
Abstract
The number of patients with neurodegenerative diseases is increasing significantly worldwide. Thus, intense research is being pursued to uncover mechanisms of disease development in an effort to identify molecular targets for therapeutic intervention. Analysis of postmortem tissue from patients has yielded important histological and biochemical markers of disease progression. However, this approach is inherently limited because it is not possible to study patient neurons prior to degeneration. As such, transgenic and knockout models of neurodegenerative diseases are commonly employed. While these animal models have yielded important insights into some molecular mechanisms of disease development, they do not provide the opportunity to study mechanisms of neurodegeneration in human neurons at risk and thus, it is often difficult or even impossible to replicate human pathogenesis with this approach. The generation of patient-specific induced pluripotent stem (iPS) cells offers a unique opportunity to overcome these obstacles. By expanding and differentiating iPS cells, it is possible to generate large numbers of functional neurons in vitro, which can then be used to study the disease of the donating patient. Here, we provide an overview of human stem cell models of neurodegeneration using iPS cells from patients with Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, frontotemporal dementia, Huntington’s disease, spinal muscular atrophy and other neurodegenerative diseases. In addition, we describe how further refinements of reprogramming technology resulted in the generation of patient-specific induced neurons, which have also been used to model neurodegenerative changes in vitro.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases include a variety of neuropsychiatric and neurological disorders with limited therapeutic options. Advanced age is the biggest risk factor for the development of neurodegenerative changes, and because people are living increasingly longer life spans, treatment of patients with neurodegenerative diseases results in an increasingly significant socio-economic impact on society. For example, the global cost of dementia comprised 604 billion US dollars in 2010 (Alzheimer’s Disease International). Hence, both medically and economically, it is urgently necessary to identify the mechanisms of neurodegeneration and to develop novel treatments to protect neurons against pathologic changes.
Directly studying living patient neurons is severely limited by the inaccessibility of the human brain. As a result, postmortem analyses of brain tissue have been performed, which yielded extensive insights into end-stage disease pathology. In order to study disease development, alternative approaches have been applied, which include the analysis of patients’ fibroblasts or transformed cell lines that harbor disease-associated mutations. Although the ability to easily culture transformed cell lines has enabled detailed mechanistic studies to be carried out, the biology of cell lines does not resemble the biology of primary neurons. As a result, it is often unclear whether the mechanisms studied are directly comparable to patients’ pathology.
Another method of studying neurodegenerative disease is the creation of transgenic animal models—especially mice. For example, human mutations that cause familial forms of neurodegenerative diseases can be inserted into mice, and mechanisms of disease development can be studied. However, these models often do not accurately recapitulate human disease, as typical phenotypes are often underrepresented or even lacking [114]. As an example, in animal models of Alzheimer’s disease (AD), overexpression of the human mutated amyloid precursor protein (APP) resulted in abundant plaque deposition and amyloid-associated pathology, but neurofibrillary tangles and significant neuronal loss as additional hallmarks of AD were not detected [69, 143]. Furthermore, animal models of the sporadic forms of neurodegenerative diseases cannot easily be established due to species-related differences in genetic susceptibility factors.
Therefore, understanding the mechanisms causing human neurodegeneration and developing effective therapies may require human-specific models, which more closely recapitulate human pathogenesis. Stem cell technologies enable the development of such models and encompassed the analysis of disease-affected human embryonic stem (ES) cells. ES cells are derived from the inner cell mass of pre-implantation stage embryos and, as pluripotent stem cells, have the ability of potentially unlimited self-renewal and of spontaneous differentiation into cells of the three germ layers: mesoderm, endoderm and ectoderm [180]. By applying sophisticated differentiation protocols, high numbers of functional neurons can be derived from undifferentiated ES cells, which behave similarly to primary neurons and which have been widely used in cell culture assays [9, 36, 44, 138, 169] (Fig. 1).

In vitro modeling of neurodegenerative diseases using human embryonic stem cells, induced pluripotent stem cells and induced neurons. I Human ES cells are derived from human blastocysts after isolation and dissection of the inner cell mass and can be differentiated into mature neurons. It is possible to genetically modify ES cells and to introduce a disease-specific mutation in order to model neurodegenerative diseases after differentiation into disease-affected neurons. An alternative, but a less suitable approach, is to analyze ES cells already carrying a genetic defect. II Skin fibroblasts of patients can be reprogrammed into induced pluripotent stem (iPS) cells by ectopic expression of the transcription factors Oct4, Sox2, Klf4 and c-Myc. These patient-specific iPS cells can be differentiated into disease-affected neuronal subtypes. In cases of known genetic abnormalities causing the neurodegenerative disease, gene targeting can be applied to repair the genetic mutation thereby establishing isogenic control iPS cell lines. In addition, these genetically corrected iPS cells can be differentiated into neurons and, at least theoretically, subsequently used for cell replacement therapy. Alternatively, iPS cells derived from healthy individuals serve as a source of healthy control neurons. III Skin fibroblasts from patients with neurodegenerative diseases or from healthy individuals can be directly transdifferentiated into neuronal cells (so-called induced neurons or iN cells) through ectopic expression of transcription factors such as Ascl1, Brn2, Myt1l and NeuroD1
The most obvious approach to modeling neurodegenerative diseases with human ES cells is to use neurons derived from stem cells carrying disease-causing mutations. However, because ES cells are derived from embryos, this would involve using pre-implantation genetic diagnostic (PGD) testing to identify embryos harboring a genetic disease, as has been done for Huntington’s disease (HD) or Down syndrome (DS). Neurons have been differentiated from these ES cells to analyze disease-associated phenotypes [19, 115, 123, 124, 186]. For instance, aggregates of Aβ42 protein or increased expression of phospho-tau have been observed in stem cell-derived neurons using this approach (Table S1). However, in addition to ethical concerns [60], the isolation of ES cells during PGD can only be used for diseases with very specific mutations and not for the vast majority of neurodegenerative diseases, which are sporadic and not clearly associated with a specific mutation. Thus, deriving human ES cells from embryos analyzed by PGD cannot be a standard procedure for modeling neurodegenerative diseases.
One alternative to PGD is to insert disease-causing mutations into existing human ES cell lines using endonuclease-mediated gene targeting (Fig. 1). For instance, zinc-finger nucleases introduce a double-strand break at a specific target sequence in the genome, which enables the insertion of specific donor sequences through homologous recombination as the cell repairs the break. Using this technique, the Parkinson’s disease (PD)-associated mutations A53T and E46K were inserted into the α-synuclein (SNCA) locus of two different ES cell lines [168] (Table S1). These modified ES cells did not carry any additional off-site genetic modifications other than the targeted alteration within the SNCA locus and thus, the parental and modified ES cell lines were isogenic and differed only by a single mutation. As a result, whole-genome expression profiling of these stem cells demonstrated that isogenic ES cell lines had a much more similar expression profile compared with unrelated ES cell lines [168]. A major disadvantage of this technology, however, is related to the fact that cells of a patient may carry specific disease-associated genetic variants in addition to a specific mutation, which cannot be established in the gene-targeted stem cell and thus, mechanisms of disease development may not be modeled properly. Furthermore, this technique could not be applied to model sporadic diseases. Therefore, while these studies highlight the usefulness of human ES cells for research on neurodegenerative diseases, an alternative approach is needed that uses cells directly derived from patients with the disease of interest. This was accomplished by the application of patient-specific induced pluripotent stem (iPS) cells or the derivation of human induced neurons (iN cells) from patients, as reviewed in this article (Fig. 1).
Human stem cell models of neurodegeneration using patient-derived iPS cells
In 2006, Takahashi and Yamanaka [175] demonstrated a revolutionary technology in which fibroblasts could be reprogrammed into iPS cells simply by expressing the pluripotency-associated transcription factors Oct4, Klf4, Sox2 and c-Myc (Fig. 1). These iPS cells share the main characteristics of pre-implantation embryo-derived ES cells including the ability of unlimited self-renewal and the potential to differentiate into cells of the three germ layers. As a result, it is possible to derive fibroblasts from a patient with a neurodegenerative disease, to reprogram them into iPS cells and to expand them into large numbers. By applying differentiation protocols used for ES cells, iPS cells can be directed to differentiate into functional neuronal subtypes such as midbrain dopaminergic (DA) neurons [36, 44, 96, 122, 138, 139], glutamatergic cortical neurons [159], striatal GABAergic neurons [9] or cholinergic motor neurons [117, 138], which are at risk in PD, AD, HD or amyotrophic lateral sclerosis (ALS), respectively (Fig. 2). Because these differentiated neurons were generated from cells of a specific patient, the resulting phenotype of these cells can be used to model the pathology of neurons within the same individual. In addition, since iPS cells can produce specialized neurons in very large quantities, it is possible to apply these models to conduct detailed mechanistic studies, to develop cell therapies, or to identify new drug candidates [46, 73, 84, 111, 133].

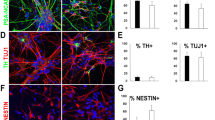
Differentiation of human iPS cells into neural subtypes in vitro. By applying improved differentiation protocols, human iPS cells can be differentiated into several mature neuronal subtypes. a Dopaminergic neurons co-expressing tyrosine hydroxylase (TH) and βIII-tubulin (TUJ1) and b midbrain-like dopaminergic neurons co-expressing TH and FoxA2. c Striatal neurons co-expressing βIII-tubulin and the dopamine and cAMP-regulated phosphoprotein DARPP-32. d Motor neurons positive for microtubule-associated protein-2 (MAP-2), choline acetyltransferase (ChAT) and non-phosphorylated neurofilament H (SMI-32). e Peripheral neurons expressing peripherin and βIII-tubulin. f Human iPS cells can also be directed to differentiate into astrocytes expressing glial fibrillary acidic protein (GFAP) and S100
The first iPS cell lines from patients with neurodegenerative diseases were described in 2008 [57, 128], and to date, more than 50 reports on iPS cell-based models of neurodegeneration have been published, most of which focus on familial forms of disease caused by specific mutations (Table S2). However, iPS cell models of sporadic forms of neurodegenerative diseases have also been established (Table S2). Interestingly, despite the usually late manifestation of degenerative changes in patients, disease phenotypes were described in patient-iPS cell-derived neurons only after a few weeks of culture and appeared either spontaneously or after challenge with stress-inducing compounds mimicking cumulative age- and disease-related insults to neurons in vivo.
Models of Alzheimer’s disease using patient-derived iPS cells
AD is the most common neurodegenerative disease and the most common cause of dementia, affecting more than 30 million people worldwide [11]. Patients suffer from severe cognitive decline with progressive loss of memory function and of abilities to perform basic daily procedures properly. Pathologic hallmarks of AD are extracellular deposits of Aβ protein forming amyloid plaques and intracellular neurofibrillary tangles composed of aggregated and hyperphosphorylated tau protein [56, 72]. These pathologic changes within and around forebrain neurons have been modeled with AD patient-derived iPS cells in different studies.
AD caused by mutated APP
AD-specific iPS cells have been derived from patients with familial forms of AD (fAD) carrying a duplication of the APP locus (APP dp) [80] or the E693Δ mutation in APP [94] (Table S2). APP is a transmembrane protein with important physiological functions including cell signaling, synapse formation and neurogenesis [100]. However, when gene dosage is increased or in the case of certain point mutations, the amyloidogenic pathway generates more pathologic Aβ protein with subsequent deposition of senile Aβ-positive plaques [177]. Similarly, APPdp-AD and E693Δ-AD patient-derived iPS neurons expressed increased levels of Aβ40 protein when compared to neurons from non-demented control individuals [80, 94]. Furthermore, APPdp-AD-iPS cell-derived neurons expressed higher amounts of active GSK-3β and phosphorylated tau protein at Thr231 indicating neurodegenerative changes within patient-derived neurons in vitro [80]. Also, APPdp-AD-derived neurons contained significantly increased numbers of RAB5-positive endosomes, which is in line with observations that RAB5-positive endosomes accumulate within neurons of a subset of AD patients [33, 34, 80]. These pathologic changes could be significantly reduced after application of β-secretase inhibitors to patient-derived neurons. However, the application of γ-secretase inhibitors only significantly reduced the level of Aβ40 protein, but not the levels of active GSK-3β and phospho-tau protein, indicating that products of APP processing other than Aβ40 protein may regulate the induction of GSK-3β activity and phospho-tau protein in AD [80].
