal cord pathology, do not induce paralysis nor inclusions when injected into G85R-SOD1:YFP mice. These findings further support the idea that misfolded conformers in the spinal cords of paralyzed mutant SOD1 mice can penetrate cells and induce the propagation of mutant SOD1 misfolding. In addition, studies have demonstrated a similarity in immunomodulatory factors expressed throughout the course of disease in patients and mouse models of Alzheimer’s disease, Parkinson’s disease, and ALS [20]. The fact that homogenates prepared from these other non-SOD1 paralyzed transgenic mice were unable to induce MND in the G85R-SOD1:YFP mouse model provides more evidence for misfolded SOD1 as the seeding factor, rather than induction of misfolding due to neurotoxic mediators.
To assess the presence of misfolded SOD1 conformers capable of inducing inclusion pathology in human tissue, we screened post-mortem spinal cord homogenates from ALS patients and controls in organotypic spinal cord slices prepared from G85R-SOD1:YFP mice. Spinal cord slices incubated with tissue homogenates from either of two A4V SOD1-fALS cases accumulated G85R-SOD1:YFP inclusions, indicating that spinal tissues from human mutant SOD1 ALS cases contain the same types of prion-like misfolded SOD1 as the mutant transgenic mice. Homogenates prepared from spinal tissues from sALS cases or controls (C9orf72 fALS, Alzheimer’s Disease, and non-neurologic control cases) were unable to induce pathology in the G85R-SOD1:YFP slice cultures following an extended incubation. The most conservative interpretation of our data is that tissues from sporadic ALS cases have very distinct strains of misfolded WT SOD1 which are less potent in inducing G85R-SOD1:YFP inclusion pathology. An alternative interpretation is that the misfolded SOD1 species capable of inducing pathology is far less abundant in sALS tissue than it is in the A4V SOD1-fALS tissue. Based on the titer of transmissible misfolded SOD1 in the A4V homogenates, it can be assumed that the transmissible species in the sALS homogenates are at least tenfold less abundant. In either case, our data reinforce the notion that SOD1-linked ALS and sALS are pathologically distinct.
A number of recent studies have demonstrated that SOD1 possesses properties similar to those observed for prions, such as its ability to move from cell-to-cell, induce the misfolding and aggregation of both mutant and WT SOD1, and propagate along neuroanatomical pathways throughout the CNS [3, 22, 28]. The idea of SOD1 possessing prion strain-like properties has been suggested previously, and recently, two SOD1 mutant proteins were shown to have distinct conformations that were maintained upon primary passage in G85R-SOD1 transgenic mice [7]. Historically, prion strains are defined by characteristic incubation periods and neuropathological features that are maintained upon experimental passage [18]. Using various SOD1 mutant proteins, prepared from either spinal homogenates or fibrillized recombinant protein, we demonstrate distinct incubation periods and inclusion pathologies following multiple passages in G85R-SOD1:YFP mice. When compared with the primary passages, the secondary passages of G93A, recWT, G37R, and L126Z homogenates all resulted in an increased penetrance in G85R-SOD1:YFP mice and a decrease in their incubation periods by ~50 %. This characteristic is referred to as “host adaptation” in the prion field and is believed to be the result of greater compatibility between the inducing seed and the host target when the sequences of each are identical [1, 10]. When comparing the incubation periods among the different second-passage homogenates, we found a significant difference between them, further implicating the existence of SOD1 strains. As for the distinct pathologies we observed, the most striking difference was between the G93A homogenates, which produced round, punctate inclusions, and the recWT homogenates, which produced fibrillar and skein-like inclusions. Importantly, these pathologies were heritable upon the second passage of the homogenates and were also observed when the homogenates were added to the spinal cord slice cultures prepared from G85R-SOD1:YFP mice. Given the extensive literature on the behavior of prion strains, the most plausible explanation for the distinct pathologies we observed as a function of inoculum, is that unique SOD1 conformations exist within the tissue homogenates, and they act as templates when injected into naïve G85R-SOD1:YFP mice; a process known as conformation-dependent templated propagation.