Since APP is encoded on chromosome 21, a duplication of the APP gene is present also in neurons from individuals with DS, which is caused by trisomy of chromosome 21. AD-like neurodegenerative changes have been described in people with DS [25], which could be due to duplication of the APP gene and/or due to duplication of other genes such as Dyrk1A kinase, known to phosphorylate tau protein at Thr212 [192]. DS-iPS cell-derived cortical neurons expressed higher amounts of both Aβ40 and Aβ42 protein with an increased Aβ42/Aβ40 ratio, formed insoluble intracellular and also extracellular amyloid aggregates, accumulated AT8-positive phosphorylated tau protein and secreted phospho-tau, thereby mimicking changes seen in patients with AD [158].
AD caused by mutant PS1 and PS2
Mutations in the presenilin 1 gene (PS1) and in the presenilin 2 gene (PS2) are common causes of fAD [48, 157] and have been described to enhance the production of Aβ protein, especially of Aβ42 [153]. AD-iPS cells have been derived from a patient with the A246E mutation in the PS1 gene and from a patient with the N141I mutation in the PS2 gene [193] (Table S2). When differentiated into neurons, both PS1-AD-iPS cells and PS2-AD-iPS cells secreted Aβ40 and Aβ42 into the medium supernatant with an increased Aβ42/Aβ40 ratio as seen in postmortem tissue from fAD patients [193]. These changes could be reversed by γ-secretase inhibition [193].
Sporadic AD
Most AD patients suffer from the sporadic, non-familial form of AD, which is not linked to a specific genetic mutation. However, genetic risk variants have been discovered in genome-wide association studies, among which apolipoprotein E isoform E4 showed the highest consistency. Other genes include Picalm, CR1, Clusterin and SORL1 [74, 99, 144, 196]. IPS cells were recently generated from two individuals with sporadic AD [80]. Neurons from one of the patients behaved like non-demented control neurons, while neurons from the other sporadic AD patient showed the same AD pathology as seen in cells from the APPdp-AD patients described above [80] (Table S2). It will be very interesting to analyze pathologic neurodegenerative changes in iPS cell-derived neurons from sporadic AD patients with different genetic risk variants for apolipoprotein E isoform E4, CR1, Clusterin and SORL1 and to use these stem cells to obtain better mechanistic insights into the pathogenesis of different forms of sporadic AD.
Models of Parkinson’s disease using patient-derived iPS cells
Among all neurodegenerative diseases, PD has been modeled most frequently with iPS cells (Fig. 3; Table S2). PD is the second most common neurodegenerative disease and affects approximately 1 % of people over 60 years of age and approximately 4 % of people over 80 years of age [50]. A minor fraction of PD patients, about 5–10 % have a familial form of PD (fPD), which is the result of inheriting specific mutations. Mutations in SNCA were the first to be linked to fPD and cause pathology in an autosomal-dominant fashion [131]. Subsequently, mutations in Leucine-Rich Repeat Kinase 2 (LRRK2) were identified as causing autosomal-dominant PD and are the most common known genetic causes of PD [43]. Mutations in PTEN-induced Putative Kinase 1 (PINK1), DJ-1 or PARKIN result in autosomal-recessive fPD (for review see [105]). In addition, duplication and triplication of the SNCA locus cause fPD [105, 163]. However, the majority of PD patients suffer from sporadic PD, which is most likely caused by a combination of environmental and genetic susceptibility factors. Indeed, genome-wide association studies revealed that variants in the gene loci of SNCA, MAPT, PARK16 and LRRK2 comprise risk factors for sporadic PD [105, 162]. Histopathologically, severe degeneration of DA neurons within the substantia nigra pars compacta is typical and Lewy bodies, which comprise intracellular aggregates of proteins including SNCA, are seen in most, though not all PD patients [43, 172].

Statistics on human iPS cell models of neurodegeneration. a Number of articles published on human iPS cell models of neurodegenerative diseases. PD Parkinson’s disease, ALS amyotrophic lateral sclerosis, HD Huntington’s disease, oTRD other triplet repeat diseases, SMA spinal muscular atrophy, SD storage diseases, FTD frontotemporal dementia, AD Alzheimer’s disease. b Number of articles describing (partial) rescue of disease phenotypes through different approaches. c Number of articles published from 2008 to 2013. Asterisk as of November 2013
Sporadic PD
The first PD-specific iPS cell lines were derived from a donor with sporadic PD [128] (Table S2). Derivation of additional iPS cell lines from five sporadic PD patients demonstrated that the neuronal differentiation potential of these cells was comparable to those of control iPS cells from healthy donors [167]. However, PD patient-derived neurons had a reduced number and length of neurites, expressed significantly increased levels of cleaved caspase 3 protein after prolonged culture in vitro and were more vulnerable to MPP+, a neurotoxin to midbrain DA neurons [147]. These findings were attributed to an accumulation of autophagic vacuoles due to an impaired maturation of autophagosomes into autophagolysosomes in patient-derived DA neurons [147].
PD caused by mutant LRRK2
Mutations in LRRK2 result in a late-onset form of PD with clinical symptoms comparable to those of patients with sporadic PD. LRRK2 encodes a multidomain protein with kinase and GTPase activities, which are mediated through a kinase domain and a ras of complex GTPase domain, respectively [43]. To date, more than 50 mutations in the LRRK2 gene have been described in PD patients, but the most common is G2019S, which occurs within the kinase domain and results in increased kinase activity [43]. Consequently, disease phenotypes were analyzed in G2019S-LRRK2-iPS cells, which had been differentiated into neural progenitor cells [108] and into DA neurons [45, 122, 126, 138, 139, 147, 148] (Table S2).
As seen in neurons from patients with sporadic PD, G2019S-LRRK2 neurons also showed impaired neurite outgrowth [122, 139, 147]. This observation is consistent with studies linking mutant LRRK2 protein to altered neurite extension [112] and linking LRRK2 overexpression to depolymerization of microtubules [107]. Furthermore, iPS cell-derived G2019S-LRRK2 DA neurons showed an increased expression of cleaved caspase 3 protein and the oxidative stress-response genes HSPB1, NOX1 and MAOB [122, 147]. These neurons revealed increased damage of mitochondrial DNA [148] and were more susceptible to toxins such as hydrogen peroxide, 6-hydroxydopamine, rotenone, MPP+, valinomycin, concanamycin A and MG-132, when compared to control cells from healthy donors [45, 122, 138, 139, 147]. These findings indicated an increased vulnerability of G2019S-LRRK2 DA neurons to stressors due to disturbances in pathways involving the mitochondrial oxidative complexes I and IV, the clearance of mitochondria via autophagosomes and the degradation of proteins through the proteasome complex, respectively. In addition, G2019S-LRRK2 DA neurons had impaired autophagosome clearance and an increased expression of stress-response genes, even when cultured over prolonged period of time and without application of oxidative stressors as similarly described for iPS cell-derived DA neurons from patients with sporadic PD [147]. An application of the antioxidant coenzyme Q10, the pro-survival AKT activator rapamycin and the LRRK2 inhibitors GW5074 and IN1 could partially rescue stress phenotypes in G2019S-LRRK2 iPS cell-derived neurons [45, 139].
iPS cell-derived G2019S-LRRK2 neurons accumulated SNCA protein, a major component in Lewy bodies in substantia nigra DA neurons of PD patients [122, 139, 147]. This is consistent with previous findings linking LRKK2 mutation to increased SNCA expression in human cells [32] and is in line with the observation that most patients with LRRK2 mutations present with α-synucleinopathy [43]. The accumulation of SNCA protein in G2019S neurons could be related to defects in autophagy [126, 139] and proteasomal function [139], which were observed in differentiated G2019S-LRRK2-iPS cells.
Further mechanistic insights into G2019S-mediated PD pathogenesis were provided by a recent publication by Reinhardt et al. [139] on patient-specific iPS cells. The expression of CPNE8, MAP7, ANXA1 and CADPS2, genes that are implicated in neurotransmission and microtubule dynamics [24], was significantly increased in G2019S-iPS cell-derived midbrain DA neurons and siRNA-mediated knockdown of these genes was followed by decreased DA neurodegeneration. Phosphorylated microtubule-associated protein tau was also significantly upregulated in G2019S-DA neurons, further supporting a critical role of LRRK2 in microtubule function and cellular integrity. In contrast, E3 ubiquitin ligase UHRF2, which is involved in clearance of polyglutamine aggregates [81], was significantly downregulated in G2019 DA neurons. UHRF2 could have a similar function in protein clearance in midbrain DA neurons, which might be impaired in G2019S neurons from patients with PD. In line with the observation that the G2019S mutation causes an increased activity of the kinase domain [43], dysregulated expression of CPNE8, UHRF2 and CADP2 in G2019S DA neurons was caused by LRRK2-dependent ERK phosphorylation [139]. Observed disease phenotypes were ameliorated by inhibition of ERK protein in G2019S neurons and by zinc finger-mediated correction of the genetic defect in G2019S-LRRK2-iPS cells. These observations are particularly important since they show that phenotypes appeared as a result of the genetic defect and not due to any unspecific confounding factors [139].
Mutations in the GTPase domain of the LRRK2 gene have also been identified in fPD [43]. When compared to control cells, R1441C-iPS cell-derived DA neurons presented with dysfunctional intracellular transport of mitochondria [45], again highlighting an important role of LRRK2 in microtubule function. Furthermore, R1441C neural cells presented with increased damage of mitochondrial DNA [148] and with increased cell death after application of valinomycin and concanamycin A, as similarly seen for G2019S iPS cell-derived neural cells. These findings indicated enhanced vulnerability of PD patient-derived R1441C cells through mechanisms, which, at least in part, are also involved in degeneration of neural cells carrying the G2019S LRRK2 mutation [45].
PD caused by mutant SNCA
SNCA point mutations A53T, A30P and E46K [106, 197] and the more frequent SNCA duplications and triplications cause an early-onset form of PD [163]. One study reported the derivation of iPS cells from a patient with the A53T SNCA mutation and was the first to demonstrate that patient-derived iPS cells can be genetically modified via zinc finger-mediated gene transfer to replace the mutation-bearing sequence with a healthy sequence thereby generating isogenic control iPS cell lines [168]. In a follow-up study, disease phenotypes were discovered in iPS cell-derived A53T cortical neurons and comprised an accumulation of nitric oxide (NO), increased levels of endoplasmic reticulum (ER) stress and an accumulation of ER-associated substrates, such as glucocerebrosidase and nicastrin [39]. These substrates were also elevated in postmortem cortical tissue from a patient carrying the A53T-SNCA mutation [39]. Elevated levels of NO and ER-associated substrates in A53T-iPS cell-derived neurons were changed to normal levels after application of NAB2, which has previously been identified as a compound to rescue SNCA-mediated toxicity [178].
Three independent groups reported the derivation of iPS cells from patients with a SNCA triplication [27, 53, 194] (Table S2). DA neurons differentiated from iPS cells with triplicated SNCA contained significantly increased amounts of SNCA protein compared to healthy controls [27, 53, 194]. In addition, these neurons expressed elevated levels of DNAJA1, HMOX2, UCHL1, HSPB1 and MAO-A mRNA [27], suggesting increased oxidative stress in the PD-derived DA neurons. Consistent with this observation, DA neurons with the SNCA triplication expressed higher levels of cleaved caspase 3 protein in response to H2O2 when compared to healthy controls [27].
PD caused by mutant PINK1 or PARKIN
Mutations in the PINK1 and PARKIN genes cause fPD in an autosomal-recessive fashion. PINK1 encodes a kinase localized at the outer mitochondrial membrane, while PARKIN encodes an E3 ubiquitin ligase found in the cytosol [119, 161, 188]. Mitochondrial damage activates PINK1 kinase activity, which recruits PARKIN protein and stimulates autophagy of the damaged mitochondria [120, 188]. Consequently, models of PD induced by mutant PINK1 or PARKIN examined mitochondrial function as well as neurodegeneration [45, 79, 85, 155] (Table S2).
DA neurons differentiated from mutant PINK1 iPS cells demonstrated significantly reduced translocation of PARKIN to mitochondria when treated with the mitochondrial stressor valinomycin [155]. Furthermore, these valinomycin-treated neurons showed an increased mitochondrial DNA copy number as well as increased biogenesis of mitochondria, which could indicate compensatory mechanisms for impaired mitochondrial function [155]. The mitochondrial disease phenotypes could be rescued by viral delivery and expression of wildtype (WT) PINK1 in patient-derived neurons [155].
In addition, iPS cells have been derived from PD patients with either a homozygous deletion of exon 3 [85], exons 2-4 [79], exons 6 and 7 [79] or with a compound heterozygous deletion of exons 3 and 5 of PARKIN [85]. DA neurons with mutant PARKIN had increased spontaneous DA release and decreased DA uptake compared to healthy controls [85]. These neurons also expressed higher levels of MAO-A and MAO-B mRNA, which produce significant amounts of reactive oxygen species through the oxidative deamination of DA [85, 160]. As demonstrated for iPS cells with mutant PINK1, the cellular phenotypes in mutant PARKIN iPS cell-derived neurons could be reversed by overexpression of WT PARKIN [85]. Together, these results indicated that PARKIN suppresses DA oxidation and controls neurotransmission in human midbrain DA neurons. Furthermore, mutant PARKIN DA neurons revealed an increased response to oxidative stress, showed altered morphology of mitochondria and impaired mitochondrial homeostasis [79], and expressed higher levels of NRF2 and NQO1 proteins, which are part of the same cytoprotective pathway and are activated in postmortem tissue of PD patients [79, 136]. Interestingly, the extent of disease phenotypes in iPS cell-derived DA neurons was directly correlated with postmortem pathology within the donor’s brain. Indeed, the accumulation of SNCA protein was only found in those iPS cell-derived neurons with mutant PARKIN, whose donor presented with SNCA-positive Lewy bodies. SNCA pathology was not detected in iPS cell-derived DA neurons generated from a family lacking Lewy body formation [79].
Models of amyotrophic lateral sclerosis using patient-derived iPS cells
ALS is a rapidly progressing neurodegenerative disease with a prevalence of 2 in 100,000 people affecting motor neurons in the spinal cord and the motor cortex. While the majority of patients suffer from the sporadic form of ALS, about 10 % of patients present with familial ALS caused by mutations in one of at least 32 known genetic loci [174] including superoxide dismutase 1 (SOD1; [145]), TAR DNA-binding protein 43 (TDP-43; [5, 121]), fused in sarcoma (FUS; [98, 185]) and C9ORF72 [51, 141].
ALS caused by mutant SOD1
Mutations in the SOD1 gene are found in about 20 % of patients with familial ALS and in approximately 2 % of all ALS patients [17, 23]. SOD1-related ALS has been the most studied form of ALS and presents with motor neuron degeneration as a result of mitochondrial dysfunction, protein misfolding, defects in axonal transport and disturbed signaling through the IGF1/PI3K/AKT pathways (for review see [88]). The first ALS-iPS cell lines were derived from two siblings, an 82- and an 89-year old, with an L144F mutation within the SOD1 gene [57] (Table S2). These iPS cells were differentiated into HB9+ and islet-1+ motor neurons and into GFAP+ astrocytes in vitro but disease phenotypes were not analyzed. In a follow-up study, the same group published additional iPS cell lines with the L144F mutation and also iPS cell lines with a G85S mutation in the SOD1 gene, and the neural differentiation potential of six ALS-iPS cell lines was compared to ten control iPS cells lines from healthy donors [18]. No differences in motor neuron differentiation were observed between these lines, although two ALS-iPS cell lines and one control iPS cell line could be differentiated into motor neurons only using an alternative differentiation protocol including dual inhibition of SMAD signaling in differentiating cells [18]. Furthermore, ALS-iPS cells carrying the L144F mutation were used to test the effect of a substance, kenpaullone, which appeared to be neuroprotective in a high-throughput screen of about 5,000 different compounds on motor neurons derived from mouse ES cells [195]. In this study, kenpaullone, an inhibitor of GSK-3 and HGK kinases, strongly improved the survival of motor neurons derived from L144F-SOD1-ALS-iPS cells and was more active than two compounds that recently failed in clinical trials on ALS patients [195]. These results highlight that patient-derived iPS cells may play a crucial role in drug discovery and in pre-clinical drug testing for the benefit of patients with neurodegenerative diseases.
As both cell-autonomous and non-cell-autonomous mechanisms of neurodegeneration have been found to contribute to ALS pathogenesis, stem cell-based models have been applied to address these effects in vitro. It is known that mutant SOD1 in astrocytes causes mitochondrial dysfunction and oxidative stress in neighboring motor neurons (non-cell-autonomous mechanism), while initiation of neuronal degeneration in SOD1-ALS is thought to be caused by an intrinsic, cell-autonomous mechanism within motor neurons themselves [15, 17, 55]. Non-cell-autonomous, mutant SOD1-mediated detrimental effects on motor neurons were shown in mouse [55] and also human [54] pluripotent stem cells that had been differentiated into mature motor neurons in vitro. Indeed, these studies demonstrated that pluripotent stem cell-derived motor neurons had an increased vulnerability and showed increased cell death when co-cultured with astrocytes derived from mice carrying the SOD1-G93A mutation [54, 55].
ALS caused by mutant TDP-43
TDP-43 is the transactive response (TAR) DNA-binding protein with a molecular weight of 43 kDa, a nuclear protein that plays a significant role in RNA metabolism including RNA transcription, splicing and transport [101, 130, 182]. Furthermore, TDP-43 is found in proteinaceous inclusion bodies in the cytosol of motor neurons in most forms of ALS, and mutations in the TDP-43 locus have been linked to the development of familial and sporadic ALS [173]. Indeed, more than 30 different mutations, including the M337V mutation, have been described in ALS patients so far [89, 173]. As seen in independent studies, motor neurons derived from M337V-TDP-43-iPS cells were more susceptible to cellular stress and showed increased vulnerability in different in vitro-culture assays including growth factor withdrawal, antagonism of the PI3K pathway or the application of the cytotoxic stressor arsenite [13, 63, 195] (Table S2). However, contradictory results were obtained regarding the viability of M337V-TDP-43 neurons under basal culture conditions eliciting discussions about clonal variations and the use of appropriate viability assays in the context of iPS cell-based disease modeling [13, 14, 63, 64]. M337V-TDP-43 motor neurons contained significantly higher amounts of soluble [13] and detergent-resistent, insoluble TDP-43 protein [13, 63] and demonstrated redistribution of TDP-43 protein from the nucleus to the cytosol with the formation of cytosolic TDP-43+ pre-inclusion-like aggregates [63]. In line with the role of TDP-43 in RNA metabolism, gene expression profiles in purified TDP-43-iPS cell-derived motor neurons revealed alterations in pathways mediating RNA processing, binding and splicing [63]. As seen in postmortem tissue of ALS patients, ALS-iPS cell-derived motor neurons expressed cytoskeletal intermediate filaments at lower levels when compared to control neurons and had impaired neurite outgrowth as similarly seen in a zebrafish model of ALS [63, 89]. Interestingly, these changes were also observed in iPS cell-derived motor neurons from ALS patients with the Q343R and the G298S TDP-43 mutations, which are also located within the glycine-rich domain of the TDP-43 protein [63, 129]. Similarly, intracellular TDP-43 aggregates were recently observed in iPS cell-derived motor neurons from patients with sporadic ALS [26]. Notably, a reversal of disease phenotypes in TDP-43-iPS cell-derived motor neurons could be achieved through the application of anacardic acid, an inhibitor of histone acetyl transferase [63].
While non-cell-autonomous effects have been demonstrated for SOD1-ALS, it is still unclear, if such mechanisms would also apply to other forms of ALS. Recently, a study by Serio et al. [156] addressed this topic and investigated the influence of M337V-TDP-43-iPS cell-derived astrocytes on healthy iPS cell-derived motor neurons. As shown for motor neurons with the M337V-TDP-43 mutation, M337V-TDP-43-iPS cell-derived astrocytes were more vulnerable and contained increased levels of soluble TDP-43 protein that was mislocalized from the nucleus to the cytoplasm [13, 156]. When co-cultured with M337V-TDP-43-iPS cell-derived astrocytes, however, healthy motor neurons did not show an increased vulnerability but behaved like neurons that had been co-cultured with healthy iPS cell-derived astrocytes. These iPS cell-based results demonstrated that neurodegeneration did not occur in a non-cell-autonomous fashion in M337V-TDP-43-ALS [156].
Models of frontotemporal dementia using patient-derived iPS cells
Pathological TDP-43 has also been linked to the development of frontotemporal lobe degeneration (FTLD-TDP), an ubiquitin-positive but tau- and SNCA-negative form of frontotemporal dementia (FTD). FTD is the second most common pre-senile dementia after AD and is clinically characterized by deficits in cognition, language and behavior due to pronounced atrophy in the frontal and temporal lobes [166]. In addition to mutations in the TDP-43 locus, mutations in the progranulin (PGRN) gene, intronic hexanucleotide repeat expansions within the C9ORF72 locus and mutations in the MAPT gene, besides others, have been linked to the development of FTD (for review see [135]).
FTD caused by mutant progranulin
PGRN mutations are seen in about 10 % of all FTD cases and cause haploinsufficiency of the PGRN gene with concomitant loss of PGRN function [6, 10, 47]. A recent publication by Almeida et al. [3] investigated pathologic alterations in iPS cell-derived cortical neurons from sporadic FTD patients and from FTD patients with the heterozygous S116X PGRN mutation (Table S2). This study demonstrated that patient-derived cortical neurons from both sporadic and S116X-PGRN-FTD patients were more susceptible to tunicamycin, an inhibitor of protein N-glycosylation within the ER, and to lactacystin, an inhibitor of the proteasome. Furthermore, S116X-PGRN-FTD-iPS cell-derived cortical neurons were more vulnerable towards staurosporine, towards the two PI3K-inhibitors wortmannin and LY294002 and towards the MEK/MAPK inhibitor PD98059 [3] demonstrating involvement of these pathways in PGRN-FTD pathogenesis. Interestingly, FTD-iPS cell-derived neurons exhibited stress-induced translocation of nuclear TDP-43 protein to the cytoplasm and this translocation of TDP-43 was observed in PGRN-FTD-iPS cell-derived neurons even in the absence of cellular stressors. These findings are in line with observations that the brains of FTD patients with PGRN mutations have TDP-43 pathology including accumulation of TDP-43 protein within the cytoplasmic compartment [6, 121]. As seen for other iPS cell models of neurodegeneration involving loss of gene function due to genetic mutations (e.g. PARKIN or PINK-1), genetic correction of PGRN haploinsufficiency through lentiviral overexpression of PGRN in PGRN-FTD-iPS cell-derived neurons resulted in reversal of most of the phenotypes described above [3].
FTD caused by mutant MAPT
Mutations in MAPT encoding the microtubule-associated protein tau constitute another cause of familial FTD and result in the accumulation of phosphorylated tau in neurons and glia in several brain areas including the frontal and temporal cortex as well as the substantia nigra [132, 165, 171]. Fong et al. [67] derived iPS cells from an individual with a heterozygous A152T MAPT mutation and generated isogenic control iPS cell lines using zinc-finger technology (Table S2). With this approach, pathologic changes were observed, which comprised increased fragmentation and enhanced phosphorylation of tau protein (Ser 202 and Thr 205) in mutated iPS cell-derived neurons, which were significantly reduced in gene-corrected cells [67]. On the other hand, introducing a second mutation in patient-derived iPS cells instead of genetic correction resulted in homozygous A152T MAPT and in even more pronounced pathologic changes in iPS cell-derived neurons, as evidenced by increased phosphorylation and enhanced fragmentation of tau protein [67].