Prior to these studies, the only demonstration of transmissibility of SOD1-mediated MND has been in transgenic mice that express the G85R variant of SOD1 [2, 7]. We present here two additional transgenic lines which upon syngeneic transmission of spinal homogenates from diseased mice produced a dramatically decreased age to paralysis when compared with the uninjected mice. Both of these lines express truncated versions of SOD1 that lack the homodimer interface and are, therefore, incapable of forming a mature and stable SOD1 dimer. Interestingly, the G85R variant has also been reported to exist mainly as a disulfide-reduced, monomeric protein [11, 40], potentially highlighting a critical factor in defining which SOD1 transgenic mouse models might be permissive for SOD1-MND transmission.
There are currently a number of therapeutic trials in SOD1-fALS patients aimed at slowing disease progression by SOD1 gene-silencing therapies that may prove to be effective in the sALS patients as well, if in fact, SOD1 has a pathogenic role in these patients. Our studies suggest that non-SOD1 ALS tissues do not harbor the same type of misfolded “transmissible” SOD1 that is present in mutant SOD1-ALS cases. However, the potential for SOD1 to adopt different strain-like conformations leaves open the potential for some type of misfolded SOD1 that transmits poorly to be involved in the pathogenesis of sporadic ALS. Clearly, the best test of whether misfolded SOD1 has any role in the pathogenesis of non-SOD1 ALS would be to have an SOD1-gene-silencing therapy, or other SOD1 targeting therapy, that is remarkably effective in the SOD1-ALS cases translate to non-SOD1-linked patients. Our demonstration that tissues from SOD1-ALS patients have the capacity to induce the misfolding of G85R-SOD1:YFP indicates the presence of conformational seeds that can spread between cells. How quickly and how broadly these seeds spread in patients before the onset of symptoms could dictate how effective the gene-silencing therapies might be in SOD1-ALS patients.
References
Aguzzi A, Heikenwalder M, Polymenidou M (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8:552–561. doi:10.1038/nrm2204
Ayers J, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, Borchelt DR (2014) Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol 128:791–803. doi:10.1007/s00401-014-1342-7
Ayers J, Fromholt SE, O’Neal VM, Diamond JH, Borchelt DR (2016) Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol 131:103–114. doi:10.1007/s00401-015-1514-0
Ayers J, Xu G, Pletnikova O, Troncoso JC, Hart PJ, Borchelt DR (2014) Conformational specificity of the C4F6 SOD1 antibody; low frequency of reactivity in sporadic ALS cases. Acta Neuropathol Commun 2:55. doi:10.1186/2051-5960-2-55
Banci L, Bertini I, Durazo A, Girotto S, Gralla EB, Martinelli M, Valentine JS, Vieru M, Whitelegge JP (2007) Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc Natl Acad Sci USA 104:11263–11267. doi:10.1073/pnas.0704307104
Bessen RA, Marsh RF (1992) Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol 73(Pt 2):329–334
Bidhendi EE, Bergh J, Zetterström P, Andersen PM, Marklund SL, Brännström T (2016) Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis–like disease. J Clin Invest. doi:10.1172/JCI84360
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien J-P, Brady ST, Brown RH (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13:1396–1403. doi:10.1038/nn.2660
Brotherton TE, Li Y, Cooper D, Gearing M, Julien J-P, Rothstein JD, Boylan K, Glass JD (2012) Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci USA. doi:10.1073/pnas.1115009109
Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H (1994) Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 343:405–411. doi:10.1098/rstb.1994.0036
Cao X, Antonyuk SV, Seetharaman SV, Whitson LJ, Taylor AB, Holloway SP, Strange RW, Doucette PA, Valentine JS, Tiwari A, Hayward LJ, Padua S, Cohlberg JA, Hasnain SS, Hart PJ (2008) Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J Biol Chem 283:16169–16177. doi:10.1074/jbc.M801522200
Chattopadhyay M, Durazo A, Sohn SH, Strong CD, Gralla EB, Whitelegge JP, Valentine JS (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci USA 105:18663–18668. doi:10.1073/pnas.0807058105
Chia R, Tattum MH, Jones S, Collinge J, Fisher EMC, Jackson GS (2010) Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS One 5:e10627. doi:10.1371/journal.pone.0010627
Deng H-X, Shi Y-Z, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung W-Y, Bigio EH, Lukas T, Dal Canto MC, O’Halloran TV, Siddique T (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA 103:7142–7147. doi:10.1073/pnas.0602046103
Dickinson AG, Meikle VM (1971) Host-genotype and agent effects in scrapie incubation: change in allelic interaction with different strains of agent. Mol Gen Genet 112:73–79
Ezzi SA, Urushitani M, Julien J-P (2007) Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem 102:170–178. doi:10.1111/j.1471-4159.2007.04531.x
Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brännström T (2010) Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 5:e11552. doi:10.1371/journal.pone.0011552
Fraser H, Dickinson AG (1968) The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78:301–311
Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34:521–533
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934. doi:10.1016/j.cell.2010.02.016
Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW (1992) Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 258:806–808
Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR (2014) Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci USA 111:3620–3625. doi:10.1073/pnas.1312245111
Graffmo KS, Forsberg K, Bergh J, Birve A, Zetterstrom P, Andersen PM, Marklund SL, Brannstrom T (2012) Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum Mol Genet 22:51–60. doi:10.1093/hmg/dds399
Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC (2001) CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol 102:293–305. doi:10.1007/s004010100399
Kerman A, Liu H-N, Croul S, Bilbao J, Rogaeva E, Zinman L, Robertson J, Chakrabartty A (2010) Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol 119:335–344. doi:10.1007/s00401-010-0646-5
Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RSG, Budka H (2002) Mutations of the prion protein gene phenotypic spectrum. J Neurol 249:1567–1582. doi:10.1007/s00415-002-0896-9
Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405. doi:10.1038/78078
Münch C, O’Brien J, Bertolotti A (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci USA 108:3548–3553. doi:10.1073/pnas.1017275108
Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB (2001) Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 10:854–863. doi:10.1110/ps.39201
Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR (2012) Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 7:e35050. doi:10.1371/journal.pone.0035050
Prudencio M, Durazo A, Whitelegge JP, Borchelt DR (2010) An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum Mol Genet 19:4774–4789. doi:10.1093/hmg/ddq408
Prudencio M, Hart PJ, Borchelt DR, Andersen PM (2009) Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet 18:3217–3226. doi:10.1093/hmg/ddp260
Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A (2004) Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem 279:15499–15504. doi:10.1074/jbc.M313295200
Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi X-F, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A (2002) Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem 277:47551–47556. doi:10.1074/jbc.M207356200
Reed LJ, Muench H (1938) A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497
Seetharaman SV, Taylor AB, Holloway S, Hart PJ (2010) Structures of mouse SOD1 and human/mouse SOD1 chimeras. Arch Biochem Biophys 503:183–190. doi:10.1016/j.abb.2010.08.014
Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR, Perrin S, Feany MB, Horwich AL (2009) Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci USA 106:1392–1397. doi:10.1073/pnas.0813045106
Wang J, Xu G, Li H, Gonzales V, Fromholt D, Karch CM, Copeland NG, Jenkins NA, Borchelt DR (2005) Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: alphaB-crystallin modulates aggregation. Hum Mol Genet 14:2335–2347. doi:10.1093/hmg/ddi236
Xu G, Ayers J, Roberts BL, Brown H, Fromholt S, Green C, Borchelt DR (2014) Direct and indirect mechanisms for wild-type SOD1 to enhance the toxicity of mutant SOD1 in bigenic transgenic mice. Hum Mol Genet. doi:10.1093/hmg/ddu517
Zetterström P, Stewart HG, Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brännström T, Oliveberg M, Marklund SL (2007) Soluble misfolded subfractions of mutant superoxide dismutase-1 s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci USA 104:14157–14162. doi:10.1073/pnas.0700477104
Acknowledgments
We would like to thank the patients and family members who contributed tissue to the study; Drs. B. Giasson and J. Lewis for donating mouse tissue for the study; and Dr. C. Janus for his help on statistical analysis. A. G. was supported by St. Mary’s University Research Grant and the Biaggini Research Program. This work was supported by an NIH Shared Instrumentation Grant (S10OD020026), a grant from the National Institutes of Neurological Disease and Stroke (1R01NA092788-01), and the Packard Center for ALS Research at Johns Hopkins University. Collection and characterization of human tissues provided by Johns Hopkins were funded by the Target ALS Multicenter Post-mortem Tissue Core.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ayers, J.I., Diamond, J., Sari, A. et al. Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol 132, 827–840 (2016). https://doi.org/10.1007/s00401-016-1623-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-016-1623-4