FTD caused by mutant C9ORF72
The most common mutation in FTD and ALS is the GGGGCC hexanucleotide repeat expansion in the noncoding region of C9ORF72 [51, 113, 141]. Recently, several research groups generated iPS cells from patients carrying expanded C9ORF72 GGGGCC repeats and described disturbed RNA metabolism as a contributing pathogenic factor in C9ORF72-FTD/ALS [2, 58, 152] (Table S2). Indeed, while GGGGCC repeats showed instability upon reprogramming and during neuronal differentiation [2], a subset of patient-derived iPS cells [2] and neurons [2, 58, 152] exhibited nuclear RNA foci containing GGGGCC repeats, which were not detected in healthy control cells but have been described in some patients with C9ORF72-FTD [51]. To characterize potential binding partners of these intranuclear GGGGCC RNA transcripts, a proteome analysis on 16,368 proteins was performed on C9ORF72-iPS cell-derived neurons [58]. This screen revealed 19 candidates and included the RNA-binding protein ADARB2, which also colocalized to GGGGCC RNA foci in postmortem CNS tissue of C9ORF72 patients [58]. C9ORF72 neurons contained non-ATG translation (RAN) di-peptide (poly-Gly-Pro) repeats [2, 58], that have recently been observed in patients with FTD/ALS [8, 118]. These data indicated that pathologic changes in C9ORF72-FTD/ALS patients could be modeled in patient-specific iPS cells. That was further supported by the fact that C9ORF72-iPS cell-derived neurons revealed dysregulation of specific genes, which were similarly misexpressed in CNS tissue of C9ORF72 patients including NEDD4L, FAM3C, CHRDL1, SEPP1 and SERPINE2 [58]. Neurons also accumulated p62, a component in neuronal inclusions in C9ORF72 patients and a substrate of the autophagy pathway [1, 2]. In line with that, neurons had compromised autophagy function, as indicated by an increased vulnerability after exposure to the autophagy inhibitors chloroquine and 3-MA [2], and revealed an increased susceptibility to excitotoxic stress induced by glutamate [58]. Importantly, C9ORF72-associated RNA toxicity could be rescued by application of antisense oligonucleotides (ASO) targeting either the intronic GGGGCC repeat sequence or a C9ORF72 downstream sequence in exon 2 and appeared to be independent of C9ORF72 mRNA levels and accumulating RAN products [58]. An ASO-mediated rescue of repeat-associated neuronal disease phenotypes was similarly described in an independent study on C9ORF72-iPS cells [152].
Models of Huntington’s disease using patient-derived iPS cells
HD is a monogenetic, autosomal-dominant disease caused by CAG trinucleotide expansions in exon 1 of the huntingtin (HTT) gene [41]. It is characterized by a degeneration of medium-sized spiny projection neurons within the striatum and, at later stages of the disease, also by degeneration of neurons within the cerebral cortex [137].
IPS cells have been generated from patients carrying enhanced numbers of CAG repeats within the HTT gene ranging from 45, 60, 72 and 109 to 180 repeats [4, 29, 35, 38, 42, 84, 128, 198] (Table S2). In addition, iPS cells have been derived from patients with rare homozygous HTT CAG extensions (39/43 and 42/44 CAG repeats, respectively; [29]). These studies revealed disease phenotypes, which have previously been tied to HD pathogenesis, validating these stem cell-based models of HD. Indeed, it has been demonstrated that mutant HTT, through interaction with other HTT-binding proteins, causes decreased levels of intracellular ATP and increased caspase activity, disturbed mitochondrial function, impaired signaling by brain-derived neurotrophic factor (BDNF), perturbation of Ca2+ signaling and eventually excitotoxicity [49, 75, 199]. Likewise, HD-iPS cell-derived neurons showed increased caspase activity and cell death after prolonged culture or upon growth factor deprivation [4, 35, 42, 198], especially after removal of BDNF from the cell culture medium [42]. Furthermore, these cells presented with a decreased oxygen consumption rate along with decreased intracellular ATP concentrations, indicating an impairment of mitochondrial bioenergetics in patient-derived cells [4, 42]. In addition, HD-iPS cell-derived neural cells were more susceptible to H2O2-mediated oxidative stress and to excitotoxicity induced by pulsatile or chronic treatment with glutamate [42]. This effect was accompanied by disturbed Ca2+ homeostasis in challenged cells [42]. Finally, HD-iPS cell-derived neurons revealed an increased basal lysosomal activity [29] and an increased susceptibility to 3-MA [42]. These findings are in line with the observation that an increased number of autophagosome-like structures and thus disturbed protein clearance is found in the brains of HD patients [49, 150].
Disease-associated changes in stress-response levels could already be detected in undifferentiated iPS cells from HD patients [4, 35]. Comparative proteomic analysis among hES cells, healthy donor-derived iPS cells and iPS cells from HD patients revealed 26 proteins that were dysregulated in patient-derived cells. Among these proteins, the antioxidant enzymes SOD1, glutathione transferase (GST) and glutathione peroxidase 1 (Gpx1) were significantly downregulated, while the oxidative stress-response proteins Prx1, Prx2 and Prx6 were found in higher quantities in undifferentiated HD-iPS cells [35]. Notably, similar findings have also been observed in the striatum of HD patients [170]. Furthermore, gene array analysis was performed on undifferentiated iPS cells from HD patients that had been genetically corrected through bacterial artificial chromosome (BAC)-mediated homologous recombination to derive isogenic control lines [4]. This study demonstrated that TGF-β pathway molecules, cadherin family members and caspase-related signaling molecules were significantly altered in non-corrected HD-iPS cells as opposed to gene-corrected HD-iPS cells [4]. These pathways were also altered in alternative models of HD and in the brain of HD patients [12, 90, 140].
Whole-genome expression analysis was also performed on control- and HD-iPS cells that had been differentiated into neural precursor cells in vitro [42]. In this study, 1,601 genes were differentially regulated between control and patient-derived neural precursor cells and gene ontology analysis revealed dysregulated pathways involved in proliferation, cell cycle regulation, cell signaling, axonal guidance and cellular assembly [42]. Consistent with alterations in postmortem tissue from HD patients and HD transgenic mice, dysregulated genes in HD-iPS cell-derived neural precursor cells included UCHL1, EGFR (epidermal growth factor receptor), TRK (tyrosine kinase) receptors, p53, Syndecan4, SRPX and also the HMG box protein 1, which was found to accumulate in the brains of patients suffering from AD [42, 176].
The onset of HD and disease severity are dependent on the number of CAG repeats within the HTT gene. Thus, an interesting question was if disease phenotypes in HD-iPS cell-derived neural cells were also dependent on the length of CAG repeats. This question was addressed by the HD iPSC Consortium by comparing disease phenotypes in iPS cells derived from HD patients with either 60, 109 or 180 CAG repeats [42]. They found that all HD-iPS cell lines shared a similar cumulative risk of cell death and an impaired energy metabolism when compared to the control lines but they also observed that certain disease phenotypes such as responses to BDNF withdrawal and toxicity upon glutamate exposure were much more pronounced and robust in the HD-iPS cell line carrying the highest number (180) of CAG repeats in the HTT gene [42]. These results were thus consistent with observations and models of HD pathogenesis.
Intracytoplasmic and intranuclear aggregates of HTT protein are pathological hallmarks of HD [7]. Hence, it was examined if such changes also occur in differentiated iPS cells from HD patients. While intracellular aggregates did not appear under normal differentiation conditions in vitro, such inclusions were induced through the application of the proteasome inhibitor MG-132 to differentiating HD-iPS cells [38, 84]. Furthermore, intracellular aggregates of HTT protein were observed 33 weeks after injection of differentiated HD-iPS cells into the lateral ventricle of postnatal P2 mice [84]. These data show that pathological hallmarks were elicited in patient-derived neural cells and underscore that genetic correction of cells prior to transplantation might be necessary if cells were to be used for cell replacement therapy in regenerative paradigms. In this context, it should be noted that gene-corrected HD-iPS cell-derived neurons survived after transplantation in the striatum of R6/2 HD mice [111] and that HD-iPS cell-derived neurons mediated functional recovery in the quinolinic acid model of HD [84].
Models of spinal muscular atrophy using patient-derived iPS cells
Spinal muscular atrophy (SMA) is an autosomal-recessive neurodegenerative disease caused by mutations in the survival motor neuron 1 gene (SMN1) [104]. These mutations result in a significant reduction of the SMN1 protein and cause a selective degeneration of lower motor neurons within the anterior horn of the spinal cord. The SMN protein has several functions, including splicing of pre-messenger RNA and the biogenesis of small nuclear ribonucleoprotein particles [68, 82]. It also interacts with the 3′-untranslated region of β-actin mRNA in the axonal compartment of motor neurons [146]. Although SMA patients carry an intact SMN2 gene encoding an SMN2 protein with overlapping function to the SMN1 protein, only about 10 % of the SMN2 protein remains functional due to alternative splicing and degradation of truncated SMN2 protein, which cannot compensate for significantly reduced SMN1 protein levels in patients. The first human iPS cell models of SMA were published in 2009 by Ebert et al. [62] (Table S2). In this study, iPS cells were derived from a 3-year-old boy suffering from SMA. IPS cells from his unaffected mother served as controls. Quantification of full-length SMN1 protein revealed significantly decreased levels in SMA-patient-derived fibroblasts and iPS cells, while the levels of alternatively spliced SMN2 gene transcripts were unchanged in these cells. Thus, these findings highlighted that the typical transcriptional regulation of the SMN1 and SMN2 genes found in SMA patients in vivo was also reflected in patient-derived iPS cells in vitro [62]. The SMA- and control iPS cells were differentiated into HB9+, SMI-32+ and ChAT+ motor neurons, and motor neuron degeneration was observed in SMA-patient-derived cultures after 8, but not 4 weeks of differentiation [62]. This was indicated by reduction of the size, percentage and synaptic coverage of SMA motor neurons when compared to control iPS cell-derived neurons. These findings could be reproduced by other groups [37, 46] and also with iPS cells from a second patient, a 23-month-old boy with SMA [37, 151]. In addition, these studies demonstrated reduced neurite outgrowth in SMA-iPS cell-derived motor neurons [37, 46] and showed that motor neuron degeneration was associated with increased expression of the pro-apoptotic marker molecules cleaved caspase-3, cleaved caspase-8 and Fas ligand but not Bax, Bcl-2, cleaved caspase-9 or the apoptosis-inducing factor AIF [151]. These findings indicated death-receptor-mediated apoptosis rather than mitochondria-associated apoptosis in SMA-iPS cell-derived motor neurons [151]. In addition, SMA-iPS cell-derived astrocytes showed morphological changes and functional impairment, supporting the hypothesis that glial pathology might contribute to SMA development [116]. Some of the observed neuronal disease phenotypes could be rescued by the application of valproic acid and tobramycin to SMA-iPS cells [62], which increased the level of SMN protein in the context of SMA [22, 190], further indicating that SMA-iPS cells may constitute a suitable cell population for in vitro drug testing and screening in SMA. An overexpression of SMN protein in SMA-iPS cells resulted in correction of impaired neurite outgrowth and improved survival of SMA-iPS cell-derived motor neurons [37]. Furthermore, genetic modification of the SMN2 locus towards a more SMN1-like state was followed by the rescue of several disease phenotypes [46]. In this report, single-stranded DNA oligonucleotides with coding sequences of the SMN1 gene were introduced into undifferentiated SMA-iPS cells to generate isogenic control iPS cell lines with increased amounts of functional full-length SMN1 protein. This approach not only resulted in prevention of iPS cell-derived motor neuron degeneration but also in increased formation of neuromuscular junctions between motor neurons and myotubes in in vitro-co-culture assays [46]. Furthermore, gene array analysis on gene-corrected cells revealed rescued expression of differentially regulated genes involved in RNA metabolism, axonal guidance and motor neuron development. Notably, gene-corrected SMA-iPS cell-derived motor neurons showed improved engraftment after intraspinal transplantation into a mouse model of SMA and caused prolonged survival of these animals when compared to the transplantation of non-modified SMA-iPS cell-derived motor neurons [46]. These data again highlight the regenerative potential of gene-corrected patient-iPS cell-derived neurons in the context of cell therapy in neurodegenerative diseases.
Models of other rare neurodegenerative diseases using patient-derived iPS cells
IPS cell models of other monogenetic neurodegenerative diseases included models of triplet repeat diseases, lysosomal storage diseases and additional rare degenerative diseases of the central or peripheral nervous system (Table S2).
Triplet repeat diseases other than Huntington’s disease
Spinocerebellar atrophy type 3 (SCA-3) or Machado–Joseph disease is a slowly progressive neurodegenerative disease caused by CAG repeat expansions in the ataxin 3 (ATXN3) gene, which lead to deficits in gait coordination and to poor control of speech, eye and hand movements [70]. SCA-3-iPS cells were derived from four patients ranging between 38 and 42 years of age and were differentiated into mature neurons along with healthy control iPS cells. This report demonstrated formation of ATXN3-positive, SDS-insoluble aggregates in patient-iPS cell-derived neurons, which are typically found in patients’ tissue. The formation of aggregates was dependent on l-glutamine-induced excitation and on functional Na+ and K+ channels in addition to functional ionotropic and voltage-gated Ca2+ channels. Indeed, formation of SDS-insoluble ATXN3 aggregates was not observed in SCA3-fibroblasts, undifferentiated SCA3-iPS cells or SCA3-iPS cell-derived glial cells, consistent with the neuron-specific phenotype in SCA3. Furthermore, this report provided mechanistic insights into aggregate formation, as excitation-induced aggregation was preceded by Ca2+-dependent proteolysis of ATXN3. Notably, these SDS-insoluble ATXN3 aggregates disappeared after inhibition of calpain, but not after caspase inhibition, confirming a significant role of calpain protease in ATXN3 aggregation in the context of SCA3-disease development [93].
Friedreich ataxia (FA) is a disease with progressive spinocerebellar neurodegeneration due to GAA·TTC repeat expansions in the first intron of the frataxin (FXN) gene [31]. This mutation causes reduced expression of FXN protein within mitochondria and leads to altered cellular iron metabolism, mitochondrial dysfunction and increased sensitivity to oxidative stress [30, 154]. Several groups derived iPS cells from FA patients [61, 76, 97, 109] and found epigenetic silencing of the FXN locus [97] and decreased levels of FXN transcripts in undifferentiated [76, 97, 109] and differentiated iPS cells [76]. In addition, neurons presented with delayed maturation and decreased mitochondrial membrane potential [76]. Furthermore, these studies demonstrated instability of GAA·TTC repeats during reprogramming with extensions [61, 76, 97, 109] and contractions [76, 109] of these repeats, and also described GAA·TTC repeat extensions during prolonged culturing of iPS cells in vitro [61, 76]. These changes were associated with increased expression of the mismatch repair enzymes MSH2 and MSH6, as shRNA-mediated silencing of these two factors resulted in reduced GAA·TTC extensions during culture in vitro [61, 97]. These findings were consistent with the fact that GAA·TTC repeats in FA are genetically highly unstable and with the observation that mismatch repair enzymes are involved in the instability of trinucleotide repeat diseases [59, 95, 110]. Notably, such instability was not found in other iPS cell models of triplet repeat diseases including models of SCA-3 [93], spinal and bulbar muscular atrophy [125] or dentato-rubral-pallido-luysian dystrophy [125] (Table S2).
Storage diseases
Gaucher’s disease (GD) is a lysosomal storage disease caused by a mutation in the GBA-1 gene encoding acid-β-glucocerebrosidase [87]. This enzyme cleaves glucosylceramide into glucose and ceramide [20]. However in GD patients, the enzymatic activity is significantly decreased leading to the accumulation of glucosylceramide and glucosylsphingosine in lysosomes as well as to cellular degeneration in multiple brain areas [191]. So far, stem cell models of the acute (type 2) and the chronic (type 3) neuronopathic form of GD have been published [128, 181] (Table S2). While iPS cells from type 3 GD patients were characterized only at the undifferentiated stage [128], Tiscornia et al. [181] derived iPS cells from type 2 GD patients and described decreased levels of acid-β-glucocerebrosidase in iPS cells and differentiated DA neurons. Decreased acid-β-glucocerebrosidase activity in neurons was rescued by enzyme overexpression in undifferentiated cells and by the application of the two chaperone compounds 6S-AdBI-NJ and NOI-NJ, which were suggested to stabilize protein levels and to facilitate protein trafficking to the lysosome [181]. Thus, such iPS cell models could be useful in finding and testing potential small molecules as alternatives to enzyme replacement and substrate reduction therapy in GD patients. Other stem cell models of storage diseases included models of Pompe disease [78], Niemann–Pick type C1 disease [183] and X-linked adrenoleukodystrophy [83, 189]. Notably, intracellular accumulation of disease-specific products such as glycogen in Pompe disease [78], cholesterol in Niemann–Pick type C1 disease [183] or very long chain fatty acids in X-linked adrenoleukodystrophy [83] was recapitulated in patient-derived iPS cells, neurons or oligodendrocytes, respectively (Table S2).
Other rare degenerative diseases of the central or peripheral nervous system
Hereditary spastic paraplegia (HSP) is a group of neurodegenerative diseases associated with spasticity of the lower limbs as a result of degeneration of corticospinal motor neurons and the pyramidal tract [66]. The most common form of HSP (SPG4) is caused by an autosomal-dominant mutation in the SPAST gene encoding spastin. Similar to neurons in the brains of HSP patients [91], HSP-iPS cell-derived forebrain neurons presented with axonopathic changes including axonal swelling and an impairment of fast axonal transport of mitochondria along axons [52]. Interestingly, axonopathic changes in iPS cell-derived neurons could be rescued by vinblastine, which has previously been applied to reduce axonal swelling in SPG4-deficient primary neurons [65].
Familial dysautonomia (FD) is associated with a degeneration of neurons of the peripheral nervous system and is caused by a mutation in the I-κ-B kinase complex-associated protein (IKBKAP) [77]. As a consequence, missplicing of IKBKAP and a decrease of normal IKBKAP transcripts lead to impaired migration of disease-affected cells [40]. Several FD-iPS cell lines were differentiated into neural crest precursor cells and peripheral neurons [102]. Interestingly, FD-iPS cell-derived neural precursor cells also showed defects in IKBKAP splicing and impairment of migration in scratch wound assays. In addition, these cells presented with impaired differentiation into peripheral neurons, altogether providing an excellent human in vitro-model of FD. In an initial experiment, the plant hormone kinetin [164] proved successful in reducing the level of misspliced IKBKAP transcripts in FD-iPS cell-derived neural crest precursor cells. Furthermore, kinetin improved the differentiation of FD-iPS cells into peripheral neurons [102]. Based on this study, the same group performed high-throughput screenings on FD-iPS cell-derived neural crest precursor cells with approximately 7,000 compounds and found eight substances that decreased the levels of misspliced IKBKAP transcripts, while increasing the levels of WT-IKBKAP transcripts [103]. This study also unraveled the molecular targets of one of these hits, compound SKF-86466, highlighting the usefulness of patient-derived iPS cells for large-scale screening of compounds for the potential treatment of neurodegenerative diseases [103].
Human cell culture models of neurodegeneration using iN cells
During the last few years, progress has been made towards generating neuronal cell types from fibroblasts without passing through a stage of pluripotency. This can be achieved by transduction of fibroblasts with the neural transcription factors Ascl1, Brn2, Myt1l and NeuroD1 resulting in the derivation of functional neurons, which were termed induced neurons or iN cells [127] (Fig. 1). These iN cells were electrophysiologically active and expressed typical neuronal marker molecules such as MAP2, NeuN and synapsin [127]. Importantly, this technology can be applied to adult human fibroblasts isolated from skin biopsies of patients or healthy control individuals and thus has been used, with slight modifications, to model neurodegenerative changes in vitro (Table S3).
One report demonstrated the derivation of iN cells from two patients with familial forms of AD, who carried the A246E mutation in the PS1 gene or the N141I mutation in the PS2 gene, respectively [134]. These AD-iN cells were generated within 3 weeks of culture through transduction with the transcription factors Ascl1, Brn2, Myt1l, Olig2 and Zic1. AD-iN cells and control iN cells expressed markers of glutamatergic forebrain neurons and formed synaptic connections in in vitro-co-culture assays [134]. In comparison to their parental fibroblasts, iN cells from the AD patients and iN cells from control individuals expressed higher levels of APP and sAPPβ, while only the AD-iN cells expressed significantly higher amounts of Aβ40 and Aβ42 protein. Importantly, the Aβ42/Aβ40 ratio was significantly enhanced in AD-iN cells when compared to control iN cells, thus mimicking findings obtained from studies on AD-iPS cell-derived neurons described above. Likewise, AD-iN cells presented with an enlarged endocytic compartment with increased numbers of APP+ intracellular punctate [134]. Notably, this disease phenotype was rescued by transfection of AD-iN cells with a plasmid encoding non-mutated PS1 [134].
iN cells have also been derived from two patients with PD [28]. One patient carried a duplication of the SNCA locus, while the other PD patient had a mutation in the PARKIN gene. This study highlighted that functional DA neurons can be derived from human fibroblasts by direct transdifferentiation, which was achieved by transduction of fibroblasts with doxycycline-inducible lentiviruses encoding Ascl1, Nurr1 and Lmx1a. These DA-iN cells expressed typical DA marker molecules such as TH, ALDH-1A1, AADC, VMAT2 and DAT and showed electrophysiological activity [28]. Since DA-iN cells from PD patients and from control individuals were derived with similar efficiencies, these cells provide a suitable cell source for an in-depth analysis of disease-related phenotypes as described above for iPS cell-based models of PD.
These studies show that important progress has been made towards modeling neurodegenerative diseases using human cell-based systems. As iN cells are generated within only weeks by direct transdifferentiation of patients’ fibroblasts, disease modeling assays can be performed in a shorter time frame when compared to assays using iPS cells. However, due to rapid cell-type conversion, it is possible that cells do not properly pass through a neuronal step-by-step maturation program and that unconverted fibroblasts and partially reprogrammed cells remain in culture, which may influence outcomes of disease modeling assays using iN cells (for reviews see [149, 187]). Current research addresses these challenges. For instance, further development of this technology led to the derivation of neural progenitor cells termed induced neural stem cells (iNSCs), which can be propagated over long periods of time and which have the potential to differentiate into mature neuroectodermal cell types such as neurons, astrocytes and oligodendrocytes at higher numbers in vitro [71, 92, 142, 179]. Although human iNSC-based stem cell models of neurodegeneration have not been established yet, this technology could constitute a very important approach towards research and therapy in neurodegenerative diseases and could complement research on patient-derived iPS cells.
Conclusions
Research on patient-derived iPS cell lines has significantly increased in the past years (Fig. 3). Other neurologic diseases have also been modeled with iPS cells and include neurodevelopmental disorders such as Rett syndrome [96] or fragile X syndrome [184] and neuropsychiatric diseases such as schizophrenia [21]. Importantly, not only early-disease phenotypes of genetic, but also of sporadic, diseases can be captured with this approach and several studies have presented significant neuropathological changes in patient-iPS cell-derived neurons. These changes point towards underlying pathogenic mechanisms of disease development, as presented in this article. It is desirable that iPS cell research translates into clinical applications, where compounds are first tested in high-throughput screening systems to modify disease-affected pathways in vitro, and where suitable candidates would then be applied in clinical trials with the goal to facilitate the development of new therapies for the benefit of patients. While further improvements of neural differentiation protocols are needed to reduce cellular heterogeneity and to continuously refine sensitivities of assays and drug responses, other challenges have to be carefully addressed, which may critically impinge the discovery of disease phenotypes in patient-specific iPS cells. For instance, the heterogeneity of the patient pool, possible differences in the quality of fibroblasts for reprogramming and potential variabilities in differentiation capacities of iPS cells have to be considered. Hence, an analysis of a single iPS cell clone from only one single patient could yield different results when compared to an analysis of several iPS cell clones from several different patients with the same disease. This would, in turn, result in erroneous conclusions in disease modeling experiments (for review see [149]). Furthermore, the choice of control cell lines has a strong influence on disease modeling assays. The use of iPS cells from gender- and age matched control individuals and from individuals genetically related to the patient might not always be the most appropriate approach. When modeling monogenetic neurodegenerative diseases, isogenic, gene-corrected control cell lines can be generated, which carry the same genetic variants as their parental patient-derived cells and which differ from these cells only by the disease-causing mutation [149]. Hence, further progress has recently been pursued to more easily derive isogenic control lines, for example through the application of transcription activator-like effector nucleases (TALENs) [16] or by taking advantage of CAS/clustered regularly interspaced short palindromic repeats (CRISPR)-mediated genomic editing [86].
Altogether, these recent studies on human stem cell models of neurodegeneration demonstrate that iPS cell technology, in particular, carries a strong potential for biomedical research on human neurodegenerative diseases.
References
Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE (2011) p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 122(6):691–702. doi:10.1007/s00401-011-0911-2
Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126(3):385–399. doi:10.1007/s00401-013-1149-y
Almeida S, Zhang Z, Coppola G, Mao W, Futai K, Karydas A, Geschwind MD, Tartaglia MC, Gao F, Gianni D, Sena-Esteves M, Geschwind DH, Miller BL, Farese RV Jr, Gao FB (2012) Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep 2(4):789–798. doi:10.1016/j.celrep.2012.09.007
An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM (2012) Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11(2):253–263. doi:10.1016/j.stem.2012.04.026
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351(3):602–611. doi:10.1016/j.bbrc.2006.10.093
Armstrong RA, Carter D, Cairns NJ (2012) A quantitative study of the neuropathology of 32 sporadic and familial cases of frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP). Neuropathol Appl Neurobiol 38(1):25–38. doi:10.1111/j.1365-2990.2011.01188.x
Arrasate M, Finkbeiner S (2011) Protein aggregates in Huntington’s disease. Exp Neurol 238(1):1–11. doi:10.1016/j.expneurol.2011.12.013
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77(4):639–646. doi:10.1016/j.neuron.2013.02.004
Aubry L, Bugi A, Lefort N, Rousseau F, Peschanski M, Perrier AL (2008) Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci USA 105(43):16707–16712. doi:10.1073/pnas.0808488105
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442(7105):916–919. doi:10.1038/nature05016
Barnes DE, Yaffe K (2011) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10(9):819–828. doi:10.1016/S1474-4422(11)70072-2
Battaglia G, Cannella M, Riozzi B, Orobello S, Maat-Schieman ML, Aronica E, Busceti CL, Ciarmiello A, Alberti S, Amico E, Sassone J, Sipione S, Bruno V, Frati L, Nicoletti F, Squitieri F (2011) Early defect of transforming growth factor beta1 formation in Huntington’s disease. J Cell Mol Med 15(3):555–571. doi:10.1111/j.1582-4934.2010.01011.x
Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, Park IH, Friedman BA, Daley GQ, Wyllie DJ, Hardingham GE, Wilmut I, Finkbeiner S, Maniatis T, Shaw CE, Chandran S (2012) Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci USA 109(15):5803–5808. doi:10.1073/pnas.1202922109
Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, Park IH, Friedman BA, Daley GQ, Wyllie DJ, Hardingham GE, Wilmut I, Finkbeiner S, Maniatis T, Shaw CE, Chandran S (2013) Comment on “Drug screening for ALS using patient-specific induced pluripotent stem cells”. Sci Transl Med 5(188):188le182. doi:10.1126/scitranslmed.3005065
Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR (2008) Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem 107(5):1271–1283. doi:10.1111/j.1471-4159.2008.05699.x
Boch J (2011) TALEs of genome targeting. Nat Biotechnol 29(2):135–136. doi:10.1038/nbt.1767
Boillee S, Vande Velde C, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52(1):39–59. doi:10.1016/j.neuron.2006.09.018
Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, Rodolfa CT, Dimos JT, Mikkilineni S, MacDermott AB, Woolf CJ, Henderson CE, Wichterle H, Eggan K (2011) A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol 29(3):279–286. doi:10.1038/nbt.1783
Bradley CK, Scott HA, Chami O, Peura TT, Dumevska B, Schmidt U, Stojanov T (2010) Derivation of Huntington’s disease-affected human embryonic stem cell lines. Stem Cells Dev 20(3):495–502. doi:10.1089/scd.2010.0120
Brady RO, Gal AE, Kanfer JN, Bradley RM (1965) The metabolism of glucocerebrosides. 3. Purification and properties of a glucosyl- and galactosylceramide-cleaving enzyme from rat intestinal tissue. J Biol Chem 240(10):3766–3770
Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH (2011) Modelling schizophrenia using human induced pluripotent stem cells. Nature 473(7346):221–225. doi:10.1038/nature09915
Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B (2003) Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet 12(19):2481–2489. doi:10.1093/hmg/ddg256
Brown RH Jr (1997) Amyotrophic lateral sclerosis. Insights from genetics. Arch Neurol 54(10):1246–1250
Brunk I, Blex C, Speidel D, Brose N, Ahnert-Hilger G (2009) Ca2+-dependent activator proteins of secretion promote vesicular monoamine uptake. J Biol Chem 284(2):1050–1056. doi:10.1074/jbc.M805328200
Burger PC, Vogel FS (1973) The development of the pathologic changes of Alzheimer’s disease and senile dementia in patients with Down’s syndrome. Am J Pathol 73(2):457–476
Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M, Garnes J, Dang V, Lievers J, Shoukat-Mumtaz U, Martinez R, Gai H, Blake R, Vaisberg E, Grskovic M, Johnson C, Irion S, Bright J, Cooper B, Nguyen L, Griswold-Prenner I, Javaherian A (2013) A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci 56C:355–364. doi:10.1016/j.mcn.2013.07.007
Byers B, Cord B, Nguyen HN, Schule B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, Palmer TD (2011) SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One 6(11):e26159. doi:10.1371/journal.pone.0026159
Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, Sotnikova TD, Menegon A, Roncaglia P, Colciago G, Russo G, Carninci P, Pezzoli G, Gainetdinov RR, Gustincich S, Dityatev A, Broccoli V (2011) Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476(7359):224–227. doi:10.1038/nature10284
Camnasio S, Delli Carri A, Lombardo A, Grad I, Mariotti C, Castucci A, Rozell B, Lo Riso P, Castiglioni V, Zuccato C, Rochon C, Takashima Y, Diaferia G, Biunno I, Gellera C, Jaconi M, Smith A, Hovatta O, Naldini L, Di Donato S, Feki A, Cattaneo E (2012) The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis 46(1):41–51. doi:10.1016/j.nbd.2011.12.042
Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Durr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M (1997) Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet 6(11):1771–1780
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271(5254):1423–1427
Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ (2010) Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal 22(5):821–827. doi:10.1016/j.cellsig.2010.01.006
Cataldo A, Rebeck GW, Ghetri B, Hulette C, Lippa C, Van Broeckhoven C, van Duijn C, Cras P, Bogdanovic N, Bird T, Peterhoff C, Nixon R (2001) Endocytic disturbances distinguish among subtypes of Alzheimer’s disease and related disorders. Ann Neurol 50(5):661–665
Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA (2000) Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 157(1):277–286. doi:S0002-9440(10)64538-5
Chae JI, Kim DW, Lee N, Jeon YJ, Jeon I, Kwon J, Kim J, Soh Y, Lee DS, Seo KS, Choi NJ, Park BC, Kang SH, Ryu J, Oh SH, Shin DA, Lee DR, Do JT, Park IH, Daley GQ, Song J (2012) Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem J 446(3):359–371. doi:10.1042/BJ20111495
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27(3):275–280. doi:10.1038/nbt.1529
Chang T, Zheng W, Tsark W, Bates S, Huang H, Lin RJ, Yee JK (2011) Brief report: phenotypic rescue of induced pluripotent stem cell-derived motoneurons of a spinal muscular atrophy patient. Stem Cells 29(12):2090–2093. doi:10.1002/stem.749
Cheng PH, Li CL, Chang YF, Tsai SJ, Lai YY, Chan AW, Chen CM, Yang SH (2013) miR-196a Ameliorates Phenotypes of Huntington Disease in Cell, Transgenic Mouse, and Induced Pluripotent Stem Cell Models. Am J Hum Genet. doi:10.1016/j.ajhg.2013.05.025
Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schule B, Lippard SJ, Tsai LH, Krainc D, Buchwald SL, Jaenisch R, Lindquist S (2013) Identification and Rescue of alpha-Synuclein Toxicity in Parkinson Patient-Derived Neurons. Science. doi:10.1126/science.1245296
Close P, Hawkes N, Cornez I, Creppe C, Lambert CA, Rogister B, Siebenlist U, Merville MP, Slaugenhaupt SA, Bours V, Svejstrup JQ, Chariot A (2006) Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell 22(4):521–531. doi:10.1016/j.molcel.2006.04.017
Consortium H (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72(6):971–983. doi:0092-8674(93)90585-E
Consortium THi (2012) Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11(2):264–278. doi:10.1016/j.stem.2012.04.027
Cookson MR (2010) The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci 11(12):791–797. doi:10.1038/nrn2935
Cooper O, Hargus G, Deleidi M, Blak A, Osborn T, Marlow E, Lee K, Levy A, Perez-Torres E, Yow A, Isacson O (2010) Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol Cell Neurosci 45(3):258–266. doi:10.1016/j.mcn.2010.06.017
Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, Romero N, Jiang H, Uitti RJ, Huang Z, Opala G, Scarffe LA, Dawson VL, Klein C, Feng J, Ross OA, Trojanowski JQ, Lee VM, Marder K, Surmeier DJ, Wszolek ZK, Przedborski S, Krainc D, Dawson TM, Isacson O (2012) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med 4 (141):141ra190. doi:10.1126/scitranslmed.3003985
Corti S, Nizzardo M, Simone C, Falcone M, Nardini M, Ronchi D, Donadoni C, Salani S, Riboldi G, Magri F, Menozzi G, Bonaglia C, Rizzo F, Bresolin N, Comi GP (2012) Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci Transl Med 4 (165):165ra162. doi:10.1126/scitranslmed.3004108
Cruts M, Kumar-Singh S, Van Broeckhoven C (2006) Progranulin mutations in ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Curr Alzheimer Res 3(5):485–491
Cruts M, van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St George-Hyslop PH, Hofman A, Van Broeckhoven C (1998) Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet 7(1):43–51. pii:ddb004
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90(3):537–548. pii:S0092-8674(00)80513-9
de Lau LM, Breteler MM (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5(6):525–535. doi:10.1016/S1474-4422(06)70471-9
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256. doi:10.1016/j.neuron.2011.09.011
Denton KR, Lei L, Grenier J, Rodionov V, Blackstone C, Li XJ (2013) Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells. doi:10.1002/stem.1569
Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T (2011) Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun 2:440. doi:10.1038/ncomms1453
Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC (2008) Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3(6):637–648. doi:10.1016/j.stem.2008.09.017
Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (2007) Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 10(5):608–614. doi:10.1038/nn1885
Dickson DW (2001) Neuropathology of Alzheimer’s disease and other dementias. Clin Geriatr Med 17(2):209–228
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K (2008) Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321(5893):1218–1221. doi:10.1126/science.1158799
Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80(2):415–428. doi:10.1016/j.neuron.2013.10.015
Dragileva E, Hendricks A, Teed A, Gillis T, Lopez ET, Friedberg EC, Kucherlapati R, Edelmann W, Lunetta KL, MacDonald ME, Wheeler VC (2009) Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis 33(1):37–47. doi:10.1016/j.nbd.2008.09.014
Draper H, Chadwick R (1999) Beware! Preimplantation genetic diagnosis may solve some old problems but it also raises new ones. J Med Ethics 25(2):114–120
Du J, Campau E, Soragni E, Ku S, Puckett JW, Dervan PB, Gottesfeld JM (2012) Role of mismatch repair enzymes in GAA.TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J Biol Chem 287(35):29861–29872. doi:10.1074/jbc.M112.391961
Ebert AD, Yu J, Rose FF Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN (2009) Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457(7227):277–280. doi:10.1038/nature07677
Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, Aoi T, Watanabe A, Yamada Y, Morizane A, Takahashi J, Ayaki T, Ito H, Yoshikawa K, Yamawaki S, Suzuki S, Watanabe D, Hioki H, Kaneko T, Makioka K, Okamoto K, Takuma H, Tamaoka A, Hasegawa K, Nonaka T, Hasegawa M, Kawata A, Yoshida M, Nakahata T, Takahashi R, Marchetto MC, Gage FH, Yamanaka S, Inoue H (2012) Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med 4(145):145ra104. doi:10.1126/scitranslmed.3004052
Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, Aoi T, Watanabe A, Yamada Y, Morizane A, Takahashi J, Ayaki T, Ito H, Yoshikawa K, Yamawaki S, Suzuki S, Watanabe D, Hioki H, Kaneko T, Makioka K, Okamoto K, Takuma H, Tamaoka A, Hasegawa K, Nonaka T, Hasegawa M, Kawata A, Yoshida M, Nakahata T, Takahashi R, Marchetto MC, Gage FH, Yamanaka S, Inoue H (2013) Response to comment on “Drug screening for ALS using patient-specific induced pluripotent stem cells”. Sci Transl Med 5(188):188lr182. doi:10.1126/scitranslmed.3005697
Fassier C, Tarrade A, Peris L, Courageot S, Mailly P, Dalard C, Delga S, Roblot N, Lefevre J, Job D, Hazan J, Curmi PA, Melki J (2012) Microtubule-targeting drugs rescue axonal swellings in cortical neurons from spastin knockout mice. Dis Model Mech 6(1):72–83. doi:10.1242/dmm.008946
Fink JK (2002) Hereditary spastic paraplegia. Neurol Clin 20(3):711–726
Fong H, Wang C, Knoferle J, Walker D, Balestra ME, Tong LM, Leung L, Ring KL, Seeley WW, Karydas A, Kshirsagar MA, Boxer AL, Kosik KS, Miller BL, Huang Y (2013) Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports 1(3):1–9. doi:10.1016/j.stemcr.2013.08.001
Gabanella F, Butchbach ME, Saieva L, Carissimi C, Burghes AH, Pellizzoni L (2007) Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One 2(9):e921. doi:10.1371/journal.pone.0000921
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F et al (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373(6514):523–527. doi:10.1038/373523a0
Gatchel JR, Zoghbi HY (2005) Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 6(10):743–755. doi:10.1038/nrg1691
Han DW, Tapia N, Hermann A, Hemmer K, Hoing S, Arauzo-Bravo MJ, Zaehres H, Wu G, Frank S, Moritz S, Greber B, Yang JH, Lee HT, Schwamborn JC, Storch A, Scholer HR (2012) Direct reprogramming of fibroblasts into neural stem cells by defined factors. Cell Stem Cell 10(4):465–472. doi:10.1016/j.stem.2012.02.021
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. doi:10.1126/science.1072994
Hargus G, Cooper O, Deleidi M, Levy A, Lee K, Marlow E, Yow A, Soldner F, Hockemeyer D, Hallett PJ, Osborn T, Jaenisch R, Isacson O (2010) Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc Natl Acad Sci USA 107(36):15921–15926. doi:10.1073/pnas.1010209107
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41(10):1088–1093. doi:10.1038/ng.440
Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE, Hackam AS, Logvinova AV, Peel AL, Chen SF, Hook V, Singaraja R, Krajewski S, Goldsmith PC, Ellerby HM, Hayden MR, Bredesen DE, Ellerby LM (2004) Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington’s disease. Cell Death Differ 11(4):424–438. doi:10.1038/sj.cdd.4401358
Hick A, Wattenhofer-Donze M, Chintawar S, Tropel P, Simard JP, Vaucamps N, Gall D, Lambot L, Andre C, Reutenauer L, Rai M, Teletin M, Messaddeq N, Schiffmann SN, Viville S, Pearson CE, Pandolfo M, Puccio H (2013) Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis Model Mech 6(3):608–621. doi:10.1242/dmm.010900
Hims MM, Ibrahim EC, Leyne M, Mull J, Liu L, Lazaro C, Shetty RS, Gill S, Gusella JF, Reed R, Slaugenhaupt SA (2007) Therapeutic potential and mechanism of kinetin as a treatment for the human splicing disease familial dysautonomia. J Mol Med (Berl) 85(2):149–161. doi:10.1007/s00109-006-0137-2
Huang HP, Chen PH, Hwu WL, Chuang CY, Chien YH, Stone L, Chien CL, Li LT, Chiang SC, Chen HF, Ho HN, Chen CH, Kuo HC (2011) Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum Mol Genet 20(24):4851–4864. doi:10.1093/hmg/ddr424
Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, Nihira T, Kobayashi T, Ohyama M, Sato S, Takanashi M, Funayama M, Hirayama A, Soga T, Hishiki T, Suematsu M, Yagi T, Ito D, Kosakai A, Hayashi K, Shouji M, Nakanishi A, Suzuki N, Mizuno Y, Mizushima N, Amagai M, Uchiyama Y, Mochizuki H, Hattori N, Okano H (2012) Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 5:35. doi:10.1186/1756-6606-5-35
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS (2012) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482(7384):216–220. doi:10.1038/nature10821
Iwata A, Nagashima Y, Matsumoto L, Suzuki T, Yamanaka T, Date H, Deoka K, Nukina N, Tsuji S (2009) Intranuclear degradation of polyglutamine aggregates by the ubiquitin–proteasome system. J Biol Chem 284(15):9796–9803. doi:10.1074/jbc.M809739200
Jablonka S, Rossoll W, Schrank B, Sendtner M (2000) The role of SMN in spinal muscular atrophy. J Neurol 247(Suppl 1):I37–I42
Jang J, Kang HC, Kim HS, Kim JY, Huh YJ, Kim DS, Yoo JE, Lee JA, Lim B, Lee J, Yoon TM, Park IH, Hwang DY, Daley GQ, Kim DW (2011) Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann Neurol 70(3):402–409. doi:10.1002/ana.22486
Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, Shim SH, Choi C, Chang DJ, Kwon J, Oh SH, Shin DA, Kim HS, Do JT, Lee DR, Kim M, Kang KS, Daley GQ, Brundin P, Song J (2012) Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 30(9):2054–2062. doi:10.1002/stem.1135
Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, Azabdaftari G, Nakaso K, Yan Z, Feng J (2012) Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun 3:668. doi:10.1038/ncomms1669
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821. doi:10.1126/science.1225829
Jmoudiak M, Futerman AH (2005) Gaucher disease: pathological mechanisms and modern management. Br J Haematol 129(2):178–188. doi:10.1111/j.1365-2141.2004.05351.x
Joyce PI, Fratta P, Fisher EM, Acevedo-Arozena A (2011) SOD1 and TDP-43 animal models of amyotrophic lateral sclerosis: recent advances in understanding disease toward the development of clinical treatments. Mamm Genome 22(7–8):420–448. doi:10.1007/s00335-011-9339-1
Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Vande Velde C, Rouleau GA, Drapeau P (2010) Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet 19(4):671–683. doi:10.1093/hmg/ddp534
Kandasamy M, Couillard-Despres S, Raber KA, Stephan M, Lehner B, Winner B, Kohl Z, Rivera FJ, Nguyen HP, Riess O, Bogdahn U, Winkler J, von Horsten S, Aigner L (2010) Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease. J Neuropathol Exp Neurol 69(7):717–728. doi:10.1097/NEN.0b013e3181e4f733
Kasher PR, De Vos KJ, Wharton SB, Manser C, Bennett EJ, Bingley M, Wood JD, Milner R, McDermott CJ, Miller CC, Shaw PJ, Grierson AJ (2009) Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J Neurochem 110(1):34–44. doi:10.1111/j.1471-4159.2009.06104.x
Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S, Lipton SA, Zhang K, Ding S (2011) Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci USA 108(19):7838–7843. doi:10.1073/pnas.1103113108
Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, Doerr J, Ladewig J, Mertens J, Tuting T, Hoffmann P, Klockgether T, Evert BO, Wullner U, Brustle O (2011) Excitation-induced ataxin-3 aggregation in neurons from patients with Machado–Joseph disease. Nature 480(7378):543–546. doi:10.1038/nature10671
Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, Takahashi K, Asaka I, Aoi T, Watanabe A, Watanabe K, Kadoya C, Nakano R, Watanabe D, Maruyama K, Hori O, Hibino S, Choshi T, Nakahata T, Hioki H, Kaneko T, Naitoh M, Yoshikawa K, Yamawaki S, Suzuki S, Hata R, Ueno S, Seki T, Kobayashi K, Toda T, Murakami K, Irie K, Klein WL, Mori H, Asada T, Takahashi R, Iwata N, Yamanaka S, Inoue H (2013) Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12(4):487–496. doi:10.1016/j.stem.2013.01.009
Kramer PR, Pearson CE, Sinden RR (1996) Stability of triplet repeats of myotonic dystrophy and fragile X loci in human mutator mismatch repair cell lines. Hum Genet 98(2):151–157
Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480(7378):547–551. doi:10.1038/nature10648
Ku S, Soragni E, Campau E, Thomas EA, Altun G, Laurent LC, Loring JF, Napierala M, Gottesfeld JM (2010) Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAATTC triplet repeat instability. Cell Stem Cell 7(5):631–637. doi:10.1016/j.stem.2010.09.014
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208. doi:10.1126/science.1166066
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41(10):1094–1099. doi:10.1038/ng.439
Lazarov O, Demars MP (2012) All in the family: how the APPs regulate neurogenesis. Front Neurosci 6:81. doi:10.3389/fnins.2012.00081
Lee EB, Lee VM, Trojanowski JQ (2011) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13(1):38–50. doi:10.1038/nrn3121
Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L (2009) Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461(7262):402–406. doi:10.1038/nature08320
Lee G, Ramirez CN, Kim H, Zeltner N, Liu B, Radu C, Bhinder B, Kim YJ, Choi IY, Mukherjee-Clavin B, Djaballah H, Studer L (2012) Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol 30(12):1244–1248. doi:10.1038/nbt.2435
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80(1):155–165 pii:0092-8674(95)90460-3
Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18 (R1):R48-59. doi:10.1093/hmg/ddp012
Li J, Uversky VN, Fink AL (2001) Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 40(38):11604–11613 pii:bi010616g
Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64(6):807–827. doi:10.1016/j.neuron.2009.11.006
Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, Wagner U, Kim A, Ren B, Li Y, Goebl A, Kim J, Soligalla RD, Dubova I, Thompson J, Yates J 3rd, Esteban CR, Sancho-Martinez I, Izpisua Belmonte JC (2012) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491(7425):603–607. doi:10.1038/nature11557
Liu J, Verma PJ, Evans-Galea MV, Delatycki MB, Michalska A, Leung J, Crombie D, Sarsero JP, Williamson R, Dottori M, Pebay A (2011) Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev 7(3):703–713. doi:10.1007/s12015-010-9210-x
Lopez Castel A, Cleary JD, Pearson CE (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11(3):165–170. doi:10.1038/nrm2854
Ma L, Hu B, Liu Y, Vermilyea SC, Liu H, Gao L, Sun Y, Zhang X, Zhang SC (2012) Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell 10(4):455–464. doi:10.1016/j.stem.2012.01.021
MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52(4):587–593. doi:10.1016/j.neuron.2006.10.008
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11(4):323–330. doi:10.1016/S1474-4422(12)70043-1
Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K (2003) The role of alpha-synuclein in Parkinson’s disease: insights from animal models. Nat Rev Neurosci 4(9):727–738. doi:10.1038/nrn1199
Mateizel I, De Temmerman N, Ullmann U, Cauffman G, Sermon K, Van de Velde H, De Rycke M, Degreef E, Devroey P, Liebaers I, Van Steirteghem A (2006) Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum Reprod 21(2):503–511. doi:10.1093/humrep/dei345
McGivern JV, Patitucci TN, Nord JA, Barabas ME, Stucky CL, Ebert AD (2013) Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 61(9):1418–1428. doi:10.1002/glia.22522
Mitne-Neto M, Machado-Costa M, Marchetto MC, Bengtson MH, Joazeiro CA, Tsuda H, Bellen HJ, Silva HC, Oliveira AS, Lazar M, Muotri AR, Zatz M (2011) Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum Mol Genet 20(18):3642–3652. doi:10.1093/hmg/ddr284
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125):1335–1338. doi:10.1126/science.1232927
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803. doi:10.1083/jcb.200809125
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8(1):e1000298. doi:10.1371/journal.pbio.1000298
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133. doi:10.1126/science.1134108
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, Palmer TD, Pera RR (2011) LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8(3):267–280. doi:10.1016/j.stem.2011.01.013
Niclis J, Trounson AO, Dottori M, Ellisdon A, Bottomley SP, Verlinsky Y, Cram D (2009) Human embryonic stem cell models of Huntington disease. Reprod Biomed Online 19(1):106–113
Niclis JC, Pinar A, Haynes JM, Alsanie W, Jenny R, Dottori M, Cram DS (2013) Characterization of forebrain neurons derived from late-onset Huntington’s disease human embryonic stem cell lines. Front Cell Neurosci 7:37. doi:10.3389/fncel.2013.00037
Nihei Y, Ito D, Okada Y, Akamatsu W, Yagi T, Yoshizaki T, Okano H, Suzuki N (2013) Enhanced aggregation of androgen receptor in induced pluripotent stem cell-derived neurons from spinal and bulbar muscular atrophy. J Biol Chem 288(12):8043–8052. doi:10.1074/jbc.M112.408211
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16(4):394–406. doi:10.1038/nn.3350
Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, Wernig M (2011) Induction of human neuronal cells by defined transcription factors. Nature 476(7359):220–223. doi:10.1038/nature10202
Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ (2008) Disease-specific induced pluripotent stem cells. Cell 134(5):877–886. doi:10.1016/j.cell.2008.07.041
Pesiridis GS, Lee VM, Trojanowski JQ (2009) Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18(R2):R156–R162. doi:10.1093/hmg/ddp303
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14(4):459–468. doi:10.1038/nn.2779
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43(6):815–825. doi:10.1002/ana.410430617
Popescu IR, Nicaise C, Liu S, Bisch G, Knippenberg S, Daubie V, Bohl D, Pochet R (2013) Neural progenitors derived from human induced pluripotent stem cells survive and differentiate upon transplantation into a rat model of amyotrophic lateral sclerosis. Stem Cells Transl Med 2(3):167–174. doi:10.5966/sctm.2012-0042
Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, Doege C, Chau L, Aubry L, Vanti WB, Moreno H, Abeliovich A (2011) Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell 146(3):359–371. doi:10.1016/j.cell.2011.07.007
Rademakers R, Neumann M, Mackenzie IR (2012) Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 8(8):423–434. doi:10.1038/nrneurol.2012.117
Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL (2007) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66(1):75–85. doi:10.1097/nen.0b013e31802d6da9
Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB (1988) Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 85(15):5733–5737
Reinhardt P, Glatza M, Hemmer K, Tsytsyura Y, Thiel CS, Hoing S, Moritz S, Parga JA, Wagner L, Bruder JM, Wu G, Schmid B, Ropke A, Klingauf J, Schwamborn JC, Gasser T, Scholer HR, Sterneckert J (2013) Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One 8(3):e59252. doi:10.1371/journal.pone.0059252
Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, Hoing S, Hargus G, Heck SA, Dhingra A, Wu G, Muller S, Brockmann K, Kluba T, Maisel M, Kruger R, Berg D, Tsytsyura Y, Thiel CS, Psathaki OE, Klingauf J, Kuhlmann T, Klewin M, Muller H, Gasser T, Scholer HR, Sterneckert J (2013) Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12(3):354–367. doi:10.1016/j.stem.2013.01.008
Reis SA, Thompson MN, Lee JM, Fossale E, Kim HH, Liao JK, Moskowitz MA, Shaw SY, Dong L, Haggarty SJ, MacDonald ME, Seong IS (2011) Striatal neurons expressing full-length mutant huntingtin exhibit decreased N-cadherin and altered neuritogenesis. Hum Mol Genet 20(12):2344–2355. doi:10.1093/hmg/ddr127
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72(2):257–268. doi:10.1016/j.neuron.2011.09.010
Ring KL, Tong LM, Balestra ME, Javier R, Andrews-Zwilling Y, Li G, Walker D, Zhang WR, Kreitzer AC, Huang Y (2012) Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 11(1):100–109. doi:10.1016/j.stem.2012.05.018
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316(5825):750–754. doi:10.1126/science.1141736
Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song YQ, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P (2007) The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39(2):168–177. doi:10.1038/ng1943
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362(6415):59–62. doi:10.1038/362059a0
Rossoll W, Jablonka S, Andreassi C, Kroning AK, Karle K, Monani UR, Sendtner M (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163(4):801–812. doi:10.1083/jcb.200304128
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, Canals JM, Memo M, Alberch J, Lopez-Barneo J, Vila M, Cuervo AM, Tolosa E, Consiglio A, Raya A (2012) Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4(5):380–395. doi:10.1002/emmm.201200215
Sanders LH, Laganiere J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, Langston JW, Gregory PD, Zhang HS, Greenamyre JT, Isacson O, Schule B (2013) LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis. doi:10.1016/j.nbd.2013.10.013
Sandoe J, Eggan K (2013) Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat Neurosci 16(7):780–789. doi:10.1038/nn.3425
Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M (1997) Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol 42(4):604–612. doi:10.1002/ana.410420411
Sareen D, Ebert AD, Heins BM, McGivern JV, Ornelas L, Svendsen CN (2012) Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 7(6):e39113. doi:10.1371/journal.pone.0039113
Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5(208):208ra149. doi:10.1126/scitranslmed.3007529
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2(8):864–870
Schmucker S, Puccio H (2010) Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum Mol Genet 19(R1):R103–R110. doi:10.1093/hmg/ddq165
Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D (2011) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31(16):5970–5976. doi:10.1523/JNEUROSCI.4441-10.2011
Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura AL, Carrasco MA, Phatnani HP, Shum C, Wilmut I, Maniatis T, Shaw CE, Finkbeiner S, Chandran S (2013) Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci USA 110(12):4697–4702. doi:10.1073/pnas.1300398110
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375(6534):754–760. doi:10.1038/375754a0
Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ (2012) A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med 4(124):124ra129. doi:10.1126/scitranslmed.3003771
Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ (2012) Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci 15(3):477–486. doi:10.1038/nn.3041 (S471)
Shih JC, Chen K, Ridd MJ (1999) Monoamine oxidase: from genes to behavior. Annu Rev Neurosci 22:197–217. doi:10.1146/annurev.neuro.22.1.197
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25(3):302–305. doi:10.1038/77060
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41(12):1308–1312. doi:10.1038/ng.487
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841. doi:10.1126/science.1090278
Slaugenhaupt SA, Mull J, Leyne M, Cuajungco MP, Gill SP, Hims MM, Quintero F, Axelrod FB, Gusella JF (2004) Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum Mol Genet 13(4):429–436. doi:10.1093/hmg/ddh046
Slowinski J, Dominik J, Uitti RJ, Ahmed Z, Dickson DD, Wszolek ZK (2007) Frontotemporal dementia and Parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology 27(1):73–80
Snowden JS, Neary D, Mann DM (2002) Frontotemporal dementia. Br J Psychiatry 180:140–143
Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, Isacson O, Jaenisch R (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136(5):964–977. doi:10.1016/j.cell.2009.02.013
Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, Zhang L, Guschin D, Fong LK, Vu BJ, Meng X, Urnov FD, Rebar EJ, Gregory PD, Zhang HS, Jaenisch R (2011) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146(2):318–331. doi:10.1016/j.cell.2011.06.019
Sonntag KC, Pruszak J, Yoshizaki T, van Arensbergen J, Sanchez-Pernaute R, Isacson O (2007) Enhanced yield of neuroepithelial precursors and midbrain-like dopaminergic neurons from human embryonic stem cells using the bone morphogenic protein antagonist noggin. Stem Cells 25(2):411–418. doi:10.1634/stemcells.2006-0380
Sorolla MA, Reverter-Branchat G, Tamarit J, Ferrer I, Ros J, Cabiscol E (2008) Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med 45(5):667–678. doi:10.1016/j.freeradbiomed.2008.05.014
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95(13):7737–7741
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840. doi:10.1038/42166
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319(5870):1668–1672. doi:10.1126/science.1154584
Sreedharan J, Brown RH Jr (2013) Amyotrophic lateral sclerosis: problems and prospects. Ann Neurol 74(3):309–316. doi:10.1002/ana.24012
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. doi:10.1016/j.cell.2006.07.024
Takata K, Kitamura Y, Tsuchiya D, Kawasaki T, Taniguchi T, Shimohama S (2004) High mobility group box protein-1 inhibits microglial Abeta clearance and enhances Abeta neurotoxicity. J Neurosci Res 78(6):880–891. doi:10.1002/jnr.20340
Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120(4):545–555. doi:10.1016/j.cell.2005.02.008
Tardiff DF, Jui NT, Khurana V, Tambe MA, Thompson ML, Chung CY, Kamadurai HB, Kim HT, Lancaster AK, Caldwell KA, Caldwell GA, Rochet JC, Buchwald SL, Lindquist S (2013) Yeast reveal a “Druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science. doi:10.1126/science.1245321
Thier M, Worsdorfer P, Lakes YB, Gorris R, Herms S, Opitz T, Seiferling D, Quandel T, Hoffmann P, Nothen MM, Brustle O, Edenhofer F (2012) Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell 10(4):473–479. doi:10.1016/j.stem.2012.03.003
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM (1998) Embryonic stem cell lines derived from human blastocysts. Science 282(5391):1145–1147
Tiscornia G, Vivas EL, Matalonga L, Berniakovich I, Barragan Monasterio M, Eguizabal C, Gort L, Gonzalez F, Ortiz Mellet C, Garcia Fernandez JM, Ribes A, Veiga A, Izpisua Belmonte JC (2012) Neuronopathic Gaucher’s disease: induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum Mol Genet 22(4):633–645. doi:10.1093/hmg/dds471
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14(4):452–458. doi:10.1038/nn.2778
Trilck M, Hubner R, Seibler P, Klein C, Rolfs A, Frech MJ (2013) Niemann–Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J Rare Dis 8(1):144. doi:10.1186/1750-1172-8-144
Urbach A, Bar-Nur O, Daley GQ, Benvenisty N (2010) Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6(5):407–411. doi:10.1016/j.stem.2010.04.005
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. doi:10.1126/science.1165942
Verlinsky Y, Strelchenko N, Kukharenko V, Rechitsky S, Verlinsky O, Galat V, Kuliev A (2005) Human embryonic stem cell lines with genetic disorders. Reprod Biomed Online 10(1):105–110
Vierbuchen T, Wernig M (2011) Direct lineage conversions: unnatural but useful? Nat Biotechnol 29(10):892–907. doi:10.1038/nbt.1946
Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107(1):378–383. doi:10.1073/pnas.0911187107
Wang XM, Yik WY, Zhang P, Lu W, Dranchak PK, Shibata D, Steinberg SJ, Hacia JG (2012) The gene expression profiles of induced pluripotent stem cells from individuals with childhood cerebral adrenoleukodystrophy are consistent with proposed mechanisms of pathogenesis. Stem Cell Res Ther 3(5):39. doi:10.1186/scrt130
Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL (2005) A non-sequence-specific requirement for SMN protein activity: the role of aminoglycosides in inducing elevated SMN protein levels. Hum Mol Genet 14(9):1199–1210. doi:10.1093/hmg/ddi131
Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, Morrison A, Lwin A, Colegial C, Allman JM, Schiffmann R (2004) Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 82(3):192–207. doi:10.1016/j.ymgme.2004.04.011
Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, Proud CG (2001) The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J 355(Pt 3):609–615
Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N (2011) Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet 20(23):4530–4539. doi:10.1093/hmg/ddr394
Yagi T, Kosakai A, Ito D, Okada Y, Akamatsu W, Nihei Y, Nabetani A, Ishikawa F, Arai Y, Hirose N, Okano H, Suzuki N (2012) Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS One 7(7):e41572. doi:10.1371/journal.pone.0041572
Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ, Ngo HD, Rosowski KA, Schein PA, Ackeifi CA, Arvanites AC, Davidow LS, Woolf CJ, Rubin LL (2013) A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12(6):713–726. doi:10.1016/j.stem.2013.04.003
Young JE, Goldstein LS (2012) Alzheimer’s disease in a dish: promises and challenges of human stem cell models. Hum Mol Genet 21(R1):R82–R89. doi:10.1093/hmg/dds319
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55(2):164–173. doi:10.1002/ana.10795
Zhang N, An MC, Montoro D, Ellerby LM (2010) Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr 2:RRN1193. doi:10.1371/currents.RRN1193
Zuccato C, Valenza M, Cattaneo E (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev 90(3):905–981. doi:10.1152/physrev.00041.2009
Acknowledgments
We would like to thank Dr. Jared Sterneckert, Dr. Holm Zaehres and Dr. Vincent Ruland for critical reading of this manuscript and Dr. Peter Reinhardt for providing images of differentiated neural cells. This work was supported by the IMF at University Hospital Münster (I-HA-111219; to GH) and the German Research Foundation (DFG; SFB-TRR128-B7; to TK).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hargus, G., Ehrlich, M., Hallmann, AL. et al. Human stem cell models of neurodegeneration: a novel approach to study mechanisms of disease development. Acta Neuropathol 127, 151–173 (2014). https://doi.org/10.1007/s00401-013-1222-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1222-6